

Abstract
Background
Cryptosporidium parvum causes an opportunistic infection in AIDS patients, and no effective treatments are yet available. This parasite possesses a single fatty acyl-CoA binding protein (CpACBP1) that is localized to the unique parasitophorous vacuole membrane (PVM). The major goal of this study was to identify inhibitors from known drugs against CpACBP1 as potential new anti-Cryptosporidium agents.
Methods
A fluorescence assay was developed to detect CpACBP1 activity and to identify inhibitors by screening known drugs. Efficacies of top CpACBP1 inhibitors against Cryptosporidium growth in vitro were evaluated using a quantitative RT–PCR assay.
Results
Nitrobenzoxadiazole-labelled palmitoyl-CoA significantly increased the fluorescent emission upon binding to CpACBP1 (excitation/emission 460/538 nm), which was quantified to determine the CpACBP1 activity and binding kinetics. The fluorescence assay was used to screen a collection of 1040 compounds containing mostly known drugs, and identified the 28 most active compounds that could inhibit CpACBP1 activity with sub-micromolar IC50 values. Among them, four compounds displayed efficacies against parasite growth in vitro with low micromolar IC50 values. The effective compounds were broxyquinoline (IC50 64.9 μM), cloxyquin (IC50 25.1 μM), cloxacillin sodium (IC50 36.2 μM) and sodium dehydrocholate (IC50 53.2 μM).
Conclusions
The fluorescence ACBP assay can be effectively used to screen known drugs or other compound libraries. Novel anti-Cryptosporidium activity was observed in four top CpACBP1 inhibitors, which may be further investigated for their potential to be repurposed to treat cryptosporidiosis and to serve as leads for drug development.
Keywords: Cryptosporidium parvum, broxyquinoline, cloxyquin, cloxacillin sodium, sodium dehydrocholate
Introduction
Cryptosporidium is well known to be a troublesome waterborne pathogen for immunocompetent and especially immunosuppressed individuals. A countless number of outbreaks, mostly caused by either Cryptosporidium parvum or Cryptosporidium hominis, occur each year around the world. Transmission is typically via contaminated water supplies and/or recreational water by the environmentally resistant and chlorine-resistant oocysts. The vulnerability of community water supplies to this parasite and increased biodefence concerns have escalated Cryptosporidium to one of the waterborne category B pathogens in the NIH and CDC biodefence research programmes.
Despite numerous investigations, there is currently no completely effective drug to treat cryptosporidiosis. Drug therapy would no doubt benefit several groups.1 Severe cases, often requiring hospitalization among immunocompetent individuals, usually occur in children and the elderly. Transplant recipients and those undergoing cancer chemotherapy are often immunocompromised. These patients usually have to temporarily halt their treatment regimens in order to combat cryptosporidiosis. Anti-cryptosporidials would certainly be beneficial to these patients, as well as to those who are HIV-positive and are at great risk of a devastating infection with Cryptosporidium.
Several suggestions have been postulated as to why this apicomplexan appears to have a natural resistance to drug therapy.1 One such factor is the apparent lack of or difference in drug targets at both molecular and structural levels, namely differences in biochemical pathways. Additionally, if drugs actually reach the parasite, they may be readily transported out via transport proteins. Perhaps the most widely discussed factor is the parasite's unique intracellular but extracytoplasmic location in humans and animals.1–3 Upon initiation of infection, the infective sporozoite is enveloped by the host cell apical membrane, forming a space between the parasite and the host cell membrane known as the parasitophorous vacuole (PV). Because parasite basal membranes fuse with the host cell membrane, the PV extends only over the apical end and its membrane covering is termed the parasitophorous vacuolar membrane (PVM). Although it is unclear at this time, preliminary data indicate that the basal membranes modulate the transport of some drugs and do not allow at least some drugs to enter the parasite from host cytoplasm.1,4 This appears to be the case for both geneticin and the clinically relevant drug paromomycin, as apical but not basolateral exposure of these drugs led to parasite inhibition.4
We hypothesize that parasite proteins located in the PVM may serve as valuable drug targets. One such protein is the C. parvum acyl-CoA binding protein (CpACBP1). Our laboratory has previously characterized this unique protein at both the molecular and the biochemical level.5 This family of proteins is critical to lipid metabolism as their main function is as an intracellular acyl-CoA transporter and pool former.6–8 Animals, plants, protists and several pathogenic bacteria have been found to contain this highly conserved protein.9 Although they are typically small (∼10 kDa) cytosolic molecules, there have been larger (≥55 kDa) ACBPs found in animals and plants. The unique CpACBP1 is a long-type ACBP containing an N-terminal ACBP domain and a C-terminal ankyrin repeat sequence. Although it differs from the typical cytosolic ACBPs, it is similar to the membrane-bound ACBPs from Arabidopsis.10,11 Our previous analysis indicates that CpACBP1 is also a membrane protein associated with the PVM, probably via interaction of its ankyrin repeats with other proteins in the PVM. It is unlikely that CpACBP1 is involved in the early stages of PVM formation as it is not expressed during initial stages of infection, but it is widely known that C. parvum must import fatty acids from the host cell or the intestinal lumen. Although C. parvum is incapable of de novo fatty acid synthesis, it is capable of elongating and utilizing long-chain fatty acids.12–14 Thus, in cooperation with an acyl-CoA synthetase, it is possible that CpACBP1 serves as a fatty acyl-CoA scavenger to facilitate fatty acid uptake at the PVM.
Here we report the development of a fluorescence-based binding assay that was more sensitive and stable, and also much safer for operators, than the traditional Lipidex radioactive assay. Using the newly developed assay, we were able to produce a set of data with much improved quality regarding enzyme kinetics and substrate preference for CpACBP1. Additionally, the assay was conveniently employed to screen a library of 1040 compounds, most of which are drugs approved for use in humans for various diseases and/or ailments, to identify novel inhibitors. Several inhibitors not only inhibited the binding of CpACBP1 to fatty acyl-CoA, but also significantly reduced C. parvum growth and development in vitro, supporting the notion that CpACBP1 could potentially serve as a novel drug target in the parasite.
Materials and methods
Expression of recombinant CpACBP1 protein
We have previously cloned and expressed CpACBP1 as maltose-binding protein (MBP) fusion proteins.5 Two forms of CpACBP1 proteins were expressed and purified in this study: the long form, containing the full-length protein including the N-terminal ACBP domain and the C-terminal ankyrin repeats domain (CpACBP1, 268 amino acids), and the short form, containing only the ACBP domain (CpACBP1ΔAnk, 107 amino acids).5 Both forms of protein were expressed in a Rosetta 2 strain of Escherichia coli cells (Novagen) and purified using amylose resin-based affinity chromatography according to the manufacturer's standard protocol (New England Biolabs) as described.5 Purified proteins were dialysed extensively against Dulbecco's PBS (Sigma) and stored at −80°C.
