

Abstract
The clinical application of cis-diamminedichloroplatinum(II) (DDP, cisplatin) for cancer therapy is limited by its non-specific biodistribution and severe side effects. Here, we have developed EGFR-targeted heparin-DDP (EHDDP) nanoparticles for tumor targeted delivery of DDP. This nanoparticle delivery system possesses the following unique properties: i) the succinic anhydride-modified heparin is biocompatible and biodegradable with no anticoagulant activity; ii) the single chain variable fragment anti-EGFR antibody (ScFvEGFR) was conjugated to the nanoparticles as an EGFR-targeting ligand. Our results showed that EHDDP nanoparticles can significantly increase the intracellular concentrations of DDP and Pt-DNA adducts in EGFR-expressing non-small cell lung cancer H292 cells via an EGFR-mediated pathway. Compared to the free DDP, significantly prolonged blood circulation time and improved pharmacokinetics and biodistribution of Pt were observed after systemic delivery of the EHDDP nanoparticles. The new EHDDP nanoparticle delivery system significantly enhanced antitumor activity of DDP without weight loss or damage to the kidney and spleen in nude mice bearing H292 cell tumors.
Keywords: cisplatin, heparin, tumor targeting, drug delivery, nanoparticles, lung cancer, EGFR
The anticancer drug cis-diamminedichloridoplatinum(II) (cisplatin, DDP) has been used in the treatment of different kinds of solid tumors including head and neck squamous cell carcinoma, ovarian and non-small cell lung cancers (NSCLC).1 However, the more extensive use of DDP is limited by its severe side effects, such as nephrotoxicity, peripheral neuropathy, and ototoxicity, which may result from its non-specific systemic organ distribution and inadequate intra-tumor concentrations.2–5 Nanoparticles have been demonstrated to significantly improve drug specificity of action due to nanoparticle-facilitated changes in tissue distribution and pharmacokinetics of drugs.6–9 Nanoparticles may reach certain solid tumors via the enhanced permeability and retention effect (EPR). Non-targeted nanoparticles are usually absent in the tumor sites due to their lack of cellular uptake, while the tumor-targeted nanoparticles can enter tumor cells via receptor-mediated internalization.10 The tumor targeted delivery of DDP has the potential to significantly reduce toxicity, improving its therapeutic efficacy.11 A variety of tumor targeting ligands, such as antibodies, peptides and small molecules, have been used to facilitate the uptake of nanoparticles into target cells.12 However, there are still many challenges in engineering tumor-targeted nanoparticles for the selective delivery of DDP in vivo. First, the nanoparticles should be biocompatible and biodegradable, non-toxic and of small size; second, the DDP must be loaded on the nanoparticles with high affinity to avoid its premature release before entering tumor cells; third, the targeted receptor should be located on the tumor cell surface and be able to mediate the endocytosis of DDP-loaded nanoparticles.
Overexpression of the epidermal growth factor receptor (EGFR) has been found in about 40–80% of NSCLC tissues, making it an ideal tumor target for constructing nanoparticles for targeted NSCLC therapy.13–16 Although an EGFR monoclonal antibody has been widely used to engineer tumor-targeted nanoparticles for either tumor imaging or selective drug delivery,17–21 the high molecular weight of the full-length antibody may limit its penetration into tumor tissue, and the specificity of tumor-targeted nanoparticles may be affected by the interaction of whole antibody with Fc receptors on normal tissues. In addition, high cost also limits the wide use of this antibody as a ligand for tumor-targeted nanoaprticles. To overcome these problems, single-chain antibodies against EGFR (ScFvEGFR, MW 25KDa), which contain the specific EGFR binding region but lack the Fc region, have been isolated and their specificity of binding and internalization demonstrated.22, 23 There are many advantages in using ScFv rather than intact antibody as a tumor targeting ligand, such as 1) low molecular weight; 2) small size; 3) less immunogenicity; 4) less accumulation in normal organs, especially bone marrow.24 Although its binding affinities are lower than that of the intact antibody, the ScFv conjugated to the nanoparticle surface demonstrates multivalent binding to receptors given the multiple copies of ScFvEGFR on the nanoparticle surface, which facilitates greater functional affinity.23, 25 Thus the antibody fragments are particularly suitable for engineering targeted nanoparticles.
In this study, we have developed a class of novel ScFvEGFR-Heparin nanoparticles for targeted delivery of DDP to EGFR-positive tumor cells. The feasibility of ScFvEGFR-Heparin-DDP (EHDDP) nanoparticles as a nano-sized drug carrier was evaluated in terms of drug loading efficiency, sustained drug release profile, and cytotoxicity in vitro. Also, the real time biodistribution of EHDDP nanoparticles in vivo was analyzed. The tumor targeting ability of EHDDP nanoparticles was demonstrated by the quantification of tumor-localized platinum (Pt) using ICP-MS in tumor-bearing mice. Finally, the antitumor efficacy and toxicity of EHDDP nanoparticles in nude mice bearing human NSCLC tumors was evaluated.
RESULTS
Formulation and Characterization of HDDP and EHDDP nanoparticles
To demonstrate that DDP and heparin are able to assemble into nanoparticles through coordination between the carboxyl groups and Pt2+, pure heparin and DDP were mixed in distilled water under gentle stirring. Dynamic light scattering (DLS) measurement was used to follow the formation of nanoparticles. Narrow dispersed nanoparticles were formed with an average size around 20±5 nm after 24 hrs as observed by DLS. The results suggest that the heparin-DDP complex forms due to the substitution of two chlorides of the DDP by the carboxyl group of the heparin. To generate EHDDP, ScFvEGFR was chemically conjugated onto the surface of the HDDP nanoparticle in the presence of EDAC and NHS (Figure 1A). The final concentration of Pt in HDDP and EHDDP nanoparticles was about 0.20 ± 0.03 mg/ml, as detected by ICP-MS. Based on the ICP-MS results, 30% of the DDP was loaded into the EHDDP nanoparticles, which demonstrated higher loading capability, and the molar ratio of ScFvEGFR:Heparin:DDP was about 0.8:100:30. The DLS showed that the size of HDDP was 20±5 nm, while that of EHDDP was 150±10 nm. There are two possible reasons for the size change: 1. since the size of the conjugates is measured by dynamic light scattering, the light scatterings properties may change after the conjugation of ScFvEGFR – which leads to large DLS size; and 2. in order to conjugate ScFvEGFR onto the surface of Heparin-Cisplatin nanoparticles, EDAC and NHS was used as catalysts. It could potentially cause the further chemical reactions between COOH and OH group on the surface of HDDP nanoparticle, which led to the size increase. . Both nanoparticles had a surface charge of about −5mV.
Figure 1. Preparation of HDDP and EHDDP nanoparticles.

(A) Schematic representation of HDDP and EHDDP nanoparticle formulations. (B) In vitro Pt release from EHDDP nanoparticles. The Pt loaded in the EHDDP nanoparticles shows sustained release in PBS (pH=7.4) at 37°C, while the nanoparticles are relatively stable in distilled water.
As shown in Figure 1B, 50% of the DDP was released within 72 hrs in PBS, suggesting sustained drug release. This led us to hypothesize that the HDDP and EHDDP are reactivated by exchanging the −COOH with the chloride in PBS. However, since the in vitro conditions are different from those in tumor cells, we further studied the mechanism of drug release from the nanoparticles in vivo.
EHDDP nanoparticles can increase Pt accumulation and inhibit the proliferation of tumor cells
It has been reported that the intracellular concentration of Pt may be related to the cytotoxicity of DDP.1, 2 To determine whether EHDDP nanoparticles could deliver more Pt to EGFR-positive H292 cells, we used ICP-MS to quantify the intracellular concentration of Pt. Figure 2A shows that the intracellular Pt level was significantly higher in EHDDP-treated cells (27.91± 2.45 ng Pt/106 cells) than in cells treated with free DDP (2.49± 0.39 ng Pt/106 cells, P=0.004) or HDDP (2.14± 0.20 ng Pt/106 cells, P=0.003) after 24 hrs, while the internalization of Pt in free DDP-treated cells was not significantly different than that in cells treated with non-targeted DDP nanoparticles (P=0.25). We speculate that free DDP enters the cells mainly by free diffusion, while EHDDP nanoparticles rapidly bind to the EGFR on the surface of H292 cells and the EGFR-EHDDP complex is then internalized into the cells via an EGFR-mediated pathway. This was supported by a competition experiment, which showed that pre-incubation of H292 cells with free ScFvEGFR inhibited the uptake of Pt in H292 cells treated with EHDDP nanoparticles from 27.91± 2.45 ng Pt/106 cells to 9.40± 1.48 ng Pt/106 cells. (P=0.011). In addition, EGFR-negative NSCLC H520 cells showed only a limited increase in Pt accumulation when treated with EHDDP nanoparticles (4.18± 0.29 ng Pt/106 cells) compared with free DDP (3.38± 0.42 ng Pt/106 cells) (P=0.28), further demonstrating that the internalization of EHDDP nanoparticles was EGFR-dependent. To further explore the targeting specificity of EHDDP nanoparticles in vitro, EGFR siRNA was used to knock down EGFR expression in H292 cells. EGFR expression was inhibited by about 90%, and the level of Pt internalization in EHDDP-treated H292/EGFR-siRNA cells was significantly decreased (4.25± 0.85 ng Pt/106cells) compared with that in H292/scrambled control siRNA cells (29.86± 3.62ng Pt/106 cells) after treatment with EHDDP for 24 hrs (Figure 2B).
