

Abstract
Oxidative stress is closely involved in age-related diseases and ageing itself. There is evidence of the leading contribution of oxidative damage to neurodegenerative disease, in contrast to other diseases where oxidative stress plays a secondary role. The 42-mer amyloid β (Aβ42) peptide is thought to be a culprit in the pathogenesis of Alzheimer's disease (AD). Aβ42 aggregates form the oligomeric assembly and show neurotoxicity, causing synaptic dysfunction. Aβ42 also induces tissue oxidation (DNA/RNA, proteins, and lipids) through trace metals (Cu, Zn, and Fe), which can be protected by antioxidant enzymes, vitamin C, and vitamin E. Superoxide dismutase catalyzes the conversion of toxic superoxide radical to less reactive hydrogen peroxide, contributing to protection from AD. Here we review the involvement of oxidative stress in AD progression induced from an imbalance between the radical formation of Aβ42 itself together with unique turn structure at positions Glu22 and Asp23 and several defense systems.
1. Oxidative Stress in Ageing—The Involvement of Superoxide Radical
Oxidative stress caused by reactive oxygen species (ROS) has been implicated in numerous age-related diseases and ageing itself [1, 2]. ROS include superoxide anions, hydrogen peroxide, hydroxyl radicals, and singlet oxygen. ROS are also involved in neurodegeneration such as Alzheimer's disease (AD), Parkinson's disease, and amyotrophic lateral sclerosis, because the brain is one of the most vulnerable tissues in the body to oxidative injuries based on its high rate of oxygen consumption [3]. The hydroxyl radical is believed to be one of the main stimuli of oxidative damage (Figure 1) and reacts with several biomolecules, leading to the formation of 8-hydroxydeoxyguanosine (8-OHdG)/8-hydroxyguanosine (8-OHG) in DNA/RNA, the formation of methionine sulfoxide, carbonylation in proteins, and lipid peroxidation. In particular, lipid peroxidation can lead to the production of 4-hydroxyl nonenal (4-HNE), malondialdehyde (MDA), and thiobarbituric acid-reacting substances (TBARS) as byproducts. The subsequent processes to hydroxyl radical could be involved in peroxynitrite formation by stimulating inducible nitric oxide synthase (i-NOS). Hydrogen peroxide (H2O2) is less reactive but is involved in the Fenton reaction (Haber-Weiss procedure, Figure 1), providing the hydroxyl radical.
Figure 1.
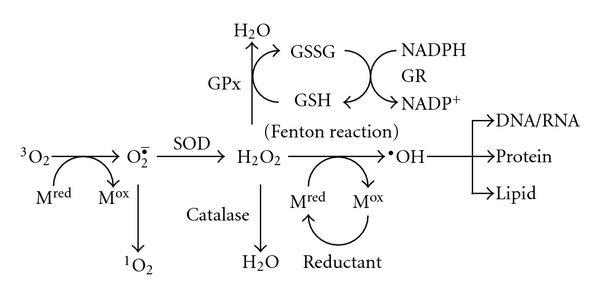
Generation of reactive oxygen species and defense systems in the cell. Mred or Mox, reduced or oxidized form of metals; SOD, superoxide dismutase; GSH, reduced glutathione; GSSG, oxidized glutathione; GR, glutathione reductase; GPx, glutathione peroxidase; NADPH or NADP+, reduced or oxidized nicotinamide adenine dinucleotide phosphate; VC, vitamin C; VE, vitamin E.
On the other hand, the superoxide radical is also biologically toxic, especially under the condition of radical-radical reactions which could occur at a diffusion-controlled rate. It presents widely in high quantity. A small fraction (0.4%–4%) of oxygen utilized in the mitochondria is reduced by single electron transfer during the initial step of the electron transport chain, followed by the generation of superoxide radical [4]. Catalase and peroxidases (such as glutathione peroxidase: GPx), which exists in ubiquitous tissues, can eliminate H2O2 generated from superoxide radicals (Figure 1).
Superoxide dismutases (SODs) are the main antioxidant enzymes that convert superoxide anions to H2O2, protecting cells and tissues from ROS generated from endogenous and exogenous sources [5]. SODs consist of three types of isoforms expressed in mammalian cells: copper/zinc SOD (CuZn-SOD, SOD1), which is located in the cytoplasm, manganese SOD (Mn-SOD, SOD2), which exists in the mitochondrial matrix, and extracellular SOD (EC-SOD, SOD3), which is also a complex of CuZn. Some CuZn-SOD is also seen in the intermembrane space of mitochondria [6].
Van Remmen is one of the most influential leaders in ageing research. She and her colleagues have reported several ageing symptoms of hepatic carcinoma [7] and muscle atrophy [8] in CuZn-SOD knockout mice. They also studied the physiological role of several antioxidant enzymes in longevity using gene-disrupting mice [9]. Notably, CuZn-SOD-deficient mice showed the multiple pathologies in several tissues with decreased lifespan compared to wild-type mice. Fujii and colleagues also indicated that hemolytic anemia was triggered by autoantibody production in CuZn-SOD-deficient mice [10].
Our group has reported that CuZn-SOD-deficient mice showed skin thinning [11] as well as increased drusen formation, which is a typical characteristic of age-related macular degeneration as neurodegeneration [12] and fatty liver [13]. Taken together, these observations demonstrate that CuZn-SOD knockout mice have the potential to be a valuable animal model for investigating human ageing. On the other hand, Shimizu and colleagues generated various tissue-specific Mn-SOD conditional knockout mice using a Cre-loxp system because total knockout of Mn-SOD induces neonatal lethality in mice [14, 15], liver-specific Mn-SOD knockout mice which show no obvious morphological abnormalities or biochemical changes in the liver [16], heart/muscle-specific Mn-SOD-deficient mice which exhibit dilated cardiomyopathy with the downregulation of specific biomolecules in the mitochondria [17], and skeletal muscle-specific Mn-SOD knockout mice which develop severe disturbance of exercise activity without muscle atrophy [18]. Furthermore, they found severe phenotypes in the brains of brain-specific Mn-SOD-deficient mice showing a spongiform encephalopathy-like pathology associated with gliosis [19]. The most abundant ROS within cells influencing synaptic plasticity, memory function, and neuronal death is considered to be the superoxide radical [20]; this suggests that SOD plays a protective role in neurodegeneration. We introduce the relevance of oxidative stress to AD in the following section.