Development of fluorometric assay for CpACBP1
A fluorescence-based assay was developed to replace the conventional radioactive assay. This was achieved by taking advantage of the unusual feature of nitrobenzoxadiazole (NBD)—that it is nearly non-fluorescent in aqueous solution, but can produce increased fluorescence in a polar environment such as in the binding pocket of an enzyme.15 In this assay, the emission of NBD-labelled palmitoyl-CoA (NBD-C16:0-CoA) upon binding to CpACBP1 was measured in a Fluoroskan Ascent fluorimeter using a pair of bandpass filters at 538 ± 12.5 nm for emission and 460 ± 9.0 nm for excitation (Thermoelectron). All reactions were set up in 96-well white plates, which offer high signal reflectance and reduced background fluorescence (Thermoelectron). The fluorimeter program was set to maintain a constant temperature of 25°C and to shake the samples for 20 s at 120 rpm (1 mm diameter rotation) prior to fluorescence measurement. An average of three to five scans was taken for each measurement, with at least three replicates for each experiment.
Enzyme kinetics and substrate preference
We also determined the major binding kinetics and substrate preference for CpACBP1 using the NBD-C16-CoA-based assay to compare with those previously reported by us using a Lipidex 1000 assay. First, the pH of the reaction was optimized using PBS at pH 5.5, 6.0, 6.5, 7.0, 7.5, 8.0 and 8.5. In addition to PBS, reaction components consisted of 0.1 μM MBP-CpACBP1 and 0.25 μM NBD-C16:0-CoA in a volume of 100 μL. Enzyme kinetics assays were performed using 0.1 μM MBP-CpACBP1, NBD-C16:0-CoA (16 nM–1 μM) and PBS, pH 7.5, to a final volume of 100 μL. We also employed a substrate competition assay to determine substrate specificity using this assay set-up. Assays included 0.1 μM MBP-CpACBP1, 0.25 μM NBD-C16:0-CoA, 0.25 μM unlabelled saturated (C2:0–C26:0) or unsaturated (C18:1, C18:3, C20:4 and C22:6) fatty acyl-CoAs and PBS, pH 7.5, in a final volume of 100 μL. In addition we also assayed the binding of palmitic acid (C16:0) to CpACBP1. For each assay, the enzyme was the final reaction component added and reactions were incubated at 25°C for 5 min to ensure maximum binding before proceeding with fluorescence measurements.
Screening of compound library against CpACBP1
We were graciously given access to a drug library consisting of 1040 compounds by Dr Friedhelm Schroeder (Texas A&M University).16 This library was purchased from Microsource Discovery Systems as the NIH and Juvenile Diabetes Research Foundation (NIH/JDRF) custom collection. Each compound was provided dissolved in DMSO at a concentration of 10 mM, and was diluted to 10 μM in PBS, pH 7.5, prior to the assay. Thus, the final concentration of DMSO in the assay was 0.0025%.
Our primary screening was carried out using a single concentration of compound. A typical reaction contained 0.1 μM MBP-CpACBP1, 0.25 μM NBD-C16:0-CoA and 0.25 μM library compound in a final volume of 100 μL of PBS (pH 7.5). MBP was used in control reactions for background subtraction. Assays were performed in duplicate. Reactions were started by adding enzyme or MBP as the final component, and fluorescence emission at 538 nm was measured after the reactions had been incubated at 25°C for 5 min.
After primary screening, the absorption spectra of compounds that displayed ≥50% inhibition on the binding of NBD-C16:0-CoA to MBP-CpACBP1 were tested. Compounds that displayed absorption spectra at 280–590 nm were excluded from subsequent kinetic analysis. Detailed inhibitory kinetic features were determined for the remaining effective compounds using a series of concentrations ranging from 20 nM to 2 μM. The IC50 values were derived from the data by appropriate non-linear regression algorithms depending on the observed curves (typically sigmoidal or hypobolic curves). The Ki values were derived from the following formula: Ki = IC50/(1 + S/Kd), where S is the concentration of NBD-C16:0-CoA (0.25 μM).
Cultivation of parasite in vitro for drug testing
Only C. parvum oocysts (Iowa strain) <3 months old since the time of harvest were used in all experiments. Oocysts were purified by Percoll gradient centrifugation and bleached as previously described.17,18 Human ileocaecal colorectal adenocarcinoma (HCT-8) cells (1 × 105/well) were seeded in 48-well tissue culture plates and allowed to grow overnight or until they reached ∼80% confluency at 37°C with 5% CO2 in RPMI 1640 medium (Sigma) containing 10% fetal bovine serum and other supplements as described previously.19,20 For drug testing, host cells were infected with 20 000 oocysts per well. In all experiments, parasites were allowed to incubate for 3–4 h at 37°C for excystation and invasion into host cells. Parasites that failed to invade host cells were removed by replacing the culture medium with one that contained specified concentrations of compounds. The treated parasite-infected cells were then incubated at 37°C with 5% CO2 for 44 h. An initial screening using high (1 mM) and low (10 μM) concentrations of compounds was first performed in order to determine the range of effective concentrations in kinetics assays. Only those that could inhibit parasite growth in the initial test were further tested for their inhibitory kinetics at concentrations ranging from 0 to 1.0 mM. Negative controls, which received no parasites and/or no treatment, were included in each experiment. Positive controls used paromomycin, the commonly used laboratory standard for in vitro drug testing. At least two wells in different plates were employed for each experimental condition, and all experiments were repeated at least in triplicate.
Quantitative analysis of drug efficacy regarding parasite growth using quantitative RT–PCR
Total RNA was isolated from parasite-infected cells at 44 h post-infection using the RNeasy isolation kit (Qiagen). The concentration and quality of the RNA in each sample were determined by measuring absorbances at 260 and 280 nm. All RNA samples were adjusted to a concentration of 20 ng/μL for use in quantitative RT–PCR (qRT–PCR). A SYBR Green-based real time qRT–PCR method was used to detect parasite 18S rRNA using a pair of previously published primers: 995F (5′ TAG AGA TTG GAG GTT GTT CCT 3′) and 1206R (5′ CTC CAC CAA CTA AGA ACG GCC 3′).12,19,21 For normalization, human 18S rRNA levels were also detected for each sample using the previously published primer pair F1373 (5′ CCG ATA ACG AAC GAG ACT CTG G 3′) and R1561 (5′ TAG GGT AGG CAC ACG CTG AGC C 3′).12,22 Reaction mixtures containing 20 ng total RNA and appropriate amounts of reagents and primers were first incubated at 48°C for 30 min to synthesize cDNA, heated at 95°C for 15 min to inactivate the reverse transcriptase, and then subjected to 40 thermal cycles (95°C for 20 s, 50°C for 30 s and 72°C for 30 s) of PCR amplification with an iCycler iQ real-time PCR detection system (Bio-Rad Laboratories). At least two reaction replicates were performed for each experimental condition, and each experiment was performed in at least triplicate.