Figure 2. Intracellular Pt accumulation, cytotoxicity and Pt-DNA adducts in vitro.

(A) Accumulation of Pt in H292 (EGFR-positive) and H520 (EGFR-negative) NSCLC cells treated with DDP, HDDP and EHDDP nanoparticles as described in materials and methods.(B) Effect of knockdown of EGFR on the internalization of EHDDP nanoparticles in NCI-H292 cells. (C) Percentage of viable cells measured by SRB assay in H292 cells treated with different concentrations of DDP, HDDP and EHDDP nanoparticles for 96hrs on 96-well plates, as described in materials and methods. (D) Quantification of Pt in Pt-DNA adducts and expression of phosphorylation of H2A.X in H292 cells treated with DDP, HDDP and EHDDP nanoparticles as described in materials and methods. The data are shown as mean ± SD (n=3), *P < 0.05, **P < 0.01
An SRB assay was used to measure the cytotoxicity of free DDP, HDDP and EHDDP in H292 cells at 96 hrs after treatment. The IC50 of EHDDP (Pt concentration: 1.08±0.12ug/ml) in H292 cells was significantly lower than that of DDP (2.12±0.20ug/ml, P=0.03) and HDDP (2.33 ±0.22ug/ml, P=0.01). As shown in Figure 2C, the inhibition of cell proliferation by EHDDP was significantly greater than that of free DDP and non-targeted HDDP nanoparticles.
EHDDP nanoparticles increase the formation of intracellular Pt-DNA adducts
The increase in intracellular Pt concentration seen in H292 cells treated with EHDDP nanoparticles, measured by ICP-MS, reflects the enhanced internalization of the total DDP loaded in the nanoparticles, not only the DDP activated from the nanoparticles, yet only this activated DDP can enter the nucleus and form Pt-DNA adducts. To quantify the levels of bioavailable DDP in the internalized EHDDP nanoparticles, we incubated H292 cells with free DDP, EGFR-targeted or non-targeted DDP nanoparticles at 37°C for 24hrs, followed by purifying the total DNA of treated cells and measuring the Pt concentration of DNA using ICP-MS. Our data showed that the Pt level associated with cellular DNA was significantly greater in EHDDP-treated cells (110.93±12.89pg Pt/ug DNA) than in cells treated with DDP (19.68±4.35pg Pt/ug DNA, P=0.003) or HDDP (22.17±3.37 pg Pt/ug DNA, P=0.002), while free DDP did not show any significant difference in DNA-associated Pt levels than HDDP (P=0.2) (Figure 2D). Phosphorylation of H2A.X can act as a highly sensitive and general marker of DNA damage induced by DDP26, therefore we assessed the phosphorylation of H2A.X in H292 cells treated with DDP, HDDP and EHDDP nanoparticle (0.4ug/ml) for 24 hrs. The expression level of phosphorylated H2A.X was significantly higher in EHDDP-treated cells than in DDP- or HDDP-treated cells (Figure 2D), which was consistent with the level of Pt-DNA adduct formation detected by ICP-MS.
EHDDP nanoparticles did not show any anticoagulant activity
Heparin exerts its anticoagulant activity by reversibly binding to antithrombin III. We modified the structure of heparin by using succinic anhydride to inactivate its anticoagulant function. The anticoagulant activities of EHDDP were determined by FXa-dependent coagulant assay. Our data showed that the EHDDP nanoparticles had undetectable anticoagulant activity, while that of unmodified heparin was 178U/mg.
EHDDP nanoparticles prolonged blood circulation time and changed the biodistribution of Pt in vivo
The plasma Pt level was measured by ICP-MS after intravenous injection of free DDP, HDDP and EHDDP nanoparticles to determine the blood half times of these three drugs as 2, 12 and 15 mins, respectively. Figure 3A shows that the HDDP and EHDDP nanoparticles significantly increased the circulation time of Pt: 30 mins after systemic administration, the Pt concentration in mice treated with free DDP was 1.17±0.12 ng/mg blood compared with 12.31±3.2 ng/mg in mice treated with EHDDP (P=0.003). After 8 hrs the Pt concentration further decreased in DDP-treated mice (0.36±0.09 ng/mg blood), while mice treated with EHDDP nanoparticles maintained a relatively higher Pt level (5.61±1.22 ng/mg blood, P=0.006). The results also showed that the ScFvEGFR on the surface of heparin nanoparticles had significant effects on the Pt concentration (12.31±3.2 ng/mg blood) in blood compared with the HDDP (17.55±0.48 ng/mg blood, P=0.016) 30 mins after iv injection, while no significant differences were found after 60 mins (P=0.07).
Figure 3. Biodistribution of Pt in mice treated with DDP, HDDP and EHDDP nanoparticles.

Pt concentrations are shown in peripheral blood (A), liver, spleen, kidney and tumor (B) of nude mice bearing H292 tumors at different time points after IV injection of a single dose of DDP, HDDP and EHDDP nanoparticles (Pt 2.5 mg/kg, 3 mice per group). The data are shown as mean ± SD, *P < 0.05, **P < 0.01.
To study the effects of the HDDP and EHDDP nanoparticles on the biodistribution of Pt in vivo, we harvested the main organs including the liver, spleen and kidney at different time points (10mins, 30mins, 4hrs and 24hrs) after the mice were injected intravenously with a single dose of free DDP and DDP-loaded nanoparticles (Pt 2.5mg/kg), and quantified the Pt concentrations in the tissue samples using ICP-MS. Figure 3B shows that significantly greater Pt accumulation was seen in the liver and spleen of HDDP and EHDDP nanoparticle-treated mice than in animals treated with free DDP, indicating that HDDP and EHDDP nanoparticles were taken up by the reticuloendothelial system (RES) after their systemic administration. In contrast, most of the free Pt is eliminated through glomerular filtration. The accumulation of Pt in the liver 4hrs following HDDP and EHDDP delivery was 2.7-fold (20.81±2.59 ng/mg dry tissue) and 4-fold (30.88±5.07 ng/mg dry tissue), respectively, greater than that of free DDP (7.71±1.61ng/mg dry tissue) (P=0.0027, P=0.0018 respectively). The accumulation of Pt in the spleen 4hrs following HDDP and EHDDP delivery was 18.6-fold (23.51±3.56 ng/mg dry tissue) and 22.1-fold (28.16±5.68 ng/mg dry tissue), respectively, greater than that of free DDP (1.27±0.19ng/mg dry tissue) (P=0.008, P=0.0017 respectively). It should be noted that the changes in biodistribution of Pt in the liver and spleen delivered by free DDP, HDDP and EHDDP were significantly different during the first 4 hrs after administration. The free DDP-induced Pt accumulation in the liver and spleen significantly decreased from 10mins to 4hrs (42.5%, P=0.033, 82.86%, P=0.0015, respectively), while EHDDP induced significantly increased Pt accumulation between 10mins and 4hrs in the liver and spleen (90.2%, P=0.0067, 88.9%, P=0.022, respectively). After 24hrs, the Pt concentration in the liver significantly decreased in both DDP-treated mice (20% decrease compared with 4 hrs, P=0.007) and EHDDP-treated mice (30% decrease, P=0.0036), and the Pt concentration in the spleen also significantly decreased in HDDP- (23.2% decrease, P=0.042) and EHDDP- (28.1% decrease, P=0.034) treated groups. We speculate that the majority of the HDDP and EHDDP nanoparticles are still intact within 4 hrs, and are degraded gradually in vivo. There were no differences in Pt concentrations delivered by DDP, HDDP and EHDDP in the liver, spleen and kidney 28 days after treatment (supplementary data, figure 1).