2. Aβ Theory in Alzheimer's Disease
AD is generally characterized by the aggregation of amyloid β (Aβ) in senile plaques. Aβ mainly consists of 40- and 42-residue amyloid β peptides (Aβ40, Aβ42), secreted from amyloid precursor protein (APP) by two proteases (β- and γ-secretases) [21, 22]. Aβ42 plays a more critical role in the pathogenesis of AD than Aβ40 because Aβ42 aggregates more extensively to form fibrils and shows stronger neurotoxicity [23]. On the other hand, there is increasing evidence that the oligomeric assembly of Aβ could induce memory decline and synaptotoxicity in AD [24], while mature plaques were reported to be nontoxic [25, 26] and to serve as a store of the toxic assembly of Aβ [27].
Studies on several kinds of Aβ oligomer associated with neurotoxicity or synaptotoxicity have been accumulated. Recently, Teplow and colleagues summarized and overviewed Aβ assembly [28]: paranucleus, protofibrils (24–700 mer) [29], Aβ-derived diffusible ligands (ADDL, ~53 kDa) [30], Aβ*56 (~56 kDa, 12-mer) [31], amylospheroid (~150–700 kDa) [32], AβO (~90 kDa, 15–20 mer), annulus (150–250 kDa), and βamyball. Selkoe and colleagues suggested that Aβ dimers are the smallest synaptotoxic species and that plaque cores are largely inactive but sequester or release dimers [33]. They developed unique oligomer specific-ELISA using 82E1 antibody, whose epitope is N-terminal, for both antigen capture and detection, to reveal a clear correlation of the oligomer levels in the plasma and brain extracts with various cognitive levels of AD patients [34]. Oligomeric molecules of Aβ are believed to consist of 2 or 3x n-multimers based on the dimer or trimer, respectively. To elucidate the mechanism of Aβ oligomerization, many scientists have developed a method or detection tools. Bitan and colleagues created a method of the photoinduced cross-linking of unmodified proteins to prepare the oligomers in large quantity [35]. Glabe and colleagues generated a conformation-dependent antibody (A11 clone) against Aβ oligomers, which does not recognize fibrils and also reacts with other types of amyloid oligomers, such as α-synuclein in Parkinson's disease, polyglutamine in Huntington's disease, and prion peptide 106–126 in prion disease [36]. Recently, they reported the fibril-specific, conformation-dependent antibody (OC clone), recognizing soluble oligomers ranging from a dimer to greater than 250 kDa [37].
3. Role of Trace Metals and Formation of Aβ Radical in Alzheimer's Disease
In 1965, Terry and Pena. first reported the relevance of aluminum to the pathology of AD; they injected aluminum salts into the rabbit brain, resulting in neurofibrillary tangle formation [38], which is another hallmark of AD. Although aluminum in the diet or drinking water had been long believed as a risk factor for AD [39], Ehmann et al. in 1986 showed that this hypothesis for AD was an artifact [40].
It is known that transition metals, such as Cu, Zn, and Fe, are enriched in senile plaques [41]. Aβ causes protein oxidation, DNA/RNA oxidation, and lipid peroxidation in vitro and in vivo, possibly by aggregating to generate radicals via a trace of metal ions (Cu and Zn) [41–44]. The imbalance of copper homeostasis is also implicated in the etiology of AD [45]. The neurotoxic effects of Aβ42 and Aβ40 in cell culture correlate with the ability to reduce Cu(II) to Cu(I) and to generate H2O2 in a cell-free system [46]. The direct interaction of metals with Aβ in the N-terminal region is essential for its aggregation and neurotoxicity. In complex formation with Cu(II) [47, 48], each of the three histidine residues at positions 6, 13, and 14 of Aβ42, Tyr10 [46, 49–52], and Asp1/Asp7 [53] may be involved. Recent ESR studies by Drew et al. suggested that the Ala2 carbonyl could be involved in the Cu(II) coordination [54]. Tyr10 is easily oxidized to the tyrosyl radical by Cu(II), leading to the production of H2O2 [55]. Quite recently, Ono et al. reported that UK (H6R) and Tottori (D7N) mutations in the N-terminal regions accelerated the ability to form oligomers and enhanced cytotoxicity [56]. These mutations might change the binding mode of metal with Aβ peptides, resulting in the increased ability to form toxic oligomers.
Based on the metal etiology in AD, therapeutics using metal chelators might be promising to prevent plaque formation by extracting the metals. Bush and colleagues treated an APP transgenic mouse with a CuZn chelator, clioquinol (8-hydroxy quinoline), showing the effective removal of plaque depositions [57]; however, it might alter the homeostasis of copper and counteract the intracellular copper-depleting effects of APP in initial clinical trials of the treatment of AD [58]. Eventually, it was removed from the market by FDA due to difficulties associated with chelation of Co(II) involved in vitamin B12. They also mentioned that the problems were also due to the large magnitude difference in affinity of Cu(II) between clioquinol and Aβ42, in which K d of Cu(II) for clioquinol and Aβ42 are nanomolar and attomolar, respectively [59]. Other processes for plaque removal by clioquinol could be involved. Recently, they advanced the chelating strategy into the second-generation clioquinol analogue, PBT2, which outperformed clioquinol by markedly decreasing soluble interstitial Aβ and rescuing cognitive impairment [60]. PBT2 was already found to reverse frontal lobe functional deficits and to decrease Aβ42 in a phase IIa clinical trial [61].