Quantitative analysis was carried out as previously described by our laboratory.22 Inhibition curves derived from quantitative analysis were subjected to non-linear regression against log-transformed compound concentrations using the Prism v4.03 program (GraphPad Software). The IC50 for each compound was derived from the sigmoidal model by determining the compound concentrations that resulted in a 50% reduction of parasite growth when compared with the growth of the controls.
In vitro cytotoxicity assay
To ensure that apparent parasite inhibition was not in fact due to the compounds actually inhibiting the host cells, we analysed host cell inhibition using an MTT-based in vitro Toxicology Assay Kit (Sigma). All four compounds were tested at both high (1 mM) and low (10 μM) concentrations. Positive controls included paromomycin at 0.8 mg/mL and 0.1 mg/mL. Negative controls that included no compound were included in each experiment as a baseline. At least two wells in different plates were employed for each experimental condition, and all experiments were repeated in at least triplicate.
Results
Determination of CpACBP1 binding activity and substrate preference
We observed increased fluorescence emission at 538 nm by NBD-C16:0-CoA upon binding to CpACBP1 protein (Figure 1a). The binding was specific, as the MBP-tag control group emitted virtually no signal. The binding was affected by pH, and the optimal condition for binding was determined to be pH 7.5 (Figure 1b). Using this fluorometric assay, we determined that CpACBP1 had a dissociation constant (Kd) of 171.2 nM towards NBD-C16:0-CoA (Figure 2). We also assayed the short form CpACBP1 without the ankyrin repeats domain (CpACBP1ΔAnk) and observed a slight reduction in its binding affinity towards NBD-C16:0-CoA (i.e. Kd = 366.7 nM) (Figure 2), which suggests that the ankyrin repeats might also contribute to the conformational stability of the protein in addition to their usual involvement in interacting with other proteins or molecules.
Figure 1.

Specific binding between CpACBP1 and NBD-labelled palmitoyl-CoA (NBD-C16:0-CoA) detected by fluorescence assay (excitation 460 nm; emission 538 nm). (a) Specific and non-specific binding of NBD-C16:0-CoA (0.25 μM) by MBP-fused CpACBP1 (0.1 μM) and the MBP tag (0.1 μM). (b) Effect of pH on CpACBP1 binding to NBD-C16:0-CoA. MBP was used as a control for background subtraction. Bars represent the SEM from triplicate experiments. a.u., arbitrary units.
Figure 2.
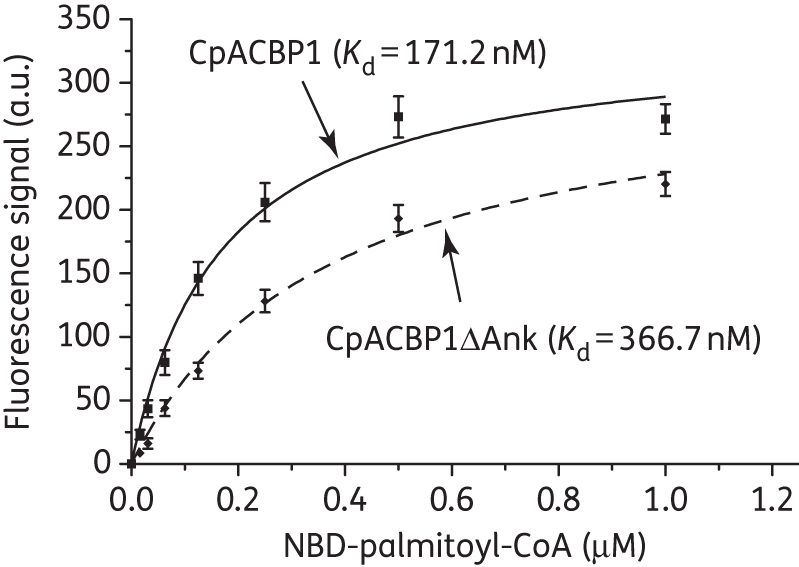
Binding kinetics of recombinant full-length CpACBP1 and short-form protein containing only the ACBP domain (CpACBPΔAnk) with NBD-C16:0-CoA as determined by the fluorescence assay. Bars represent the SEM from triplicate experiments. a.u., arbitrary units.
When various unlabelled fatty acyl-CoA esters were included in the reactions to compete with NBD-C16:0-CoA at an equal molar concentration of 25 μM, CpACBP1 displayed preferential binding towards short- to long-chain fatty acyl CoA esters (i.e. C4:0 to C16:0) (Figure 3). CpACBP1 had decreased binding affinities towards very long chain esters (C18:0 to C24:0) and little or no binding affinity towards the four unsaturated fatty acyl esters under investigation (C18:1, C18:3, C20:4 and C22:6). It had no binding affinity at all towards palmitic acid at 0.1, 0.25 and 1.0 μM, which was consistent with the ACBP family of proteins (Figure 3).
Figure 3.

Substrate preference of CpACBP1 as determined by competition assay. Saturated fatty acyl-CoAs from C2:0 to C26:0 and select unsaturated fatty acyl-CoAs were used to compete with NBD-C16:0-CoA at the same molar concentration (0.25 μM) for binding to CpACBP1. Additionally, palmitic acid (C16:0) at three concentrations (0.1, 0.25 and 1.0 μM) was used to determine whether CpACBP1 bound to fatty acids. Values are represented as the percentage activity of CpACBP1 relative to controls containing only NBD-C16:0-CoA. Bars represent the SEM from triplicate experiments.
Presence of CpACBP1 inhibitors in the compound library and their inhibition kinetics
Primary screening of the NIH/JDRF compound library using the fluorometric assay identified 37 (3.56%) out of the 1040 compounds that displayed ≥50% inhibition of binding between CpACBP1 and NBD-C16:0-CoA. Among them, nine displayed absorption spectra in the 580–600 nm range, which could interfere with the assay, and were thus excluded from subsequent analysis. The remaining 28 (2.7%) top compounds were further tested, and their IC50 values were determined to range between 0.018 μM (acetazolamide) and 0.811 μM (homatropine methylbromide) (Table 1). As shown in Table 1, these top compounds possess a diverse range of bioactivities, ranging from antiviral, antibacterial, antifungal and antiparasitic to anti-inflammatory and antidepressant. The observed anti-ACBP activity was novel for these drugs.