Since the kidney is the main organ that eliminates Pt, and the renal toxicity induced by DDP is due to damage caused by Pt to proximal tubular cells and the dilated tubular lumen, we investigated the accumulation of Pt in the kidney at different time points following delivery of free DDP, HDDP and EHDDP nanoparticles. As shown in Figure 3B, 10 minutes after systemic administration of DDP, HDDP, and EHDDP, the Pt concentrations in mice kidney were 35.25±5.70, 39.48±3.52 (P=0.079), and 41.74±2.93 (P=0.18) ng/mg dry tissue, respectively. The Pt concentrations decreased in the kidney over time; after 4hrs, the HDDP- and EHDDP nanoparticle-treated groups showed significantly higher Pt concentrations (22.1.6±2.82 and 31.7± 4.03 ng/mg dry tissue) than free DDP (11. 9±0.79 ng/mg dry tissue) (P=0.013, P=0.019, respectively), and after 24hrs, the HDDP- and EHDDP nanoparticle-treated groups still showed significantly higher Pt concentrations (19.6±3.44 and 16.09± 1.96 ng/mg dry tissue) than in the free DDP group (6.39±0.67 ng/mg dry tissue) (P=0.014, P=0.024, respectively). Whether the accumulation of Pt in the kidney relates to renal toxicity remains unknown;27, 28 it has been reported that the Pt-DNA adduct but not the total tissue Pt levels could provide a more useful correlation with the renal damage induced by Pt.29
One of the main advantages of using targeted nanoparticles for drug delivery is that they can selectively accumulate in the tumor sites. We collected tumor tissues at different time points (10mins, 30mins, 4hrs and 24hrs) after iv injection of DDP, HDDP and EHDDP nanoparticles, and measured the total Pt concentrations. As Figure 3B shows, after 10 mins the tumor Pt levels were significantly higher in mice treated with EHDDP nanoparticles (10.01±1.21 ng/mg dry tissue) than with free DDP (3.58±0.91ng/mg dry tissue, P=0.034) or HDDP (1.55±0.18ng/mg dry tissue, P=0.004), while there was no significant difference between the DDP and HDDP groups (P=0.084). After 4 hrs, the tumor levels of Pt increased in mice treated with HDDP and EHDDP nanoparticles, but decreased in mice treated with DDP; furthermore, the targeted EHDDP nanoparticles induced greater Pt tumor accumulation (31.3±4.7ng/mg dry tissue) than the non-targeted HDDP nanoparticles (14.68±2.52ng/mg dry tissue, P=0.0056), and HDDP delivered significantly higher levels of Pt to the tumor mass than free DDP (P=0.008). After 24 hrs, the Pt levels in tumors of all groups decreased, however the accumulation of Pt in the EHDDP group (13.45±2.15ng/mg dry tissue) was still significantly higher than that in the HDDP group (4.87±0.67ng/mg dry tissue, P=0.026) or the DDP group (1.48±0.52ng/mg dry tissue, P=0.02).
Since only the activated Pt can form intracellular Pt-DNA adducts, we purified DNA from the kidney at 4 and 24 hrs after treatment, and quantified the Pt level in the total DNA. As shown in Figure 4, the Pt concentrations after 4 and 24 hours were significantly higher in DNA purified from mice kidney treated with DDP (71.78±9.02, 25.72±2.78 pg/ug DNA, respectively) than those treated with HDDP (32.91±5.07, 14.88±1.71pg/ug DNA, P=0.003, P=0.0039 respectively) or EHDDP (38.61±6.01, 5.87±1.30 pg/ug DNA, P=0.042, P=0.001 respectively). We speculated that most of the Pt in HDDP and EHDDP nanoparticles was not released after 24 hrs in vivo, but will release gradually. We further quantified the level of Pt in Pt-DNA adducts in the NSCLC xenograft tumors at 4 and 24 hours after treatment with DDP, HDDP and EHDDP nanoparticles (single dose iv injection, Pt 2.5mg/kg), and our results (Figure 4) showed that the formation of Pt-DNA adducts in DDP-treated tumors (92.21±8.96pg/ugDNA) was significantly greater than that in HDDP- (26.09±5.15pg/ugDNA, P=0.0029) and EHDDP-treated (28.21±2.06pg/ugDNA, P=0.0038) groups after 4 hrs. After 24hrs, the EHDDP nanoparticle-treated tumors showed significantly increased formation of Pt-DNA adducts (163.12±26.59 pg/ug DNA) than that of DDP-(94.68±15.01pg/ugDNA, P=0.0039) and HDDP-treated (55.01±7.72 pg/ugDNA, P=0.0061) tumors.
Figure 4. Pt-DNA adducts in kidney and tumor.

Pt concentration in Pt-DNA adducts in the kidney and tumor of nude mice bearing H292 tumors at 4 and 24 hrs after IV injection of a single dose of DDP, HDDP and EHDDP nanoparticles (Pt 2.5mg/kg, 3 mice per group). The data are shown as mean ± SD, *P < 0.05, **P < 0.01.
EHDDP nanoparticles enhance antitumor effects while significantly reducing the toxicity to kidney and spleen in vivo
Nude mice bearing human NSCLC H292 tumors received treatment with free DDP, HDDP and EHDDP nanoparticles (Pt: 0.3mg/kg, equal to 2.5mg/kg DDP, 5 i.v doses, 3-day intervals). EHDDP nanoparticles showed a significant decrease in the tumor growth rate as compared with the HDDP (P<0.01) and free DDP (P<0.05) groups, while the non-targeted HDDP nanoparticles did not enhance the anti-tumor effects of DDP (Figure 5A). Although the HDDP nanoparticles carried more Pt into tumor cells than the free DDP, the level of bioavailable Pt was postulated to be less than with free DDP. Although the majority of the non-targeted HDDP nanoparticles will diffuse away from the tumor site, some nanoparticles can enter the tumor cells by passive diffusion (EPR effect) and release a relatively lower concentration of Pt, which may have some effect on tumor growth.
Figure 5. Effects of DDP, HDDP and EHDDP nanoparticles on tumor growth and body weight.
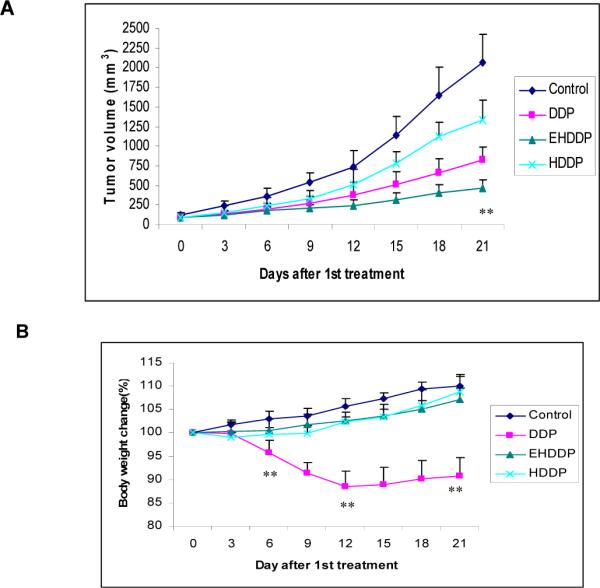
Tumor growth rate (A) and body weight change (%) (B) of nude mice treated with DDP, HDDP and EHDDP nanoparticles (Pt 2.5 mg/kg, 5 injections, 3-day intervals, 6 mice per group). The data are shown as mean ± SD, *P < 0.05, **P < 0.01.
Mice receiving the free DDP showed a significant loss of body weight, with about 5% weight loss by the 6th day (p=0.002), and 12.7% by the 12th day (P<0.001) after treatment. In contrast, mice treated with HDDP and EHDDP nanoparticles did not show any significant weight loss compared with the control group which received saline injection (Figure 5B).
All the mice were sacrificed after 21 days of treatment, since the tumor volumes in the saline control group had reached 2000 cm3. The mice blood, liver, spleen and kidney were collected. A significant shrinkage of the spleen was found in mice treated with free DDP; the spleen weight (104.94± 21.92mg) was 47.2% lower than that of the control group (197.83± 31.05mg, P=0.0005), while spleen weights in the HDDP (190.72± 19.62mg) and EHDDP (206.82± 35.89mg) groups did not show any significant change (P=0.35, P=0.66, respectively) (Figure 6A, B). Damage to the spleen was further confirmed by histopathological changes, which showed a marked hypocellularity in the white pulp and aggregation of neutrophils in the red pulp with minor necrosis in mice treated with DDP but not HDDP or EHDDP (Figure 6C, D, E, F).