Butterfield and colleagues pioneered the contribution of Met35 to the neurotoxicity and oxidative effects of Aβ [44, 62]. The oxidized form of Met35 was detected both in the brains of AD patients [63] and the APP transgenic mouse model [64]. They suggested the reactive form of Met35 in Aβ42 as an S-oxidized radical cation, abstracting an allylic hydrogen of phospholipid acyl chains to give allyl radicals, followed by lipid peroxidation [43]. The methionine sulfoxide reductase is known to reverse methionine oxidation. Moskovitz and colleagues reported that a knockout mouse of one isoform of this enzyme caused enhanced neurodegeneration in the brain hippocampus, implying that the oxidation of Met residue plays a role in brain pathology [65].
The S-oxidized radical cation in Met35 is generally too unstable to cause oxidative damage continuously [66]. A stabilization mechanism for long-lasting oxidative stress in AD progression is required. We have proposed the emerging role of the turn formation at Glu22 and Asp23 in the pathogenesis of AD [67, 68] and its contribution to oligomer formation [69] following intracellular amyloidgenesis [70]. Our continuous studies using a systematic proline replacement, solid-state NMR, and ESR have elucidated Aβ42-mediated neurotoxicity in vitro; the central turn formation could bring Tyr10 radical generated through trace metals accompanied by the generation of H2O2, which are moved close to Met35, resulting in the production of the S-oxidized radical cation (Figure 2(a)). The systematic proline replacement of Aβ42 proposed that not only the turn formation at Glu22 and Asp23 but the turn at Gly38 and Val39 increases aggregation and neurotoxicity [71]. This additional C-terminal turn could enable the carboxylate anion at Ala42 to interact with the S-oxidized radical cation by forming S-O bonding through an intramolecular β-sheet at positions 35–37 and 40–42 (Figure 2(a)). The resultant hydrophobic core in the C-terminus would enhance Aβ42 aggregation, sequestering or releasing the radical species for long-lasting oxidative stress in AD. If considered for the lower toxicity of Aβ40 toxicity, the S-oxidized radical cation of Met35 might not be fully stabilized by the incomplete association of Met35 radical with the carboxylate anion at Val40 (Figure 2(a)) or by the labile electrostatic interaction between the sulfur atom of Met35 and the amide carbonyl group of Ile31 under the condition of α-helix formation in the C-terminal region [72]. Collectively, the formation of toxic Aβ radicals generated through trace metals could induce the malfunction of signal transduction pathways after the interaction with membranes. This mechanism (Figure 2(a)) can in part explain why Aβ42 is more neurotoxic than Aβ40 [73]. The following generation of superoxide radical and hydroxyl radical occasionally accompanied with the stabilization of Aβ42 radical could attack the membranes and other macromolecules (Figure 2(b)). At least two Aβ42-mediated pathways are assumed.
Figure 2.
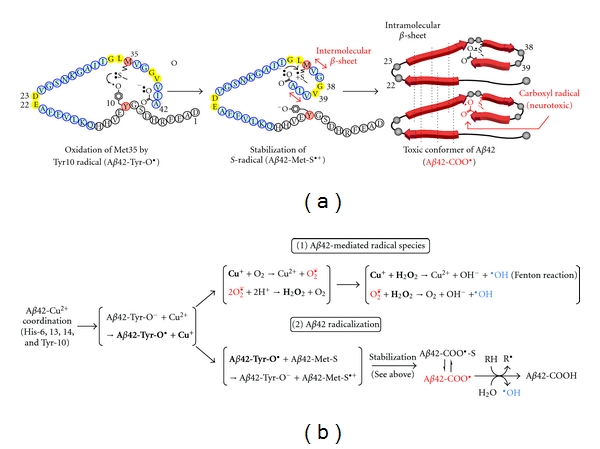
(a) Proposed mechanism of formation and stabilization of Aβ42 radical for long-lasting oxidative stress in AD and the toxic conformer of Aβ42 [73]. (b) Scheme of generation of Aβ42-mediated radical species (superoxide radical and hydroxyl radical) and the long-lasting Aβ42 radical in the pathogenesis of AD.
Recently, Butterfield and colleagues advanced the Met35 theory into in vivo analysis using APP transgenic mice with V717F (Indiana) and M631L mutations corresponding to the substitution of Met35 with Leu in the Aβ sequence [74], which showed the prevention of oxidative damages in tissues and senile plaque in the brain. Unexpectedly, M35L mutation in mice exhibited almost no effects on memory and learning impairments in the Morris water maze [74], indicating that oxidative stress may be neither required nor sufficient for memory loss. Quite recently, Bitan and colleagues suggested that Met35 is not necessary for Aβ toxicity despite its significant role in aggregation [75]. Other mechanisms in addition to Met35 will occur for the complete explanation of Aβ42-induced neurotoxicity.
A relationship between Aβ oligomers and oxidative stress has been noted; Klein and colleagues proposed that ADDL induce long-term potentiation associated with oxidative damage in vitro [76]. Barnham and colleagues proposed that Aβ generated dityrosine cross-linked dimers through oxidation of the phenolic hydroxyl group at Tyr10 under oxidative conditions [55], and that generic dityrosine levels were increased in the AD brain [77]; however, it is unclear whether Aβ-mediated oxidative damage observed in vitro is relevant to in vivo disease.
4. Oxidative Stress and Antioxidants in Alzheimer's Disease
There is increasing evidence that oxidative stress is a prominent and early feature of AD [78]. The Fenton reaction mediated by iron or copper can result in the oxidative damage of nucleic acids. Smith and colleagues proposed that 8-OHdG is an established marker of nuclear DNA oxidation for AD pathology [78]. Butterfield and colleagues proposed that HNE is produced by Aβ-induced lipid peroxidation [79]. Redox proteomics using human AD brains showed that the elevation of TBARS was associated with the numbers of neuritic but not diffuse plaques [80].
On the other hand, glutathione (GSH), a tripeptide, is biosynthesized in the cytoplasm and normally exists in the mitochondrial matrix as a reduced form [81] because glutathione reductase (GR) plays a role in maintaining the ratio of GSH to GSSG through the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH) (Figure 1). GSH maintains the integrity of the plasma membrane and adenosine triphosphate (ATP) in the synaptosomes as an antioxidant. Under severe oxidative stress, the accumulation of GSSG occurs together with protein modification. Studies using AD brains by Balaz and Leon revealed almost no changes in the levels of glutathione and catalase [82], which is an important enzyme to convert H2O2 into H2O and O2 (Figure 1). In contrast, Gsell et al. reported the decreased activity of catalase in AD brains [83]. Some markers of oxidative stress will be vulnerable or not to the formation of Aβ radical in AD pathology. Alternatively, the technical artifact during the isolation of proteins may affect the results for oxidative levels among different research groups.