Table 1.
IC50 and Ki values of the top 28 compounds for inhibition of CpACBP1 binding to NBD-palmitoyl-CoAa
| Compound | IC50 (μM) | Ki (μM) | Major bioactivity |
|---|---|---|---|
| 1S,2R-phenylpropanolamine HCl | 0.202 | 0.082 | decongestant, anorexic |
| Acetazolamideb | 0.018 | 0.007 | carbonic anhydrase inhibitor, diuretic, anti-glaucoma |
| Bithionolb | 0.117 | 0.048 | anthelmintic, antiseptic |
| Broxyquinolinec | 0.132 | 0.054 | anti-infectant, disinfectant |
| Chlorpromazineb | 0.144 | 0.059 | anti-emetic, antipsychotic |
| Cloxacillin sodiumc | 0.123 | 0.05 | antibacterial |
| Cloxyquinc | 0.097 | 0.039 | antibacterial, antifungal |
| Curcuminb | 0.216 | 0.088 | antibacterial, antifungal, lipo/cyclooxygenase inhibitor |
| Gambogic acid | 0.207 | 0.084 | anti-inflammatory, cytotoxic, inhibits HeLa cells in vitro |
| Homatropine methylbromide | 0.811 | 0.33 | anti-cholinergic (ophthalmic) |
| Hydralazine HClb | 0.163 | 0.066 | anti-hypertensive |
| Hydrocortisone acetate | 0.362 | 0.147 | glucocorticoid, anti-inflammatory |
| Isoxicam | 0.176 | 0.072 | anti-inflammatory |
| Meclocycline sulfosalicylate | 0.153 | 0.062 | antibacterial |
| Mitoxantrone HCl | 0.128 | 0.052 | antineoplastic |
| Oxacillin sodium | 0.272 | 0.111 | antibacterial |
| Phenazopyridine HCl | 0.144 | 0.059 | analgesic |
| Phenelzine sulphate | 0.199 | 0.081 | antidepressant |
| Phenothrin | 0.14 | 0.057 | ectoparasiticide |
| Phenytoin sodium | 0.327 | 0.133 | anticonvulsant, anti-epileptic |
| Pregnenolone | 0.259 | 0.105 | glucocorticoid, anti-inflammatory |
| Pristimerin | 0.168 | 0.068 | antineoplastic, anti-inflammatory |
| Quinalizarin | 0.141 | 0.057 | antiviral, HIV-1 integrase inhibitor |
| Rifampicinb | 0.227 | 0.092 | antibacterial |
| Rifaximin | 0.269 | 0.109 | antibacterial, RNA synthesis inhibitor |
| Sodium dehydrocholatec | 0.116 | 0.047 | choleretic |
| Streptozocin | 0.304 | 0.124 | antineoplastic, alkylating agent |
| Tyrothricinb | 0.039 | 0.016 | topical antibacterial |
aCompound names and major bioactivities are provided in the NIH/JDRF library.
bCompounds that were used to test their antiparasitic protozoal activities by other investigators (see Table 2 for more detail).
cCompounds that displayed anti-cryptosporidial activities in this study (see Figure 4 for more detailed kinetics).
Inhibition of parasite growth in vitro by CpACBP1 inhibitors
The top 28 compounds shown to inhibit the binding of NBD-C16:0-CoA to CpACBP1 were further tested for their effect on C. parvum growth in vitro. Our initial in vitro drug testing with two concentrations of drugs (10 μM and 1 mM) first identified four compounds that were effective against parasite growth in vitro. None of the other 24 compounds displayed any inhibition at either low or high concentration. More detailed drug testing revealed that the four compounds could inhibit parasite growth in vitro with low micromolar IC50 values. These effective compounds included broxyquinoline (IC50 64.9 μM), cloxyquin (IC50 25.1 μM) and cloxacillin sodium (IC50 36.2 μM), which are known antibacterial or antifungal agents, as well as sodium dehydrocholate (IC50 53.2 μM), which is a semi-synthetic bile salt and a choleretic agent (Figure 4). These IC50 values for parasite growth in vitro were ∼250–500 times higher than those for CpACBP1 binding to fatty acyl-CoA. Additionally, these four compounds displayed little or no toxicity towards host HCT-8 cells at IC50 concentrations, although low levels of cytotoxicity were observed at much higher concentrations (10 μM to 1 mM) (Figure 5). As a positive control, paromomycin displayed expected efficacy with an IC50 value of 112.3 μM, which is close to values reported previously.22
Figure 4.

Inhibition kinetics of the four effective compounds on in vitro growth of C. parvum. Paromomycin was included as a positive control. Values are represented as percentage inhibition relative to no-drug control groups. Bars represent the SEM from triplicate experiments. *The IC50 value for nitazoxanide was taken from an earlier publication.22
Figure 5.
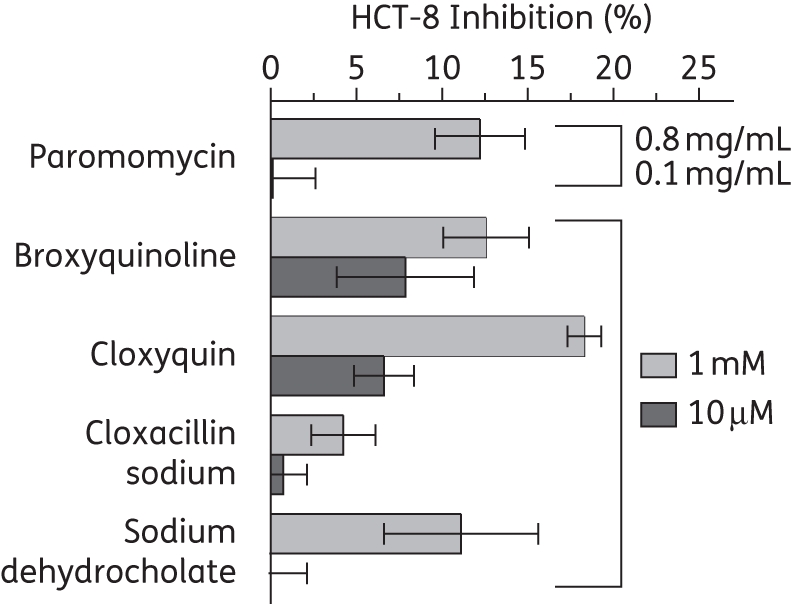
Cytotoxicity of the four effective compounds towards host HCT-8 cells as determined by the MTT assay. All compounds were tested at relatively high (1 mM) and low (10 μM) concentrations. Paromomycin was used as a control at high (0.8 mg/mL) and low (0.1 mg/mL) concentrations. Bars represent the SEM from triplicate experiments.