Figure 6. Comparison of side effects of DDP, HDDP and EHDDP nanoparticles in the spleen.

The weight change (A), actual size and shape of the spleen (B), and the histopathological results in the spleen 21 days after treatment with saline (C), DDP (D), HDDP (E), and EHDDP (F) nanoparticles (Pt 2.5 mg/kg, 5 injections, 3-day intervals, 6 mice per group), original magnification, ×200. The data are shown as mean ± SD, **P < 0.01.
Serum glutamic pyruvate transaminase (GPT) and glutamic oxaloacetic transaminase (GOT) levels were quantified to evaluate mice hepatic function. The GPT levels did not change significantly in any of the treated groups (DDP: 48± 7.04U/I; HDDP: 41.33± 11.64U/I; EHDDP: 44± 9.64U/I) compared with the control group (40.66± 10.24U/I). The GOT level in DDP-treated mice was 148.6± 32.71U/I, which was not significantly different from that in the control group (138 ± 30.08U/I) (P=0.35), or the HDDP (154.6± 22.75U/I) and EHDDP (144.3± 28.1U/I) groups (P=0.41, P=0.43, respectively). Consistent with this finding, our histopathological study did not show any significant changes in any of the treated groups (Supplementary data, Figure 2) compared with the control group under the microscope.
Serum blood urea nitrogen (BUN) and creatinine (CRE) levels were measured to assess renal function, and the DDP-treated mice showed a significantly higher concentration of BUN (62.67± 14.37mg/dl) than the control group (19.67± 3.73mg/dl, P=0.0076), while levels of BUN in mice treated with HDDP (19.33± 3.24mg/dl) and EHDDP (17.67± 2.084mg/dl) nanoparticles were not significantly increased compared with the control group (P=0.29, P=0.35, respectively, Figure 7A). Serum CRE levels did show an increase in DDP-treated mice (0.36± 0.05 mg/dl) compared with the control (0.233± 0.057 mg/dl,), although the difference was not significant, and the HDDP (0.25± 0.053mg/dl) and EHDDP (0.23± 0.046mg/dl) groups did not show any significant changes in CRE levels compared with the control group (P=0.18, P=0.39, respectively, Figure 7B). The significantly elevated BUN in DDP-treated mice indicated damage to renal function, although the serum CRE level did not increase significantly compared with the control group, and such impaired renal excretion of urea may be due to dehydration resulting from the DDP treatment. Kidney toxicity was evidenced by the significant loss of kidney weight in the DDP-treated mice (277.21± 45.27mg), 25.56% lower than that in the control mice (372.4± 37.68mg, P=0.013), while no kidney weight loss was seen in HDDP (390.33± 19.08mg) and EHDDP (384.84± 54.95mg) groups compared with the control group (P=0.24, P=0.63. respectively), as shown in Figure 7C, D. Histopathology analysis showed the structure of glomeruli in the DDP-treated group did not change noticeably, but the epithelial cells in the convoluted proximal tubule became swollen and enlarged, and some of the tubular lumen disappeared (Figure 7F). In contrast, the HDDP (Figure 7G) and EHDDP nanoparticles (Figure 7H) did not induce any significant changes in kidney histopathology compared with the control group (Figure 7E).
Figure 7. Comparison of side effects of DDP, HDDP and EHDDP nanoparticles in the kidney.

Plasma BUN (A) and CRE (B) levels, the weight change (C) and actual size of the kidney (D), the histopathological results in the kidney of nude mice bearing H292 tumors 21 days after treatment with saline (E), DDP (F), HDDP (G) and EHDDP (H), original magnification, ×200. The data are shown as mean ± SD, *P < 0.05, **P < 0.01.
DISCUSSION
Many different types of nanoparticle have been studied as carriers for DDP delivery, with the majority being non-targeted nanoparticles9, 30–38 that can increase the local concentration of Pt in the tumor mass mainly via the EPR effect. However, these delivery systems only allow a proportion of the drug-loaded nanoparticles to enter the tumor cells, due to their lack of targeting. Once free drug molecules dissociate from nanoparticles in the extracellular space, some may diffuse freely between the tumor and the tumor vessel. Tumor-targeted nanoparticles can be internalized via the receptor-mediated pathway, thus resulting in higher intracellular nanoparticle accumulation than that of non-targeted nanoparticles.39, 40 The development of tumor-targeted nanoparticles for selective DDP delivery in vivo remains challenging.
In the present study, we have developed a novel EGFR-targeted nanoparticle system based on heparin for DDP delivery. The formation of nanoparticles was demonstrated by chemical and biological assessments both in vitro and in vivo. There are many advantages of our novel nanoparticle, 1) the nanoparticle backbone, heparin, is biocompatible, biodegradable, and water-soluble, and this chemically modified heparin did not show any anticoagulant activity in our previous studies;41 2) the relatively small size and low molecular weight ScFvEGFR is easy to obtain at lower cost, and its binding specificity to EGFR has been demonstrated;25 the simple covalent attachment of ScFvEGFR to nanoparticles produced a conjugate that could be internalized into EGFR-positive cells; 4) DDP was conjugated to the nanoparticles via exchange reaction between a −COOH group in the nanoparticle and chloride ions in DDP, and was able to be released from the nanoparticles.32, 42 In addition, the Pt concentration can be quantified by inductively coupled plasma–mass spectrometry (ICP-MS),43–45 which makes it possible to measure the exact Pt concentration in samples (cells, blood, DNA and organs). This assay possesses many advantages, which include; 1) high sensitivity, levels as low as 2ppb Pt can be measured, 2) high specificity due to the absence of background Pt in samples, 3) consistency and reproducibility.
Studies of the biodistribution of targeted nanoparticles have yielded controversial results regarding the accumulation and internalization of nanoparticles in tumors. Some studies have suggested that the targeted nanoparticles did not accumulate to higher concentrations in tumor sites, but could be internalized more by tumor cells,46, 47 while others demonstrated that the tumor-targeted nanoparticles did selectively accumulate in the tumor mass at significantly higher concentrations compared with non-targeted nanoparticles.12, 25, 39, 40, 48 These discrepancies may be explained by the different nanoparticles and ligands used. In our system, we expected that the tumor-targeted DDP-loaded nanoparticles could be delivered to the tumor sites quickly and be further taken up by tumor cells, so that the drug would be released intracellularly but not extracellularly, avoiding the unnecessary accumulation of nanoparticles in the body. We therefore did not modify the surface of HDDP and EHDDP nanoparticles with PEG. We found that both the EHDDP and HDDP nanoparticles led to increased Pt concentrations in tumor sites compared with the free DDP, while there were significant differences between EHDDP and HDDP, and HDDP had less effect on the tumor growth rate compared with free DDP. We postulated that the majority of the non-targeted HDDP nanoparticles were located extracellularly rather than being internalized by the tumor cells, while only the EGFR-targeted EHDDP nanoparticles were substantially taken up by tumor cells, enabling the Pt to target the nucleus to form Pt-DNA adducts. The first step involved in nanoparticles reaching the tumor site depends mainly on their blood circulation time and the EPR effect, while active targeting only occurs after the passive accumulation of nanoparticle in tumor sites. The longer the circulation time, the more opportunity for the nanoparticles to find leaky sites in the blood vessels, therefore the HDDP and EHDDP nanoparticles could accumulate in the tumor at relatively high concentrations due to their longer blood circulation time than free DDP. Once the non-targeted nanoparticles extravasate, they would huddle around the leaky sites of tumor blood vessels, further inhibiting the continuous accumulation of nanoparticle in tumor sites, and so the majority of the nanoparticles would be located in the tumor extracellular fluid but not inside the tumor cells. In contrast, the EGFR-targeted nanoparticles are internalized by the tumor cells via the EGFR-mediated pathway, allowing the greater accumulation of nanoparticles in the tumor.
Little is known about how the DDP enters tumor cells and forms Pt-DNA adducts, however both passive diffusion and active transport are speculated to be involved. It is accepted that DDP is inactive when dissolved in 0.9% saline due to the relatively high concentration of chloride ions, and that once it has entered cells, it is activated because of the relatively lower concentration of chloride ions (3–20mM), and is then available to form Pt-DNA adducts.1, 2 However, one study has shown that there are no significant differences in anti-tumor effect between saline- and water-dissolved DDP.49 In this study, we loaded DDP into the heparin nanoparticles by the exchange reaction between the −COOH in the heparin and chloride ions in the DDP, the loading efficacy was about 30%, and the nanoparticles were dissolved in distillated water. Our hypothesis is that the DDP may be reactivated in an environment with higher chloride ion concentration (100mM), this was supported by our observation of the sustained release of Pt in PBS (50% released at 72 hours). Since the blood half-time is only 12 mins for EHDDP and 15 mins for HDDP nanoparticles, the nanoparticles would be still intact before entering the tumor site, thus avoiding premature drug release. We expect that the heparin backbone of the nanoparticles may be biodegraded within the tumor cells, allowing Pt to be released in the cells.