Table 1 summarizes in vivo studies on the involvement of these enzymes in AD. The cytochrome c oxidase (COX) is involved in respiratory electron transport in the mitochondrial inner membrane. There are several studies on the correlation of the reduced activity of COX and increased oxidative stress in AD brains [93, 94]. Fukui et al. crossed an AD transgenic mouse with a neuron-specific COX10 knockout mouse and reported that COX deficiency failed to increase both senile plaques and oxidative damage in AD progression in contrast to their expectations [90]. NADPH oxidase, believed to be one of the major ROS sources in the brain, participates in the generation of superoxide radical by transferring electrons across the membrane into molecular oxygen [95]. Genetic inactivation of Nox2, an isozyme of the catalytic subunit of NADPH oxidase, prevented oxidative stress, Aβ-derived neurovascular dysfunction, and behavioral impairment without affecting Aβ assembly [91]. Binding of the transcription factor nuclear factor E2-related factor 2 (Nrf2) to the antioxidant response element (ARE) enhancer sequence is known to induce the endogenous defense system against oxidative stress. The Nrf2-ARE pathway is activated in response to ROS, triggering the expression of antioxidant enzymes. Kanninen et al. reported that intrahippocampal injection of Nrf2 mitigated the spatial impairment of AD mice (APP/PS1 mice) associated with increased plaque formation and heme oxygenase-1 levels [96].
Table 1.
Studies on involvement of oxidative stress in AD in vivo.
| Objected mice | APP mice | Behavior | Aβ-dependent pathology | ROS marker | References |
|---|---|---|---|---|---|
| CuZn-SOD KO | Tg2576 | Early memory loss | Oligomer↑, P-tau↑ | 8-OHdG↑, Protein carbonyls↑ | Submitted |
| CuZn-SOD Tga | Tg1130H | NT | Cerebral dysfunction↓ | NT | [84]a |
| Mn-SOD hetero KO | Tg2576b, J20c, Tg19959d | Early memory lossc | Aβ depositions↑c,d, P-tau↑b, | NT | [85]b, [86]c, [87]d |
| Mn-SOD Tg | Tg2576e, Tg19959f | Improved memory losse,f | Aβ42/ Aβ40↓e, Plaque↓f, | DHE↓e, Catalase↑f, Protein carbonyls↓f, | [88]e, [89]f |
| COX10 KO | APPswe/PSEN1ΔE9 | NT | Plaque↓, Aβ42↓ | 8-OHdG↓, Protein carbonyls↓ | [90] |
| Nox2 KO | Tg2576 | Improved abnormal behavior | Unchanged (Aβ42, Aβ42, plaque) | DHE↓ | [91] |
| α-tocophenol transfer protein KO | Tg2576 | NT | Aβ40↑, IDE↓ | Unchanged | [92] |
Abbreviations: Aβ, amyloid β; AD, Alzheimer's disease; APP, amyloid precursor protein; COX, cytochrome c oxidase; DHE, dihydroethidium; IDE, insulin-degrading enzyme; KO, knock out; Nox, NADPH oxidase; NT, not tested; 8-OHdG, 8-hydroxydeoxyguanosine; PSEN, presenilin; P-tau, phosphorylated tau; ROS, reactive oxygen species; SOD, superoxide dismutase; Tg, transgenic; ↑, increased; ↓, decreased.
Glutathione peroxidase (GPx) is also a key modulator in the neuronal system, participating in the elimination of H2O2. Overexpression of GPx4, an isoform expressed in the membrane, reduced the lipid peroxidation of mice after exposure to diquat, known as a herbicide, and induced mice resistant to apoptosis from oxidants [97]. Embryonic fibroblasts of catalase transgenic mice are more resistant to toxic H2O2 [98]. Thioredoxin plays a role in repairing the oxidation of cysteine residues in proteins [99]. Yodoi and colleagues generated transgenic mice overexpressing human thioredoxin, which reduced oxidative stress and extended its lifespan [100]. The therapeutic effects of these antioxidative enzymes against AD are expected although no studies on their role in AD pathology have been reported.
5. Therapeutic Role of Superoxide Dismutase in Alzheimer's Disease
The role of SOD in AD pathogenesis has long been controversial. Several studies have shown decreased SOD in the frontal cortex of AD patients [101] whereas a slight elevation of SOD was documented in the caudate nucleus of AD patients [102]. Alternatively, other researchers have suggested that almost no changes in SOD levels are found in AD brains [83]. Quite recently, Ansari and Scheff reported a strong correlation between several oxidative damage levels using various dementia subjects with negligible levels of premortem hypoxia in order to eliminate the possibility of affecting protein integrity [103]. As shown in Table 1, Melov et al. suggested that mitochondrial oxidative stress could induce the hyperphosphorylation of tau at Ser396 using Tg2576 transgenic AD mouse model [85]. There have also been reports on the role of Mn-SOD in AD pathology; AD transgenic mouse models crossed with Sod2+/− resulted in increased accelerated behavioral deficits [86] or senile plaques [87] (Table 1). Quite recently, our group proposed the involvement of CuZn-SOD in AD progression; the superoxide radical in the cytoplasma induced Aβ oligomerization and early cognitive impairment in Tg2576, and these phenomena notably preceded oxidative damage (Murakami, K. et al., submitted) (Table 1). Our findings do not contradict the implication by Marlatt et al. that oxidative damage occurs primarily within the cytoplasm rather than the mitochondria [3].