Discussion
The binding kinetics between ACBP and fatty acyl-CoA have traditionally been determined with a Lipidex 1000 assay.5,23,24 Because this conventional assay uses radioactive materials, it is less safe and unfriendly for high-throughput screening (HTS). Lipidex resin also competes with ACBP when used to remove unbound substrates, which compromises the assay's sensitivity and accuracy. In contrast, the newly developed NBD-based fluorometric assay overcomes these weaknesses, particularly when used for HTS of CpACBP1 inhibitors. The fluorescence emission of NBD is highly environmentally sensitive. This feature has been explored to study various fatty acid and fatty acyl-CoA binding proteins from other organisms using NBD-labelled fatty acids or stearic CoA.6,15,25 A more recent example is the use of NBD-C16:0-CoA to study in vitro acyl CoA:diacylglycerol acyltransferase activity.26 In the present study, we developed a fluorescence assay using NBD-C16:0-CoA to study the binding properties and HTS of inhibitors of CpACBP1, which to our knowledge is the first such study of an ACBP family protein.
The new fluorescence assay produced a Km value of 171.2 nM for the binding of full-length CpACBP1 to radioactive C16:0-CoA, which is ∼2.4-fold lower than, but comparable to, the value obtained in our previous study using the Lipidex 1000 assay (i.e. 405 nM).5 The newly obtained substrate preference data were also similar to those obtained previously, except that CpACBP1 was unable to bind acyl-CoA esters with ≥20 carbons using the Lipidex 1000 assay.5 These slight differences in both Km values and substrate preferences are likely due to the binding competition between ACBP and Lipidex 1000 resin during the extraction step of the assay.5,27 It is also possible that the presence of the NBD group in the fatty acyl-CoA might slightly change the binding affinity.
Using the fluorescence assay, we also performed HTS of 1040 compounds and identified 28 top candidates that could inhibit CpACBP1 binding activity by ≥50% at 0.25 μM. Subsequent in vitro drug testing identified 4 of the 28 compounds that displayed efficacy against parasite growth in vitro at low micromolar levels. These included β-lactamase-resistant penicillin (cloxacillin sodium) and two quinoline derivatives (broxyquinoline and cloxyquin), which are known for their antibacterial, antifungal and antiprotozoal activities,28–30 as well as a semisynthetic bile salt (sodium dehydrocholate), which is primarily used as a choleretic agent.31,32 These data are the first to show that these four drugs could target an ACBP protein and inhibit the growth of an apicomplexan parasite in vitro. Their in vitro IC50 values range from around 25 to 65 μM, which are about 4- to 65-fold higher than the values for the FDA-approved anti-cryptosporidial drug nitazoxanide (IC50 ∼1–4 μM), but about 1.7–4.5× lower than that of paromomycin (IC50 ∼112.3) as determined in this study or reported previously (IC50 125–170 μM).22
It is noticeable that acetazolamide and tyrothricin were the two most effective compounds in inhibiting CpACBP1 binding activity (IC50 0.018 and 0.039 μM, respectively) (Table 1). However, these two compounds displayed no efficacy against parasite growth in vitro, which ruled out their potential as anti-cryptosporidial drugs. On the other hand, their extremely high affinity to CpACBP1 indicates that their analogues are worth examining.
We studied at least 8 out of the top 28 compounds in greater detail for their effects on several other parasitic protozoa (Table 2). Only three of the drugs presented here have previously been tested as treatments for cryptosporidiosis. Among them, bithionol was shown to be relatively inactive in in vitro studies.33 In another study, rifampicin reduced the number of parasites by only 17.4% when used at a concentration of 8 μg/μL and by 74.4% when used in combination with 50 μM ranalexin.34 These observations are congruent with our in vitro drug testing data on the ineffectiveness of bithionol and rifampicin on C. parvum growth. On the other hand, in two uncontrolled studies with a small number of patients with HIV infection, treatments with rifaximin resulted in resolution of clinical symptoms and the clearance of infection.35 In the present study, rifaximin could effectively inhibit CpACBP1 binding activity (IC50 0.227 μM) (Table 1). However, it displayed no effect on parasite growth in our primary in vitro drug testing at both 10 μM and 1.0 mM. If rifaximin is truly effective in treating cryptosporidial infection in patients, our data imply that CpACBP1 could be its target, or at least one of its targets, whereas the ineffectiveness of rifaximin under in vitro conditions might be explained as follows. First, rifaximin may have different absorption kinetics between in vitro and in vivo conditions. Second, this drug may be more effective on the later sexual development and/or oocyst formation stages of C. parvum, which could not be detected by the current 2 day in vitro drug assay.
Table 2.
CpACBP1 inhibitors from this study that have been examined for their antiparasitic protozoal activities
| Parasitic protozoa | Compoundreference(s) |
|---|---|
| Cryptosporidium spp. | bithionol33 |
| rifampicin34 | |
| rifaximin35 | |
| Plasmodium spp. | acetazolamide36–38 |
| chlorpromazine39–42 | |
| curcumin43,44 | |
| rifampicin45,46 | |
| tyrothricin47 | |
| Giardia lamblia | bithionol48 |
| curcumin49 | |
| Trichomonas vaginalis | bithionol48 |
| Entamoeba histolytica | bithionol50 |
| chlorpromazine51 | |
| Trypanosoma spp. | chlorpromazine52–54 |
| hydralazine55 | |
| Schistosoma mansoni | chlorpromazine56 |
| Leishmania spp. | chlorpromazine57,58 |
| curcumin59 | |
| rifampicin60,61 |
ACBP has been characterized as a family of housekeeping proteins that plays a critical and regulatory role in lipid metabolism.27 These proteins bind and sequester fatty acyl-CoA esters and act as an intracellular acyl-CoA transporter and pool former. The parasite C. parvum possesses only a single ACBP (CpACBP1). It is localized to the unique host cell membrane-derived PVM, which separates the extracytoplasmic parasite from the extracellular environment, such as the gut. The present study not only identified a number of known drugs that could serve as CpACBP1 inhibitors, but also indicated that several of them could in fact inhibit the growth of C. parvum in vitro by targeting CpACBP1, probably by making the parasite incapable of further processing fatty acids. These observations suggest that CpACBP1 and PVM may serve as novel drug targets in the treatment of cryptosporidial infection.