Our study showed that the targeted delivery of DDP by EHDDP nanoparticles significantly reduced related toxicity to the kidney and spleen. The accumulation of DDP-loaded nanoparticles in organs (liver, spleen and kidney) and the tumor may be affected by different factors. We suspect that the accumulation of EHDDP in tumor sites depends mainly on active targeting, as the specific binding may increase the nanoparticle retention in the tumor sites, while free DDP can be eliminated by the renal system very quickly (<5mins).50, 51 There are many factors involved in the distribution of nanoparticles and their toxicity in vivo, including size, surface charge, blood circulation time, drug release profile and metabolism process. The liver and spleen uptake of targeted- and non-targeted nanoparticles play important roles in their biodistribution, and the drug-loaded nanoparticles showed a significantly increased accumulation of DDP in both liver and spleen. We speculated that the majority of the nanoparticles were intact and may be captured by macrophages. Renal toxicity is considered to depend on the peak urinary DDP concentration and on the maximum DDP level in the uriniferous tubules. Our results showed that the retention of Pt in the kidney delivered by HDDP, EHDDP and DDP was significantly different. Since ICP-MS can only measure the concentration of Pt but not that of the intact HDDP and EHDDP nanoparticles, we speculated that the higher concentration of Pt in HDDP- and EHDDP-treated mice resulted mainly from inactivated HDDP and EHDDP nanoparticles. As shown in Figure 4A, the significant decrease in Pt accumulation in the kidney following free DDP treatment occurs within 4hrs (65.1% decrease), while HDDP and EHDDP showed decreases of 42.7% and 33.15%, respectively, in kidney Pt levels. HDDP and EHDDP may circulate to the blood and be caught mainly by the liver and spleen again rather than being eliminated quickly by the kidneys, thus the slow and sustained release of Pt from nanoparticles significantly reduced toxicity.
In conclusion, we have developed tumor-targeted heparin nanoparticles for DDP delivery to lung cancer cells. The EGFR-targeted nanoparticles led to significantly higher accumulation of DDP in tumor cells, and enhanced the antitumor effect both in vitro and in vivo, while significantly reducing the toxicity of DDP to the spleen and kidney. Since both DDP and heparin have been widely used in clinical applications for many years, our novel nanoparticle delivery system has great potential to be used in the clinic in the future.
MATERIAL AND METHODS
Chemical materials
Cisplatin (cis-diaminedichloroplatinum (II), DDP) was purchased from Sigma (St. Louis, MO) and used without further purification. Heparin (sodium salt form) was purchased from Celsus Laboratories (Cincinati, OH). All other chemicals were of analytical grade and were used without further purification.
Purification of ScFvEGFR from E. coli cells
Recombinant ScFvEGFR protein with a molecular weight of 25 kDa was purified from the bacterial lysates of scFv B10 transformed TG1 competent cells using Ni-NTA-agarose column separation under native conditions (Qiagen, Valencia, CA) as described in Yang et al.25
Preparation and characterization of Heparin-DDP (HDDP) and ScFvEGFR-Heparin-DDP (EHDDP) nanoparticles
To further increase the DDP loading capacity and reduce the undesirable anticoagulant property of heparin which could potentially lead to internal bleeding, we modified the heparin (Mw = 12,000) with succinic anhydride according to the previously reported procedure. The succinyl ester content is 20 wt% of the resulting modified heparin (Heparin-Su), which was quantified by proton NMR with the presence of a predetermined amount of pyridine as an internal standard. For the preparation of HDDP, Heparin-Su (10 mg) and DDP (10 mg) were mixed in distilled water (10 ml) under gentle stirring at room temperature for 72 hrs in the dark. The heterogeneous solutions slowly became homogeneous over a period of 24 hrs. The free DDP was removed by dialyses against distilled water (Spectroa/Por6, MWCO= 6–8K) for 24 hrs. The control experiment, in which saturated free DDP in aqueous solution was dialyzed under the same conditions, showed that less than 0.5% DDP was retained in the dialysis bag. To prepare EHDDP, Heparin-Su (10 mg) and DDP (10 mg) were mixed in distilled water (10 ml) under gentle stirring at room temperature for 72 hrs in the dark. The resulting HDDP nanoparticles were negative charged and covered by hydrophilic functional groups such as −COOH and −HSO3−. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDAC) (1mg) and N-hydroxysuccinimide (NHS) (1mg) were added into HDDP nanoparticles solution containing Heparin-Su (10 mg) and DDP (3.0 mg) (determined by ICPMS) to activate carboxylic acid groups for 30 min, then ScFvEGFR (0.2 mg) was added. The mixture was stirred at room temperature for 2 hrs, then at 4°C for another 48 hrs. The free DDP, other impurities such as EDAC and NHS, were removed by dialysis as mentioned above. ScFvEGFR labeled with the fluorescent dye Oregon-green 488 (Invitrogen) was used under the same conditions to determine the reaction yield. The resulting mixture was placed in a NANOSEP filter device (MWCO 100K, Pall Life Science). After centrifugation, the fluorescence of the filtrate was measured by fluorometer (Fluoromax-2, Jobin Yvon-Spex, Horiba Group).
Characterization of the EGFR-targeted heparin-DDP (EHDDP) nanoparticles
The mean diameter of HDDP and EHDDP nanoparticles was evaluated by dynamic light scattering (DLS) measurements using a model 127–35 laser (Spectra Physics, CA) operating at 633 nm and 25 °C. The zeta potential of the nanoparticles was determined using microelectrophoresis. The loading of nanoparticles with DDP was determined using ICP-MS.
Study of the in vitro release of DDP from EHDDP nanoparticles
Release of Pt from EHDDP nanoparticles in PBS at 37 °C was evaluated as follows: 20 ml of EHDDP nanoparticle solution was placed into dialysis bags and immersed into 480 ml of PBS. At definite time intervals, 1 ml of the solution outside the dialysis bag was analyzed by ICP-MS to determine the amount of DDP released from EHDDP nanoparticles.
Cell culture
Two cell lines (EGFR-positive H292,52 and EGFR-negative H52053) were selected for our study to represent NSCLC cells. Both cells were maintained in RPMI1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (streptomycin, penicillin G and amphotericin B) in a 37°C, 5% CO2 humidified incubator.
In vitro cytotoxicity assay
To test the effects of free DDP, HDDP and EHDDP nanoparticles on cell growth of tumor cells, sulforhodamine B (SRB) cytotoxicity assays were adapted from Skehan et al.54 H292 and H520 cells maintained in medium with 2% FBS were seeded in 96-well plates at a density of 5,000 cells/well overnight prior to drug treatment. DDP, HDDP, or EHDDP nanoparticles were added in various concentrations, followed by incubation at 37°C and 5% CO2 for 96hrs. Cells were fixed for 1hr with 10% cold trichloroacetic acid. Plates were washed 5 times in water, air-dried and then stained with 0.4% SRB for 10min. After washing 4 times in 1% acetic acid and air-drying, bound SRB was dissolved in 10 mM Tris base (pH 10.5). Plates were read in a microplate reader by measuring absorbance at 492 nm. The percent survival was then calculated based upon the absorbance values relative to untreated samples. The experiment was repeated 3 times.