In the therapy of AD by SODs, cerebral endothelial dysfunction in the AD mouse model can be improved by overexpression of Sod1 [84] (Table 1) or the administration of SOD [104]. Bayer et al. proposed that dietary intake of Cu stabilizes CuZn-SOD activity and decreases Aβ production in the APP transgenic mouse model [105]. On the other hand, overexpression of Sod2 rescued several markers for oxidative stress associated with AD-like pathologies in two representative lines of AD model mice (Tg2576 [88] and Tg19959 [89]) (Table 1). Under the excessive reduced redox-acitve metal ions, the adverse effects due to hydroxyl radical formation should be taken into account. The therapeutic treatment of both SOD and catalase mimetics (e.g., EUK-8 [106]) could be one of promising approaches.
Breteler and colleagues performed a clinical survey of the dietary intake of antioxidants and the risk of AD based on over 5,000 participants in the Netherlands [107]. It was suggested that high dietary intake of vitamin C and vitamin E might lower the risk of AD. Dementia control by vitamin C and vitamin E has long been discussed [108–112]. Interestingly, Rinaldi et al. suggested the correlation of vitamin C and SOD levels with the dementia status [113]. SOD might be one of the most vulnerable indicators as an antioxidant enzyme in AD and cognitive dementia. Alternatively, it was reported that environmental enrichment prevented AD-like pathology associated with elevated CuZn-SOD and Mn-SOD levels [114].
6. Conclusions
One of the most accepted knowledge in the etiology of AD is thought to be the free-radical theory; however, it remains to be determined whether oxidative stress is a cause or effect in AD. Aβ42 aggregates (oligomerizes) induce neurotoxins by interacting with trace metals at Tyr10 or in the N-terminal region, leading to tissue oxidation by an S-oxidized radical cation in Met35. There are three means of defense from Aβ42-dependent AD pathology: (1) to slow the rate of Aβ42 aggregation, (2) to decrease the production of Aβ42 by downregulating the activity of β- or γ-secretase, and (3) to enhance protease activity (such as neprilysin [115], an insulin-degrading enzyme [116]) against Aβ42. Oxidative stress may affect one or all of the protective mechanisms. Antioxidant enzymes including SOD or dietary supplements of vitamin C and vitamin E could counteract these dysfunctions. Food treatments for prevention are a better choice to maintain the quality of life. There are increasing reports on the inhibitory effects of natural products such as several flavonoids [117], vitamin A [118], and vitamin E [119] on AD pathology in vivo. Nishida et al. reported α-tocophenol transfer protein-knockout mice, in which Aβ deposits accumulated by decreasing the clearance of Aβ peptide from the brain and blood [92] (Table 1). Quite recently, we discovered the potential of silymarin [120], the active ingredient of milk thistle extract which is long used as a hepatoprotective medicine, and vitamin C (Murakami, K. et al., submitted) for AD prevention. Further research on structural analysis of the inhibitory mechanism is under investigation to effectively develop inhibitors with few adverse effects.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research, the Program for the Promotion of Basic Research Activities for Innovative Biosciences, and funds for the Promotion of Science for Young Scientists from The Ministry of Education, Culture, Sports, Science, and Technology of the Japanese Government.
References
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nature Medicine. 2004;10:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 3.Marlatt M, Lee HG, Perry G, Smith MA, Zhu X. Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer’s disease. Acta Neurobiologiae Experimentalis. 2004;64(1):81–87. doi: 10.55782/ane-2004-1493. [DOI] [PubMed] [Google Scholar]
- 4.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 5.Wallace DC, Melov S. Radicals r’aging. Nature Genetics. 1998;19(2):105–106. doi: 10.1038/448. [DOI] [PubMed] [Google Scholar]
- 6.Valentine JS, Doucette PA, Potter SZ. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annual Review of Biochemistry. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 7.Elchuri S, Oberley TD, Qi W, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24(3):367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 8.Muller FL, Song W, Liu Y, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radical Biology and Medicine. 2006;40(11):1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Van Remmen H, Jones DP. Current thoughts on the role of mitochondria and free radicals in the biology of aging. Journals of Gerontology A. 2009;64(2):171–174. doi: 10.1093/gerona/gln058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iuchi Y, Okada F, Onuma K, et al. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochemical Journal. 2007;402(2):219–227. doi: 10.1042/BJ20061386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami K, Inagaki J, Saito M, et al. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochemical and Biophysical Research Communications. 2009;382(2):457–461. doi: 10.1016/j.bbrc.2009.03.053. [DOI] [PubMed] [Google Scholar]
- 12.Imamura Y, Noda S, Hashizume K, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(30):11282–11287. doi: 10.1073/pnas.0602131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchiyama S, Shimizu T, Shirasawa T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. Journal of Biological Chemistry. 2006;281(42):31713–31719. doi: 10.1074/jbc.M603422200. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 15.Lebovitz RM, Zhang H, Vogel H, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochemical and Biophysical Research Communications. 2002;296(3):729–736. doi: 10.1016/s0006-291x(02)00933-6. [DOI] [PubMed] [Google Scholar]
- 17.Nojiri H, Shimizu T, Funakoshi M, et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. Journal of Biological Chemistry. 2006;281(44):33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- 18.Kuwahara H, Horie T, Ishikawa S, et al. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radical Biology and Medicine. 2010;48(9):1252–1262. doi: 10.1016/j.freeradbiomed.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Shimizu T, Nojiri H, Kawakami S, Uchiyama S, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase gene. Geriatrics and Gerontology International. 2010;10, supplement 1:S70–S79. doi: 10.1111/j.1447-0594.2010.00604.x. [DOI] [PubMed] [Google Scholar]