Because these effective compounds are known drugs, it is worth further testing their potential effects against cryptosporidial infection in vivo. If satisfactory efficacies could be observed, these drugs might be quickly repurposed to treat cryptosporidiosis, for which effective treatments are currently unavailable for immunocompromised patients. Additionally, the four top drugs may also serve as leads for synthesizing new analogues in future studies to identify more effective anti-cryptosporidial drugs.
Funding
The project described was supported by grant number R01AI44594 from the National Institute of Allergy and Infectious Diseases (NIAID) (to G. Z.).
Transparency declarations
None to declare.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of NIAID or the NIH.
Acknowledgements
We thank Dr Friedhelm Schroeder for giving us access to the drug library consisting of 1040 compounds from the Microsource Discovery Systems as the NIH/JDRF custom collection. The use of the compound library was also authorized by NIH/JDRF.
References
- 1.Mead JR. Cryptosporidiosis and the challenges of chemotherapy. Drug Resist Updat. 2002;5:47–57. doi: 10.1016/s1368-7646(02)00011-0. doi:10.1016/S1368-7646(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 2.Tzipori S, Griffiths JK. Natural history and biology of Cryptosporidium parvum. Adv Parasitol. 1998;40:5–36. doi: 10.1016/s0065-308x(08)60116-5. doi:10.1016/S0065-308X(08)60116-5. [DOI] [PubMed] [Google Scholar]
- 3.Huang BQ, Chen XM, LaRusso NF. Cryptosporidium parvum attachment to and internalization by human biliary epithelia in vitro: a morphologic study. J Parasitol. 2004;90:212–21. doi: 10.1645/GE-3204. doi:10.1645/GE-3204. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths JK, Balakrishnan R, Widmer G, et al. Paromomycin and geneticin inhibit intracellular Cryptosporidium parvum without trafficking through the host cell cytoplasm: implications for drug delivery. Infect Immun. 1998;66:3874–83. doi: 10.1128/iai.66.8.3874-3883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng B, Cai X, Zhu G. Functional characterization of a fatty acyl-CoA-binding protein (ACBP) from the apicomplexan Cryptosporidium parvum. Microbiology. 2006;152:2355–63. doi: 10.1099/mic.0.28944-0. doi:10.1099/mic.0.28944-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gossett RE, Frolov AA, Roths JB, et al. Acyl-CoA binding proteins: multiplicity and function. Lipids. 1996;31:895–918. doi: 10.1007/BF02522684. doi:10.1007/BF02522684. [DOI] [PubMed] [Google Scholar]
- 7.Knudsen J, Neergaard TB, Gaigg B, et al. Role of acyl-CoA binding protein in acyl-CoA metabolism and acyl-CoA-mediated cell signaling. J Nutr. 2000;130:294S–8S. doi: 10.1093/jn/130.2.294S. [DOI] [PubMed] [Google Scholar]
- 8.Schroeder F, Jolly CA, Cho TH, et al. Fatty acid binding protein isoforms: structure and function. Chem Phys Lipids. 1998;92:1–25. doi: 10.1016/s0009-3084(98)00003-6. doi:10.1016/S0009-3084(98)00003-6. [DOI] [PubMed] [Google Scholar]
- 9.Burton M, Rose TM, Faergeman NJ, et al. Evolution of the acyl-CoA binding protein (ACBP) Biochem J. 2005;392:299–307. doi: 10.1042/BJ20050664. doi:10.1042/BJ20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chye ML, Huang BQ, Zee SY. Isolation of a gene encoding Arabidopsis membrane-associated acyl-CoA binding protein and immunolocalization of its gene product. Plant J. 1999;18:205–14. doi: 10.1046/j.1365-313x.1999.00443.x. doi:10.1046/j.1365-313X.1999.00443.x. [DOI] [PubMed] [Google Scholar]
- 11.Leung KC, Li HY, Xiao S, et al. Arabidopsis ACBP3 is an extracellularly targeted acyl-CoA-binding protein. Planta. 2006;223:871–81. doi: 10.1007/s00425-005-0139-2. doi:10.1007/s00425-005-0139-2. [DOI] [PubMed] [Google Scholar]
- 12.Fritzler JM, Millership JJ, Zhu G. Cryptosporidium parvum long-chain fatty acid elongase. Eukaryot Cell. 2007;6:2018–28. doi: 10.1128/EC.00210-07. doi:10.1128/EC.00210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fritzler JM, Zhu G. Functional characterization of the acyl-[acyl carrier protein] ligase in the Cryptosporidium parvum giant polyketide synthase. Int J Parasitol. 2007;37:307–16. doi: 10.1016/j.ijpara.2006.10.014. doi:10.1016/j.ijpara.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu G. Current progress in the fatty acid metabolism in Cryptosporidium parvum. J Eukaryot Microbiol. 2004;51:381–8. doi: 10.1111/j.1550-7408.2004.tb00384.x. doi:10.1111/j.1550-7408.2004.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 15.Schroeder F, Myers-Payne SC, Billheimer JT, et al. Probing the ligand binding sites of fatty acid and sterol carrier proteins: effects of ethanol. Biochemistry. 1995;34:11919–27. doi: 10.1021/bi00037a033. doi:10.1021/bi00037a033. [DOI] [PubMed] [Google Scholar]
- 16.Hostetler HA, Syler LR, Hall LN, et al. A novel high-throughput screening assay for putative antidiabetic agents through PPARα interactions. J Biomol Screen. 2008;13:855–61. doi: 10.1177/1087057108323127. doi:10.1177/1087057108323127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nesterenko MV, Upton SJ. A rapid microcentrifuge procedure for purification of Cryptosporidium sporozoites. J Microbiol Methods. 1996;25:87–9. doi:10.1016/0167-7012(95)00083-6. [Google Scholar]
- 18.Zhu G, Keithly JS. Molecular analysis of a P-type ATPase from Cryptosporidium parvum. Mol Biochem Parasitol. 1997;90:307–16. doi: 10.1016/s0166-6851(97)00168-0. doi:10.1016/S0166-6851(97)00168-0. [DOI] [PubMed] [Google Scholar]
- 19.Cai X, Lancto CA, Abrahamsen MS, et al. Intron-containing β-tubulin transcripts in Cryptosporidium parvum cultured in vitro. Microbiology. 2004;150:1191–5. doi: 10.1099/mic.0.26897-0. doi:10.1099/mic.0.26897-0. [DOI] [PubMed] [Google Scholar]