Quantification of intracellular platinum accumulation and DNA-adduct formation in vitro
1×106 H292 cells were incubated for 24hrs at 37°C with free DDP or HDDP, EHDDP nanoparticles (0.4ug/ml). EGFR-negative H520 cells were used as negative control. To study the internalization of DDP in the presence of free ScFvEGFR, H292 cells were incubated with different concentrations of ScFvEGFR at 37°C for 20mins, and then EHDDP nanoparticles were added and incubated for another 24 hrs. To remove surface-bound DDP, cells were washed three times with ice-cold PBS, and incubated with 1.5 ml of 0.15 M sodium chloride (pH 3.0 was adjusted by acetic acid) for 3 min at 4°C, then rinsed with 2 ml of cold PBS, harvested by scraping in ice-cold PBS, centrifuged, and cell numbers were counted.55 The cell pellet was digested in 65% (v/v) nitric acid at 75°C for 2 h. The Pt content was analyzed by ICP-MS. Cellular Pt levels were expressed in ng Pt/106 cells. To further check the targeting specificity of EHDDP nanoparticles in vitro, EGFR was knocked down by EGFR small interfering RNAs (siRNA). H292 cells were transfected with EGFR siRNA (sc-29301 from Santa Cruz Bio) (100 pmol) or scrambled siRNA (sc-37007 from Santa Cruz Bio) (100 pmol) as a nonspecific control) according to the manufacturer's protocols. Briefly, in a 6-well tissue culture plate, 2 × 105 cells per well were seeded in 2ml antibiotic-free RPMI 1640 medium supplemented with 10% FCS, and on the following day transfected with siRNAs at the final concentration of 100 pmol. Cells were harvested 24h later, whole cell lysates were extracted using lysis buffer, and 15 μg of protein was separated on 8–12% SDS-PAGE, then transferred onto a polyvinylidene difluoride membrane (Millipore Corp., Billerica, MA) and immunoblotted with specific antibodies against EGFR (sc-03 from Santa Cruz Bio). Mouse anti-β-actin antibody (Trevigen, Gaithersburg, MD) was used as a sample loading control. Immunostained protein bands were detected with an enhanced chemiluminescence kit (Thermo Scientific, Rockfield, IL). 24 h after transfection of EGFR siRNA, cells were treated with DDP, HDDP and EHDDP (0.4ug/ml) for 24 h and Pt concentration was quantified by ICP-MS as described above. The data are shown as mean ± SD (n=3). DDP-DNA-adduct formation was measured using 1 × 107 cells per condition. DNA was isolated by using DNA purification kit (Invitrogen, USA) and the DNA was digested with DNase I before analysis of Pt content by ICP-MS56, 57. Total DNA-adducts were expressed in pg Pt/ug DNA. In addition, the expression level of the phosphorylation of H2A.X was detected by Western Blot as described above using the Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb (Cell Signaling Technology, Inc.).
Coagulation assays
The anticoagulant activities of EHDDP were determined by FXa-dependent coagulant assay using Coatest Heparin (Helena Laboratories, Beaumont, TX).58 Briefly, 200μl of standard samples of heparin (concentrations of heparin from 0.01 to 0.07 unit/ml) and EHDDP were incubated at 37°C for 3–4 minutes then 100μl of FXa (0.355nkat) was added and mixed well. The mixture was incubated at 37°C for 30 seconds and 200μl 1mM of chromogenic substrate S-2222 was added. The mixture was incubated at 37°C for 3 minutes. The reaction was stopped by adding 300μl 20% (v/v) acetic acid. The sample was transferred to a semi-micro plate, and the absorbance of the samples at 405nm was detected. The anticoagulant activity was calculated based on the standard curve.
Concentration profiles of Pt in mice peripheral blood
DDP, HDDP and EHDDP (Pt concentration 2.5mg/kg) nanoparticles were injected into nude mice by the tail vein. Blood (30ul) was collected from the retro-orbital plexus at different time points (2, 5, 15, 30, 60, 120, 480 and 1440mins). The whole blood samples were dissolved in 65% (v/v) nitric acid at 75°C for 2 hrs, and Pt concentration was measured by ICP-MS.
Biodistribution of EHDDP nanoparticles and formation of Pt-DNA adducts in vivo
To evaluate the biodistribution of DDP, HDDP and EHDDP nanoparticles in tumor-bearing mice, the agents were injected into mice (3 mice each group) by tail vein, and 10mins, 30mins, 4hrs and 24hrs later, mice were sacrificed. Blood, organs (liver, spleen, kidney and lung) and tumor tissue were harvested, half of the organs were dissolved in 65% (v/v) nitric acid at 75°C for 2 hrs, and Pt concentrations were measured by ICP-MS. The blood and other half of the organs were used to extract DNA, and the Pt concentration in DNA was quantified as described above.
In vivo antitumor efficacy of EHDDP nanoparticles
The animal experiments were approved by the Animal Care and Use Committee of Emory University. Twenty-four nude mice (athymic nu/nu, Taconic, NY), aged 4 to 6 weeks (about 20 g weight), were randomly divided into 4 groups. H292 tumor cells (1 × 106 cells per mouse) were injected subcutaneously into the left flank of male nude mice. When the tumor volume was approximately 100 mm3, the mice were injected intravenously via the tail vein 5 times at 3 day intervals with free DDP (200 μl of aqueous DDP solution) at a dose of 2.5 mg/kg or DDP-loaded nanoparticles at the same dose. The control group was administered saline. The tumor size was measured 3 times a week. The antitumor activity was evaluated in terms of the tumor size at different times post-administration, which was estimated by the following equation: V =6 × larger diameter × (smaller diameter)2 /π, as reported previously.59 Growth curves were plotted using the average tumor size within each experimental group at the set time points. The whole group of mice was sacrificed once the size of any tumor in that group reached 2 cm in diameter. To evaluate the tolerance of nude mice to EHDDP nanoparticles, the body weight and physical state of the mice were measured simultaneously as an indicator of systemic toxicity.
Peripheral blood analysis
After treatment with DDP, HDDP and EHDDP nanoparticles (5 injections, Pt 2.5mg/kg, 21 days), the mice were sacrificed by CO2 and 1ml blood was immediately harvested in a heparinized tube, centrifuged at 3000 rpm for 10 minutes, and the serum separated. Serum samples were subjected to chemistry analysis of alkaline phosphatase (ALT), alanine aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE) levels using Theoretical Tests Per Kit, on the P-Modular Analytics System manufactured by Roche Diagnostics Inc (Indianapolis, IN).
Histopathology evaluation
As described above, the spleen, liver, and kidney were dissected from the mice (21 days after treatment) for histopathological analysis. The paraffin-embedded tissues were cut at 5 μm thickness. The tissues were stained with hematoxylin and eosin (Sigma) to assess histological alterations by microscope.
Statistical analysis
Paired t-test was conducted to compare differences in the values of same outcome variables (Pt concentration, viable cells, tumor volume, body weight change, spleen weight, etc.) from the same samples at two different time points. Two sample t-test was used for different samples. One-way analysis of variance (ANOVA) was employed to test the overall significance across multiple groups (such as control, DDP, HDDP, and EHDDP). We further evaluated pairwise differences by using Tukey's method when the overall difference was significant at the significance level of 0.05. Two-way ANOVA was further conducted to estimate the effects of tumor site (liver vs spleen) and cancer therapy type (DDP vs HDDP vs EHDDP). Finally, a mixed model was used to deal with longitudinal data and compare the overall difference over time across multiple groups. The SAS statistical package (SAS Institute, Inc., Cary, North Carolina) was used for all data management and analyses.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grants from the NCI Centers of Cancer Nanotechnology Excellence (CCNE) Program (U54CA119338) and the Specialized Program of Excellence (SPORE) in Head and Neck Cancer (P50CA128613), NCI Cancer Nanotechnology Platform Partner (CNPP, U01 CA151802), and the Emory University Head & Neck Cancer SPORE Career Development Award to XH Peng. D.M.S., Z.G.C, and S.M.N. are Distinguished Cancer Scholars of the Georgia Cancer Coalition (GCC). We thank Dr. Anthea Hammond for her assistance in critical reading and editing of the manuscript.