- 20.Resende R, Moreira PI, Proença T, et al. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radical Biology and Medicine. 2008;44(12):2051–2057. doi: 10.1016/j.freeradbiomed.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 22.Masters CL, Simms G, Weinman NA. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nature Reviews Molecular Cell Biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 24.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 25.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-β in Alzheimer disease: the null versus the alternate hypotheses. Journal of Pharmacology and Experimental Therapeutics. 2007;321(3):823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 26.Gómez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Annals of Neurology. 1997;41(1):17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 27.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451(7179):720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer disease. Journal of Biological Chemistry. 2009;284(8):4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chemistry and Biology. 1997;4(2):119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 30.Klein WL, Krafft GA, Finch CE. Targeting small A β oligomers: the solution to an Alzheimer’s disease conundrum? Trends in Neurosciences. 2001;24(4):219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 31.Lesné S, Ming TK, Kotilinek L, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 32.Hoshi M, Sato M, Matsumoto S, et al. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β . Proceedings of the National Academy of Sciences of the United States of America. 2003;100(11):6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shankar GM, Li S, Mehta TH, et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia W, Yang T, Shankar G, et al. A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer Disease. Archives of Neurology. 2009;66(2):190–199. doi: 10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bitan G, Teplow DB. Rapid photochemical cross-linking—a new tool for studies of metastable, amyloidogenic protein assemblies. Accounts of Chemical Research. 2004;37(6):357–364. doi: 10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 36.Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 37.Kayed R, Head E, Sarsoza F, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Molecular Neurodegeneration. 2007;2(1):p. 18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terry RD, Pena C. Experimental production of neurofibrillary degeneration 2. electron microscopy, phosphatase histochemistry and electron probe analysis. Journal of Neuropathology and Experimental Neurology. 1965;24:200–210. doi: 10.1097/00005072-196504000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Frisardi V, Solfrizzi V, Capurso C, et al. Aluminum in the diet and alzheimer’s disease: from current epidemiology to possible disease-modifying treatment. Journal of Alzheimer’s Disease. 2010;20(1):17–30. doi: 10.3233/JAD-2009-1340. [DOI] [PubMed] [Google Scholar]
- 40.Ehmann WD, Markesbery WR, Alauddin M. Brain trace elements in Alzheimer’s disease. NeuroToxicology. 1986;7(1):197–206. [PubMed] [Google Scholar]
- 41.Bush AI. The metallobiology of Alzheimer’s disease. Trends in Neurosciences. 2003;26(4):207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 42.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidatives stress. Nature Reviews Drug Discovery. 2004;3(3):205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 43.Butterfield DA. Amyloid β-peptide [1–42]-assosiated free radical-induced oxidative stress and neurodegeneration in Alzheimer’s disease brain: mechanisms and consequences. Current Medicinal Chemistry. 2003;10(24):2651–2659. doi: 10.2174/0929867033456422. [DOI] [PubMed] [Google Scholar]
- 44.Hensley K, Hall N, Subramaniam R, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. Journal of Neurochemistry. 1995;65(5):2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 45.Bayer TA, Schäfer S, Breyhan H, Wirths O, Treiber C, Multhaup G. A vicious circle: role of oxidative stress, intraneuronal Aβ and Cu in Alzheimer’s disease. Clinical Neuropathology. 2006;25(4):163–171. [PubMed] [Google Scholar]
- 46.Huang X, Cuajungco MP, Atwood CS, et al. Cu(II) potentiation of Alzheimer Aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. Journal of Biological Chemistry. 1999;274(52):37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 47.Karr JW, Kaupp LJ, Szalai VA. Amyloid-β binds Cu2+ in a mononuclear metal ion binding site. Journal of the American Chemical Society. 2004;126(41):13534–13538. doi: 10.1021/ja0488028. [DOI] [PubMed] [Google Scholar]
- 48.Karr JW, Akintoye H, Kaupp LJ, Szalai VA. N-terminal deletions modify the Cu2+ binding site in amyloid-β . Biochemistry. 2005;44(14):5478–5487. doi: 10.1021/bi047611e. [DOI] [PubMed] [Google Scholar]
- 49.Curtain CC, Ali F, Volitakis I, et al. Alzheimer’s disease amyloid-β binds Copper and Zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. Journal of Biological Chemistry. 2001;276(23):20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 50.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-β peptide with membrane lipid. Journal of Biological Chemistry. 2003;278(5):2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 51.Tickler AK, Smith DG, Ciccotosto GD, et al. Methylation of the imidazole side chains of the Alzheimer disease amyloid-β peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. Journal of Biological Chemistry. 2005;280(14):13355–13363. doi: 10.1074/jbc.M414178200. [DOI] [PubMed] [Google Scholar]
- 52.Antzutkin ON. Amyloidosis of Alzheimer’s Aβ peptides: solid-state nuclear magnetic resonance, electron paramagnetic resonance, transmission electron microscopy, scanning transmission electron microscopy and atomic force microscopy studies. Magnetic Resonance in Chemistry. 2004;42(2):231–246. doi: 10.1002/mrc.1341. [DOI] [PubMed] [Google Scholar]
- 53.Syme CD, Nadal RC, Rigby SEJ, Viles JH. Copper binding to the amyloid-β (Aβ) peptide associated with Alzheimer’s disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Aβ-(1–28): insights from a range of complementary spectroscopic techniques. Journal of Biological Chemistry. 2004;279(18):18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- 54.Drew SC, Masters CL, Barnham KJ. Alanine-2 carbonyl is an oxygen ligand in Cu2+ coordination of Alzheimer’s disease amyloid-β peptide—relevance to N-terminally truncated forms. Journal of the American Chemical Society. 2009;131(25):8760–8761. doi: 10.1021/ja903669a. [DOI] [PubMed] [Google Scholar]
- 55.Barnham KJ, Haeffner F, Ciccotosto GD, et al. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease β-amyloid. FASEB Journal. 2004;18(12):1427–1429. doi: 10.1096/fj.04-1890fje. [DOI] [PubMed] [Google Scholar]
- 56.Ono K, Condron MM, Teplow DB. Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid beta-protein assembly and toxicity. Journal of Biological Chemistry. 2010;285(30):23186–23197. doi: 10.1074/jbc.M109.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cherny RA, Atwood CS, Xilinas ME, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30(3):665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 58.Treiber C, Simons A, Strauss M, et al. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer’s disease. Journal of Biological Chemistry. 2004;279(50):51958–51964. doi: 10.1074/jbc.M407410200. [DOI] [PubMed] [Google Scholar]