- 20.Upton SJ, Tilley M, Brillhart DB. Effects of select medium supplements on in vitro development of Cryptosporidium parvum in HCT-8 cells. J Clin Microbiol. 1995;33:371–5. doi: 10.1128/jcm.33.2.371-375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abrahamsen MS, Schroeder AA. Characterization of intracellular Cryptosporidium parvum gene expression. Mol Biochem Parasitol. 1999;104:141–6. doi: 10.1016/s0166-6851(99)00081-x. doi:10.1016/S0166-6851(99)00081-X. [DOI] [PubMed] [Google Scholar]
- 22.Cai X, Woods KM, Upton SJ, et al. Application of quantitative real-time reverse transcription-PCR in assessing drug efficacy against the intracellular pathogen Cryptosporidium parvum in vitro. Antimicrob Agents Chemother. 2005;49:4437–42. doi: 10.1128/AAC.49.11.4437-4442.2005. doi:10.1128/AAC.49.11.4437-4442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasmussen JT, Borchers T, Knudsen J. Comparison of the binding affinities of acyl-CoA-binding protein and fatty-acid-binding protein for long-chain acyl-CoA esters. Biochem J. 1990;265:849–55. doi: 10.1042/bj2650849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosendal J, Ertbjerg P, Knudsen J. Characterization of ligand binding to acyl-CoA-binding protein. Biochem J. 1993;290:321–6. doi: 10.1042/bj2900321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrescu AD, Hertz R, Bar-Tana J, et al. Ligand specificity and conformational dependence of the hepatic nuclear factor-4α (HNF-4α) J Biol Chem. 2002;277:23988–99. doi: 10.1074/jbc.M201241200. doi:10.1074/jbc.M201241200. [DOI] [PubMed] [Google Scholar]
- 26.McFie PJ, Stone SJ. A fluorescent assay to quantitatively measure in vitro acyl CoA:diacylglycerol acyltransferase activity. J Lipid Res. 2011;52:1760–4. doi: 10.1194/jlr.D016626. doi:10.1194/jlr.D016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen JT, Faergeman NJ, Kristiansen K, et al. Acyl-CoA-binding protein (ACBP) can mediate intermembrane acyl-CoA transport and donate acyl-CoA for β-oxidation and glycerolipid synthesis. Biochem J. 1994;299:165–70. doi: 10.1042/bj2990165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawrence SL, Roth V, Slinger R, et al. Cloxacillin versus vancomycin for presumed late-onset sepsis in the Neonatal Intensive Care Unit and the impact upon outcome of coagulase negative staphylococcal bacteremia: a retrospective cohort study. BMC Pediatr. 2005;5:49. doi: 10.1186/1471-2431-5-49. doi:10.1186/1471-2431-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma CS. Letter: Effect of broxyquinoline and broxaldine in leprosy. Lancet. 1975;1:405. doi: 10.1016/s0140-6736(75)91334-3. doi:10.1016/S0140-6736(75)91334-3. [DOI] [PubMed] [Google Scholar]
- 30.Hongmanee P, Rukseree K, Buabut B, et al. In vitro activities of cloxyquin (5-chloroquinolin-8-ol) against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1105–6. doi: 10.1128/AAC.01310-06. doi:10.1128/AAC.01310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yousef IM, Mignault D, Weber AM, et al. Influence of dehydrocholic acid on the secretion of bile acids and biliary lipids in rats. Digestion. 1990;45:40–51. doi: 10.1159/000200223. doi:10.1159/000200223. [DOI] [PubMed] [Google Scholar]
- 32.Bobowiec R, Studzinski T, Sikorska M. Effects of sodium taurocholate and sodium dehydrocholate on bile flow, lipid and bilirubin secretion in sheep. Acta Physiol Pol. 1990;41:146–54. [PubMed] [Google Scholar]
- 33.Woods KM, Nesterenko MV, Upton SJ. Efficacy of 101 antimicrobials and other agents on the development of Cryptosporidium parvum in vitro. Ann Trop Med Parasitol. 1996;90:603–15. doi: 10.1080/00034983.1996.11813090. [DOI] [PubMed] [Google Scholar]
- 34.Giacometti A, Cirioni O, Barchiesi F, et al. In vitro anticryptosporidial activity of ranalexin alone and in combination with other peptides and with hydrophobic antibiotics. Eur J Clin Microbiol Infect Dis. 1999;18:827–9. doi: 10.1007/s100960050410. doi:10.1007/s100960050410. [DOI] [PubMed] [Google Scholar]
- 35.Amenta M, Dalle Nogare ER, Colomba C, et al. Intestinal protozoa in HIV-infected patients: effect of rifaximin in Cryptosporidium parvum and Blastocystis hominis infections. J Chemother. 1999;11:391–5. doi: 10.1179/joc.1999.11.5.391. [DOI] [PubMed] [Google Scholar]
- 36.Krungkrai J, Scozzafava A, Reungprapavut S, et al. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: towards antimalarials with a novel mechanism of action? Bioorg Med Chem. 2005;13:483–9. doi: 10.1016/j.bmc.2004.10.015. doi:10.1016/j.bmc.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 37.Krungkrai SR, Suraveratum N, Rochanakij S, et al. Characterisation of carbonic anhydrase in Plasmodium falciparum. Int J Parasitol. 2001;31:661–8. doi: 10.1016/s0020-7519(01)00172-2. doi:10.1016/S0020-7519(01)00172-2. [DOI] [PubMed] [Google Scholar]
- 38.Reungprapavut S, Krungkrai J, Krungkrai SR. Plasmodium falciparum carbonic anhydrase is a possible target for malaria chemotherapy. J Enzyme Inhib Med Chem. 2004;19:249–56. doi: 10.1080/14756360410001689577. doi:10.1080/14756360410001689577. [DOI] [PubMed] [Google Scholar]
- 39.Geary TG, Divo AA, Jensen JB. Effect of calmodulin inhibitors on viability and mitochondrial potential of Plasmodium falciparum in culture. Antimicrob Agents Chemother. 1986;30:785–8. doi: 10.1128/aac.30.5.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalkanidis M, Klonis N, Tilley L, et al. Novel phenothiazine antimalarials: synthesis, antimalarial activity, and inhibition of the formation of β-haematin. Biochem Pharmacol. 2002;63:833–42. doi: 10.1016/s0006-2952(01)00840-1. doi:10.1016/S0006-2952(01)00840-1. [DOI] [PubMed] [Google Scholar]