Footnotes
Note: X-H Peng and Y Wang contributed equally to the study
REFERENCES
- 1.Klein AV, Hambley TW. Platinum Drug Distribution in Cancer Cells and Tumors. Chem Rev. 2009;109:4911–4920. doi: 10.1021/cr9001066. [DOI] [PubMed] [Google Scholar]
- 2.Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. The Role of Cellular Accumulation in Determining Sensitivity to Platinum-Based Chemotherapy. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 3.Stewart DJ, Dulberg CS, Mikhael NZ, Redmond MD, Montpetit VA, Goel R. Association of Cisplatin Nephrotoxicity with Patient Characteristics and Cisplatin Administration Methods. Cancer Chemother Pharmacol. 1997;40:293–308. doi: 10.1007/s002800050661. [DOI] [PubMed] [Google Scholar]
- 4.Barabas K, Milner R, Lurie D, Adin C. Cisplatin: A Review of Toxicities and Therapeutic Applications. Vet Comp Oncol. 2008;6:1–18. doi: 10.1111/j.1476-5829.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 5.Nagai N, Ogata H. Quantitative Relationship Between Pharmacokinetics of Unchanged Cisplatin and Nephrotoxicity in Rats: Importance of Area Under the Concentration-Time Curve (AUC) as the Major Toxicodynamic Determinant in Vivo. Cancer Chemother Pharmacol. 1997;40:11–18. doi: 10.1007/s002800050618. [DOI] [PubMed] [Google Scholar]
- 6.Nie S, Xing Y, Kim GJ, Simons JW. Nanotechnology Applications in Cancer. Annu Rev Biomed Eng. 2007;9:257–288. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 7.Davis ME, Chen ZG, Shin DM. Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 8.Wang MD, Shin DM, Simons JW, Nie S. Nanotechnology for Targeted Cancer Therapy. Expert Rev Anticancer Ther. 2007;7:833–837. doi: 10.1586/14737140.7.6.833. [DOI] [PubMed] [Google Scholar]
- 9.Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K. Novel Cisplatin-Incorporated Polymeric Micelles Can Eradicate Solid Tumors in Mice. Cancer Res. 2003;63:8977–8983. [PubMed] [Google Scholar]
- 10.Bae YH. Drug Targeting and Tumor Heterogeneity. J Control Release. 2009;133:2–3. doi: 10.1016/j.jconrel.2008.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen TM, Cullis PR. Drug Delivery Systems: Entering the Mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 12.Peng XH, Qian X, Mao H, Wang AY, Chen ZG, Nie S, Shin DM. Targeted Magnetic Iron Oxide Nanoparticles for Tumor Imaging and Therapy. Int J Nanomedicine. 2008;3:311–321. doi: 10.2147/ijn.s2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bublil EM, Yarden Y. The EGF Receptor Family: Spearheading a Merger of Signaling and Therapeutics. Curr Opin Cell Biol. 2007;19:124–134. doi: 10.1016/j.ceb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal Growth Factor-related Peptides and Their Receptors in Human Malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 15.Merrick DT, Kittelson J, Winterhalder R, Kotantoulas G, Ingeberg S, Keith RL, Kennedy TC, Miller YE, Franklin WA, Hirsch FR. Analysis of C-ErbB1/Epidermal Growth Factor Receptor and C-ErbB2/HER-2 Expression in Bronchial Dysplasia: Evaluation of Potential Targets for Chemoprevention of Lung Cancer. Clin Cancer Res. 2006;12:2281–2288. doi: 10.1158/1078-0432.CCR-05-2291. [DOI] [PubMed] [Google Scholar]
- 16.Hirsch FR, Varella-Garcia M, Cappuzzo F. Predictive Value of EGFR and HER2 Overexpression in Advanced Non-Small-Cell Lung Cancer. Oncogene. 2009;28:S32–37. doi: 10.1038/onc.2009.199. [DOI] [PubMed] [Google Scholar]
- 17.Nida DL, Rahman MS, Carlson KD, Richards-Kortum R, Follen M. Fluorescent Nanocrystals for Use in Early Cervical Cancer Detection. Gynecol Oncol. 2005;99:S89–94. doi: 10.1016/j.ygyno.2005.07.050. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Eom K, Lim EK, Park J, Kang Y, Yoon DS, Na S, Koh EK, Suh JS, Huh YM, et al. In Situ Detection of Live Cancer Cells by Using Bioprobes Based on Au Nanoparticles. Langmuir. 2008;24:12112–12115. doi: 10.1021/la802184m. [DOI] [PubMed] [Google Scholar]
- 19.Melancon MP, Lu W, Yang Z, Zhang R, Cheng Z, Elliot AM, Stafford J, Olson T, Zhang JZ, Li C. In Vitro and In Vivo Targeting of Hollow Gold Nanoshells Directed at Epidermal Growth Factor Receptor for Photothermal Ablation Therapy. Mol Cancer Ther. 2008;7:1730–1739. doi: 10.1158/1535-7163.MCT-08-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, et al. Targeted Delivery of Gemcitabine to Pancreatic Adenocarcinoma Using Cetuximab As a Targeting Agent. Cancer Res. 2008;68:1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 21.Wu G, Barth RF, Yang W, Kawabata S, Zhang L, Green-Church K. Targeted Delivery of Methotrexate to Epidermal Growth Factor Receptor-Positive Brain Tumors by Means of Cetuximab (IMC-C225) Dendrimer Bioconjugates. Mol Cancer Ther. 2006;5:52–59. doi: 10.1158/1535-7163.MCT-05-0325. [DOI] [PubMed] [Google Scholar]
- 22.Horak E, Heitner T, Robinson MK, Simmons HH, Garrison J, Russeva M, Furmanova P, Lou J, Zhou Y, Yuan QA, et al. Isolation of ScFvs to in Vitro Produced Extracellular Domains of EGFR Family Members. Cancer Biother Radiopharm. 2005;20:603–613. doi: 10.1089/cbr.2005.20.603. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, Marks JD. Impact of Single-Chain Fv Antibody Fragment Affinity on Nanoparticle Targeting of Epidermal Growth Factor Receptor-Expressing Tumor Cells. J Mol Biol. 2007;371:934–947. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlinkova G, Colcher D, Booth BJ, Goel A, Batra SK. Pharmacokinetics and Biodistribution of a Light-Chain-Shuffled CC49 Single-Chain Fv Antibody Construct. Cancer Immunol Immunother. 2000;49:267–275. doi: 10.1007/s002620000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Mao H, Wang YA, Cao Z, Peng X, Wang X, Duan H, Ni C, Yuan Q, Adams G, et al. Single Chain Epidermal Growth Factor Receptor Antibody Conjugated Nanoparticles for in Vivo Tumor Targeting and Imaging. Small. 2009;5:235–243. doi: 10.1002/smll.200800714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clingen PH, Wu JY, Miller J, Mistry N, Chin F, Wynne P, Prise KM, Hartley JA. Histone H2AX Phosphorylation As a Molecular Pharmacological Marker for DNA Interstrand Crosslink Cancer Chemotherapy. Biochem Pharmacol. 2008;76:19–27. doi: 10.1016/j.bcp.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 27.Stewart DJ, Mikhael NZ, Nanji AA, Nair RC, Kacew S, Howard K, Hirte W, Maroun JA. Renal and Hepatic Concentrations of Platinum: Relationship to Cisplatin Time, Dose, and Nephrotoxicity. J Clin Oncol. 1985;3:1251–1256. doi: 10.1200/JCO.1985.3.9.1251. [DOI] [PubMed] [Google Scholar]
- 28.Hanigan MH, Lykissa ED, Townsend DM, Ou CN, Barrios R, Lieberman MW. Gamma-Glutamyl Transpeptidase-Deficient Mice Are Resistant to the Nephrotoxic Effects of Cisplatin. Am J Pathol. 2001;159:1889–1894. doi: 10.1016/s0002-9440(10)63035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welters MJ, Fichtinger-Schepman AM, Baan RA, Jacobs-Bergmans AJ, Kegel A, van der Vijgh WJ, Braakhuis BJ. Pharmacodynamics of Cisplatin in Human Head and Neck Cancer: Correlation Between Platinum Content, DNA Adduct Levels and Drug Sensitivity in Vitro and in Vivo. Br J Cancer. 1999;79:82–88. doi: 10.1038/sj.bjc.6690015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeong YI, Kim ST, Jin SG, Ryu HH, Jin YH, Jung TY, Kim IY, Jung S. Cisplatin-Incorporated Hyaluronic Acid Nanoparticles Based on Ion-Complex Formation. J Pharm Sci. 2008;97:1268–1276. doi: 10.1002/jps.21103. [DOI] [PubMed] [Google Scholar]