- 59.Crouch PJ, Barnham KJ, Bush AI, White AR. Therapeutic treatments for Alzheimer’s disease based on metal bioavailability. Drug News and Perspectives. 2006;19(8):469–474. doi: 10.1358/dnp.2006.19.8.1021492. [DOI] [PubMed] [Google Scholar]
- 60.Adlard PA, Cherny RA, Finkelstein DI, et al. Rapid restoration of cognition in alzheimer’s transgenic mice with 8-Hydroxy quinoline analogs is associated with decreased interstitial Aβ . Neuron. 2008;59(1):43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 61.Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. The Lancet Neurology. 2008;7(9):779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 62.Varadarajan S, Yatin S, Kanski J, Jahanshahi F, Butterfield DA. Methionine residue 35 is important in amyloid β-peptide-associated free radical oxidative stress. Brain Research Bulletin. 1999;50(2):133–141. doi: 10.1016/s0361-9230(99)00093-3. [DOI] [PubMed] [Google Scholar]
- 63.Naslund J, Schierhorn A, Hellman U, et al. Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(18):8378–8382. doi: 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuo YM, Kokjohn TA, Beach TG, et al. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. Journal of Biological Chemistry. 2001;276(16):12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- 65.Pal R, Oien DB, Ersen FY, Moskovitz J. Elevated levels of brain-pathologies associated with neurodegenerative diseases in the methionine sulfoxide reductase A knockout mouse. Experimental Brain Research. 2007;180(4):765–774. doi: 10.1007/s00221-007-0903-6. [DOI] [PubMed] [Google Scholar]
- 66.Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s Aβ(1–42) and Aβ(25–35) Journal of the American Chemical Society. 2001;123(24):5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 67.Irie K, Murakami K, Masuda Y, et al. The toxic conformation of the 42-residue amyloid β peptide and its relevance to oxidative stress in Alzheimer’s disease. Mini-Reviews in Medicinal Chemistry. 2007;7(10):1001–1008. doi: 10.2174/138955707782110187. [DOI] [PubMed] [Google Scholar]
- 68.Murakami K, Masuda Y, Shirasawa T, Shimizu T, Irie K. The turn formation at positions 22 and 23 in the 42-mer amyloid beta peptide: the emerging role in the pathogenesis of Alzheimer’s disease. Geriatrics and Gerontology International. 2010;10(supplement 1):S169–S179. doi: 10.1111/j.1447-0594.2010.00598.x. [DOI] [PubMed] [Google Scholar]
- 69.Masuda Y, Uemura S, Ohashi R, et al. Identification of physiological and toxic conformations in Aβ42 aggregates. Chembiochem. 2009;10(2):287–295. doi: 10.1002/cbic.200800411. [DOI] [PubMed] [Google Scholar]
- 70.Murakami K, Horikoshi-Sakuraba Y, Murata N, et al. Monoclonal antibody against the turn of the 42-residue amyloid beta protein at positions 22 and 23. ACS Chemical Neuroscience. 2010;1(11):747–756. doi: 10.1021/cn100072e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morimoto A, Irie K, Murakami K, et al. Analysis of the secondary structure of beta-amyloid (Abeta42) fibrils by systematic proline replacement. Journal of Biological Chemistry. 2004;279(50):52781–52788. doi: 10.1074/jbc.M406262200. [DOI] [PubMed] [Google Scholar]
- 72.Kanski J, Aksenova M, Schöneich C, Butterfield DA. Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide. Free Radical Biology and Medicine. 2002;32(11):1205–1211. doi: 10.1016/s0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 73.Murakami K, Irie K, Ohigashi H, et al. Formation and stabilization model of the 42-mer Aβ radical: implications for the long-lasting oxidative stress in Alzheimer’s disease. Journal of the American Chemical Society. 2005;127(43):15168–15174. doi: 10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- 74.Butterfield DA, Galvan V, Lange MB, et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: requirement for methionine 35 in amyloid β-peptide of APP. Free Radical Biology and Medicine. 2010;48(1):136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maiti P, Lomakin A, Benedek GB, Bitan G. Despite its role in assembly, methionine 35 is not necessary for amyloid β-protein toxicity. Journal of Neurochemistry. 2010;113(5):1252–1262. doi: 10.1111/j.1471-4159.2010.06692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Felice FG, Velasco PT, Lambert MP, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. Journal of Biological Chemistry. 2007;282(15):11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 77.Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer’s disease amyloid βpeptide. Biochimica et Biophysica Acta. 2007;1768(8):1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 78.Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2001;60(8):759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 79.Lauderback CM, Hackett JM, Keller JN, et al. Vulnerability of synaptosomes from ApoE knock-out mice to structural and oxidative modifications induced by Aβ(1–40): implications for Alzheimer’s disease. Biochemistry. 2001;40(8):2548–2554. doi: 10.1021/bi002312k. [DOI] [PubMed] [Google Scholar]
- 80.Keller JN, Schmitt FA, Scheff SW, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64(7):1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 81.Starke DW, Chen Y, Bapna CP, Lesnefsky EJ, Mieyal JJ. Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radical Biology and Medicine. 1997;23(3):373–384. doi: 10.1016/s0891-5849(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 82.Balazs L, Leon M. Evidence of an oxidative challenge in the Alzheimer’s brain. Neurochemical Research. 1994;19(9):1131–1137. doi: 10.1007/BF00965146. [DOI] [PubMed] [Google Scholar]
- 83.Gsell W, Conrad R, Hickethier M, et al. Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of Alzheimer type. Journal of Neurochemistry. 1995;64(3):1216–1223. doi: 10.1046/j.1471-4159.1995.64031216.x. [DOI] [PubMed] [Google Scholar]