- 41.Miki A, Tanabe K, Nakayama T, et al. Plasmodium chabaudi: association of reversal of chloroquine resistance with increased accumulation of chloroquine in resistant parasites. Exp Parasitol. 1992;74:134–42. doi: 10.1016/0014-4894(92)90040-h. doi:10.1016/0014-4894(92)90040-H. [DOI] [PubMed] [Google Scholar]
- 42.Satayavivad J, Wongsawatkul O, Bunnag D, et al. Flunarizine and verapamil inhibit chloroquine-resistant Plasmodium falciparum growth in vitro. Southeast Asian J Trop Med Public Health. 1987;18:253–8. [PubMed] [Google Scholar]
- 43.Cui L, Miao J. Cytotoxic effect of curcumin on malaria parasite Plasmodium falciparum: inhibition of histone acetylation and generation of reactive oxygen species. Antimicrob Agents Chemother. 2007;51:488–94. doi: 10.1128/AAC.01238-06. doi:10.1128/AAC.01238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy RC, Vatsala PG, Keshamouni VG, et al. Curcumin for malaria therapy. Biochem Biophys Res Commun. 2005;326:472–4. doi: 10.1016/j.bbrc.2004.11.051. doi:10.1016/j.bbrc.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 45.Pukrittayakamee S, Prakongpan S, Wanwimolruk S, et al. Adverse effect of rifampin on quinine efficacy in uncomplicated falciparum malaria. Antimicrob Agents Chemother. 2003;47:1509–13. doi: 10.1128/AAC.47.5.1509-1513.2003. doi:10.1128/AAC.47.5.1509-1513.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pukrittayakamee S, Viravan C, Charoenlarp P, et al. Antimalarial effects of rifampin in Plasmodium vivax malaria. Antimicrob Agents Chemother. 1994;38:511–4. doi: 10.1128/aac.38.3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rautenbach M, Vlok NM, Stander M, et al. Inhibition of malaria parasite blood stages by tyrocidines, membrane-active cyclic peptide antibiotics from Bacillus brevis. Biochim Biophys Acta. 2007;1768:1488–97. doi: 10.1016/j.bbamem.2007.01.015. doi:10.1016/j.bbamem.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi T, Kobayashi S, Tanabe M, et al. In vitro inhibition of Giardia lamblia and Trichomonas vaginalis growth by bithionol, dichlorophene, and hexachlorophene. Antimicrob Agents Chemother. 1985;27:65–70. doi: 10.1128/aac.27.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez-Arriaga L, Mendoza-Magana ML, Cortes-Zarate R, et al. Cytotoxic effect of curcumin on Giardia lamblia trophozoites. Acta Trop. 2006;98:152–61. doi: 10.1016/j.actatropica.2006.03.005. doi:10.1016/j.actatropica.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 50.Takeuchi T, Kobayashi S, Kawasaki H. Entamoeba histolytica: inhibition in vitro by bithionol of respiratory activity and growth. Exp Parasitol. 1984;58:1–7. doi: 10.1016/0014-4894(84)90015-8. doi:10.1016/0014-4894(84)90015-8. [DOI] [PubMed] [Google Scholar]
- 51.Ondarza RN, Hernandez E, Iturbe A, et al. Inhibitory and lytic effects of phenothiazine derivatives and related tricyclic neuroleptic compounds, on Entamoeba histolytica HK9 and HM1 trophozoites. Biotechnol Appl Biochem. 2000;32:61–7. doi: 10.1042/ba19990099. doi:10.1042/BA19990099. [DOI] [PubMed] [Google Scholar]
- 52.De Castro SL, Soeiro MN, De Meirelles Mde N. Trypanosoma cruzi: effect of phenothiazines on the parasite and its interaction with host cells. Mem Inst Oswaldo Cruz. 1992;87:209–15. doi: 10.1590/s0074-02761992000200007. doi:10.1590/S0074-02761992000200007. [DOI] [PubMed] [Google Scholar]
- 53.Faerman CH, Savvides SN, Strickland C, et al. Charge is the major discriminating factor for glutathione reductase versus trypanothione reductase inhibitors. Bioorg Med Chem. 1996;4:1247–53. doi: 10.1016/0968-0896(96)00120-4. doi:10.1016/0968-0896(96)00120-4. [DOI] [PubMed] [Google Scholar]
- 54.Gutierrez-Correa J, Stoppani AO. Myeloperoxidase-generated phenothiazine cation radicals inactivate Trypanosoma cruzi dihydrolipoamide dehydrogenase. Rev Argent Microbiol. 2002;34:83–94. [PubMed] [Google Scholar]
- 55.Kalu AU, Haruna E. Effects of vasopressor drugs on number of Trypanosoma congolense in ruminant blood. Vet Parasitol. 1985;17:287–94. doi: 10.1016/0304-4017(85)90019-6. doi:10.1016/0304-4017(85)90019-6. [DOI] [PubMed] [Google Scholar]
- 56.Boyle JP, Zaide JV, Yoshino TP. Schistosoma mansoni: effects of serotonin and serotonin receptor antagonists on motility and length of primary sporocysts in vitro. Exp Parasitol. 2000;94:217–26. doi: 10.1006/expr.2000.4500. doi:10.1006/expr.2000.4500. [DOI] [PubMed] [Google Scholar]
- 57.Pearson RD, Manian AA, Hall D, et al. Antileishmanial activity of chlorpromazine. Antimicrob Agents Chemother. 1984;25:571–4. doi: 10.1128/aac.25.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Werbovetz KA, Lehnert EK, Macdonald TL, et al. Cytotoxicity of acridine compounds for Leishmania promastigotes in vitro. Antimicrob Agents Chemother. 1992;36:495–7. doi: 10.1128/aac.36.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan MM, Adapala NS, Fong D. Curcumin overcomes the inhibitory effect of nitric oxide on Leishmania. Parasitol Res. 2005;96:49–56. doi: 10.1007/s00436-005-1323-9. doi:10.1007/s00436-005-1323-9. [DOI] [PubMed] [Google Scholar]
- 60.Berman JD, Lee LS. Activity of oral drugs against Leishmania tropica in human macrophages in vitro. Am J Trop Med Hyg. 1983;32:947–51. doi: 10.4269/ajtmh.1983.32.947. [DOI] [PubMed] [Google Scholar]
- 61.Choi CM, Lerner EA. Leishmaniasis: recognition and management with a focus on the immunocompromised patient. Am J Clin Dermatol. 2002;3:91–105. doi: 10.2165/00128071-200203020-00003. doi:10.2165/00128071-200203020-00003. [DOI] [PubMed] [Google Scholar]