- 31.Mizumura Y, Matsumura Y, Hamaguchi T, Nishiyama N, Kataoka K, Kawaguchi T, Hrushesky WJ, Moriyasu F, Kakizoe T. Cisplatin-Incorporated Polymeric Micelles Eliminate Nephrotoxicity, while Maintaining Antitumor Activity. Jpn J Cancer Res. 2001;92:328–336. doi: 10.1111/j.1349-7006.2001.tb01099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-Incorporating Polymeric Micelles (NC-6004) Can Reduce Nephrotoxicity and Neurotoxicity of Cisplatin in Rats. Br J Cancer. 2005;93:678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gryparis EC, Hatziapostolou M, Papadimitriou E, Avgoustakis K. Anticancer Activity of Cisplatin-Loaded PLGA-mPEG Nanoparticles on LNCaP Prostate Cancer Cells. Eur J Pharm Biopharm. 2007;67:1–8. doi: 10.1016/j.ejpb.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Xu P, Van Kirk EA, Murdoch WJ, Zhan Y, Isaak DD, Radosz M, Shen Y. Anticancer Efficacies of Cisplatin-Releasing pH-Responsive Nanoparticles. Biomacromolecules. 2006;7:829–835. doi: 10.1021/bm050902y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Staffhorst RW, van der Born K, Erkelens CA, Hamelers IH, Peters GJ, Boven E, de Kroon AI. Antitumor Activity and Biodistribution of Cisplatin Nanocapsules in Nude Mice Bearing Human Ovarian Carcinoma Xenografts. Anticancer Drugs. 2008;19:721–727. doi: 10.1097/CAD.0b013e328304355f. [DOI] [PubMed] [Google Scholar]
- 36.Kim JH, Kim YS, Park K, Lee S, Nam HY, Min KH, Jo HG, Park JH, Choi K, Jeong SY, et al. Antitumor Efficacy of Cisplatin-Loaded Glycol Chitosan Nanoparticles in Tumor-bearing Mice. J Control Release. 2008;127:41–49. doi: 10.1016/j.jconrel.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 37.Kettering M, Zorn H, Bremer-Streck S, Oehring H, Zeisberger M, Bergemann C, Hergt R, Halbhuber KJ, Kaiser WA, Hilger I. Characterization of Iron Oxide Nanoparticles Adsorbed with Cisplatin for Biomedical Applications. Phys Med Biol. 2009;54:5109–5121. doi: 10.1088/0031-9155/54/17/003. [DOI] [PubMed] [Google Scholar]
- 38.Gryparis EC, Mattheolabakis G, Bikiaris D, Avgoustakis K. Effect of Conditions of Preparation on the Size and Encapsulation Properties of PLGA-mPEG Nanoparticles of Cisplatin. Drug Deliv. 2007;14:371–380. doi: 10.1080/10717540701202937. [DOI] [PubMed] [Google Scholar]
- 39.Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, Karna P, Zhang X, Wood WC, Gao X, et al. Receptor-Targeted Nanoparticles for in Vivo Imaging of Breast Cancer. Clin Cancer Res. 2009;15:4722–4732. doi: 10.1158/1078-0432.CCR-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, Yang L, Young AN, Wang MD, Nie S. In Vivo Tumor Targeting and Spectroscopic Detection with Surface-Enhanced Raman Nanoparticle Tags. Nat Biotechnol. 2008;26:83–90. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Li J, Wang Y, Cho KJ, Kim G, Gjyrezi A, Koenig L, Giannakakou P, Shin HJ, Tighiouart M, et al. HFT-T, A Targeting Nanoparticle, Enhances Specific Delivery of Paclitaxel to Folate Receptor-Positive Tumors. ACS Nano. 2009;3:3165–3174. doi: 10.1021/nn900649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tseng CL, Su WY, Yen KC, Yang KC, Lin FH. The Use of Biotinylated-EGF-Modified Gelatin Nanoparticle Carrier to Enhance Cisplatin Accumulation in Cancerous Lungs via Inhalation. Biomaterials. 2009;30:3476–3485. doi: 10.1016/j.biomaterials.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 43.McKay K. New Techniques in the Pharmacokinetic Analysis of Cancer Drugs. II. The Ultratrace Determination of Platinum in Biological Samples by Inductively Cupled Plasma-Mass Spectrometry. Cancer Surv. 1993;17:407–414. [PubMed] [Google Scholar]
- 44.Bonetti A, Apostoli P, Zaninelli M, Pavanel F, Colombatti M, Cetto GL, Franceschi T, Sperotto L, Leone R. Inductively Coupled Plasma Mass Spectroscopy Quantitation of Platinum-DNA Adducts in Peripheral Blood Leukocytes of Patients Receiving Cisplatin- or Carboplatin-Based Chemotherapy. Clin Cancer Res. 1996;2:1829–1835. [PubMed] [Google Scholar]
- 45.Brouwers EE, Tibben MM, Pluim D, Rosing H, Boot H, Cats A, Schellens JH, Beijnen JH. Inductively Coupled Plasma Mass Spectrometric Analysis of the Total Amount of Platinum in DNA Extracts From Peripheral Blood Mononuclear Cells and Tissue from Patients Treated with Cisplatin. Anal Bioanal Chem. 2008;391:577–585. doi: 10.1007/s00216-008-2034-8. [DOI] [PubMed] [Google Scholar]
- 46.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody Targeting of Long-circulating Lipidic Nanoparticles Does Not Increase Tumor Localization But Does Increase Internalization in Animal Models. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 47.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of Tumor-Specific Targeting on the Biodistribution and Efficacy of SiRNA Nanoparticles Measured by Multimodality in Vivo Imaging. Proc Natl Acad Sci U S A. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang L, Mao H, Cao Z, Wang YA, Peng X, Wang X, Sajja HK, Wang L, Duan H, Ni C, et al. Molecular Imaging of Pancreatic Cancer in an Animal Model Using Targeted Multifunctional Nanoparticles. Gastroenterology. 2009;136:1514–1525. doi: 10.1053/j.gastro.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mannel RS, Stratton JA, Moran G, Rettenmaier MA, Liao SY, DiSaia PJ. Intraperitoneal Cisplatin: Comparison of Antitumor Activity and Toxicity as a Function of Solvent Saline Concentration. Gynecol Oncol. 1989;34:50–53. doi: 10.1016/0090-8258(89)90105-4. [DOI] [PubMed] [Google Scholar]
- 50.Bodenner DL, Dedon PC, Keng PC, Katz JC, Borch RF. Selective Protection Against Cis-Diamminedichloroplatinum(II)-Induced Toxicity in Kidney, Gut, and Bone Marrow by Diethyldithiocarbamate. Cancer Res. 1986;46:2751–2755. [PubMed] [Google Scholar]
- 51.Siddik ZH, Newell DR, Boxall FE, Harrap KR. The Comparative Pharmacokinetics of Carboplatin and Cisplatin in Mice and Rats. Biochem Pharmacol. 1987;36:1925–1932. doi: 10.1016/0006-2952(87)90490-4. [DOI] [PubMed] [Google Scholar]
- 52.Akashi Y, Okamoto I, Iwasa T, Yoshida T, Suzuki M, Hatashita E, Yamada Y, Satoh T, Fukuoka M, Ono K, et al. Enhancement of The Antitumor Activity of Ionising Radiation by Nimotuzumab, a Humanised Monoclonal Antibody to the Epidermal Growth Factor Receptor, in Non-Small Cell Lung Cancer Cell Lines of Differing Epidermal Growth Factor Receptor Status. Br J Cancer. 2008;98:749–755. doi: 10.1038/sj.bjc.6604222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen H, Yuan Y, Sun J, Gao W, Shu YQ. Combined Tamoxifen and Gefitinib in Non-Small Cell Lung Cancer Shows Antiproliferative Effects. Biomed Pharmacother. 2010;64:88–92. doi: 10.1016/j.biopha.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 54.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt-Glenewinkel H, Reinz E, Eils R, Brady NR. Systems Biological Analysis of Epidermal Growth Factor Receptor Internalization Dynamics for Altered Receptor Levels. J Biol Chem. 2009;284:17243–17252. doi: 10.1074/jbc.M809586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fichtinger-Schepman AM, Vendrik CP, van Dijk-Knijnenburg WC, de Jong WH, van der Minnen AC, Claessen AM, van der Velde-Visser SD, de Groot G, Wubs KL, Steerenberg PA, et al. Platinum Concentrations and DNA Adduct Levels in Tumors and Organs of Cisplatin-Treated LOU/M Rats Inoculated with Cisplatin-Sensitive or -Resistant Immunoglobulin M Immunocytoma. Cancer Res. 1989;49:2862–2867. [PubMed] [Google Scholar]
- 57.Hamelers IH, Staffhorst RW, Voortman J, de Kruijff B, Reedijk J, van Bergen en Henegouwen PM, de Kroon AI. High Cytotoxicity of Cisplatin Nanocapsules in Ovarian Carcinoma Cells Depends on Uptake by Caveolae-Mediated Endocytosis. Clin Cancer Res. 2009;15:1259–1268. doi: 10.1158/1078-0432.CCR-08-1702. [DOI] [PubMed] [Google Scholar]
- 58.Liu S, Zhou F, Hook M, Carson DD. A Heparin-Binding Synthetic Peptide of Heparin/Heparan Sulfate-Interacting Protein Modulates Blood Coagulation Activities. Proc Natl Acad Sci U S A. 1997;94:1739–1744. doi: 10.1073/pnas.94.5.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Chen ZG, Choe MS, Lin Y, Sun SY, Wieand HS, Shin HJ, Chen A, Khuri FR, Shin DM. Tumor Growth Inhibition by Simultaneously Blocking Epidermal Growth Factor Receptor and Cyclooxygenase-2 in a Xenograft Model. Clin Cancer Res. 2005;11:6261–6269. doi: 10.1158/1078-0432.CCR-04-2102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.