- 84.Iadecola C, Zhang F, Niwa K, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nature Neuroscience. 1999;2(2):157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 85.Melov S, Adlard PA, Morten K, et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLos One. 2007;2(6, article e536) doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Esposito L, Raber J, Kekonius L, et al. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. Journal of Neuroscience. 2006;26(19):5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li F, Calingasan NY, Yu F, et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. Journal of Neurochemistry. 2004;89(5):1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 88.Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(32):13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB Journal. 2009;23(8):2459–2466. doi: 10.1096/fj.09-132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(35):14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park L, Zhou P, Pitstick R, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(4):1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nishida Y, Ito S, Ohtsuki S, et al. Depletion of vitamin E increases amyloid beta accumulation by decreasing its clearances from brain and blood in a mouse model of Alzheimer disease. Journal of Biological Chemistry. 2009;284(48):33400–33408. doi: 10.1074/jbc.M109.054056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parker WD, Mahr NJ, Filley CM, et al. Reduced platelet cytochrome c oxidase activity in Alzheimer’s disease. Neurology. 1994;44(6):1086–1090. doi: 10.1212/wnl.44.6.1086. [DOI] [PubMed] [Google Scholar]
- 94.Mutisya EM, Bowling AC, Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. Journal of Neurochemistry. 1994;63(6):2179–2184. doi: 10.1046/j.1471-4159.1994.63062179.x. [DOI] [PubMed] [Google Scholar]
- 95.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 96.Kanninen K, Heikkinen R, Malm T, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(38):16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ran Q, Liang H, Gu M, et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. Journal of Biological Chemistry. 2004;279(53):55137–55146. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- 98.Mele J, Van Remmen H, Vijg J, Richardson A. Characterization of transgenic mice that overexpress both copper zinc superoxide dismutase and catalase. Antioxidants and Redox Signaling. 2006;8(3-4):628–638. doi: 10.1089/ars.2006.8.628. [DOI] [PubMed] [Google Scholar]
- 99.Lillig CH, Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxidants and Redox Signaling. 2007;9(1):25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 100.Mitsui A, Hamuro J, Nakamura H, et al. Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxidants and Redox Signaling. 2002;4(4):693–696. doi: 10.1089/15230860260220201. [DOI] [PubMed] [Google Scholar]
- 101.Richardson JS. Free radicals in the genesis of Alzheimer’s disease. Annals of the New York Academy of Sciences. 1993;695:73–76. doi: 10.1111/j.1749-6632.1993.tb23031.x. [DOI] [PubMed] [Google Scholar]
- 102.Marklund SL, Adolfsson R, Gottfries CG, Winblad B. Superoxide dismutase isoenzymes in normal brains and in brains from patients with dementia of Alzheimer type. Journal of the Neurological Sciences. 1985;67(3):319–325. doi: 10.1016/0022-510x(85)90156-x. [DOI] [PubMed] [Google Scholar]
- 103.Ansari MA, Scheff SW. Oxidative stress in the progression of alzheimer disease in the frontal cortex. Journal of Neuropathology and Experimental Neurology. 2010;69(2):155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niwa K, Carlson GA, Iadecola C. Exogenous a beta1–40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. Journal of Cerebral Blood Flow and Metabolism. 2000;20(12):1659–1668. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- 105.Bayer TA, Schäfer S, Simons A, et al. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):14187–14192. doi: 10.1073/pnas.2332818100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baudry M, Etienne S, Bruce A, Paluck M, Jacobsen E, Malfroy B. Salen-manganese complexes are superoxide dismutase mimics. Biochemical and Biophysical Research Communications. 1993;192(2):964–968. doi: 10.1006/bbrc.1993.1509. [DOI] [PubMed] [Google Scholar]
- 107.Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Dietary intake of antioxidants and risk of Alzheimer disease. Journal of the American Medical Association. 2002;287(24):3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 108.Rivière S, Birlouez-Aragon I, Nourhashémi F, Vellas B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. International Journal of Geriatric Psychiatry. 1998;13(11):749–754. doi: 10.1002/(sici)1099-1166(1998110)13:11<749::aid-gps860>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 109.Sinclair AJ, Bayer AJ, Johnston JO, Warner C, Maxwell SRJ. Altered plasma antioxidant status in subjects with Alzheimer’s disease and vascular dementia. International Journal of Geriatric Psychiatry. 1998;13(12):840–845. doi: 10.1002/(sici)1099-1166(1998120)13:12<840::aid-gps877>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 110.Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS. Vitamin E and cognitive decline in older persons. Archives of Neurology. 2002;59(7):1125–1132. doi: 10.1001/archneur.59.7.1125. [DOI] [PubMed] [Google Scholar]
- 111.Paleologos M, Cumming RG, Lazarus R. Cohort study of vitamin C intake and cognitive impairment. American Journal of Epidemiology. 1998;148(1):45–50. doi: 10.1093/oxfordjournals.aje.a009559. [DOI] [PubMed] [Google Scholar]
- 112.Bourdel-Marchasson I, Delmas-Beauviex MC, Peuchant E, et al. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age and Ageing. 2001;30(3):235–241. doi: 10.1093/ageing/30.3.235. [DOI] [PubMed] [Google Scholar]
- 113.Rinaldi P, Polidori MC, Metastasio A, et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiology of Aging. 2003;24(7):915–919. doi: 10.1016/s0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 114.Herring A, Blome M, Ambrée O, Sachser N, Paulus W, Keyvani K. Reduction of cerebral oxidative stress following environmental enrichment in mice with alzheimer-like pathology. Brain Pathology. 2010;20(1):166–175. doi: 10.1111/j.1750-3639.2008.00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iwata N, Tsubuki S, Takaki Y, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292(5521):1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 116.Qiu WQ, Walsh DM, Ye Z, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid β- protein by degradation. Journal of Biological Chemistry. 1998;273(49):32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 117.Hamaguchi T, Ono K, Murase A, Yamada M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-beta aggregation pathway. American Journal of Pathology. 2009;175(6):2557–2565. doi: 10.2353/ajpath.2009.090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ding Y, Qiao A, Wang Z, et al. Retinoic acid attenuates β-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. Journal of Neuroscience. 2008;28(45):11622–11634. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sung S, Yao Y, Uryu K, et al. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. The FASEB Journal. 2004;18(2):323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 120.Murata N, Murakami K, Ozawa Y, et al. Silymarin attenuated the aAmyloid beta plaque burden and improved behavioral abnormalities in an Alzheimer’s disease mouse model. Bioscience, Biotechnology and Biochemistry. 2010;74(11):2299–2306. doi: 10.1271/bbb.100524. [DOI] [PubMed] [Google Scholar]