

Abstract
Despite the high number of genes identified in hereditary polyneuropathies/Charcot–Marie–Tooth (CMT) disease, the genetic defect in many families is still unknown. Here we report the identification of a new gene for autosomal dominant axonal neuropathy in a large three-generation family. Linkage analysis identified a 5 Mb region on 9q33–34 with a LOD score of 5.12. Sequence capture and next-generation sequencing of the region of interest identified five previously unreported non-synonymous heterozygous single nucleotide changes or indels, four of which were confirmed by Sanger sequencing. Two sequence variants co-segregated with the disease, and one, a 2 bp insertion in the last exon of LRSAM1, was also absent in 676 ethnicity-matched control chromosomes. This frameshift mutation (p.Leu708Argfx28) is located in the C-terminal RING finger motif of the encoded protein. Ubiquitin ligase activity in transfected cells with constructs carrying the patient mutation was affected as measured by a higher level of abundance of TSG101, the only reported target of LRSAM1. Injections of morpholino oligonucleotides in zebrafish embryos directed against the ATG or last splice site of zebrafish Lrsam1 disturbed neurodevelopment, showing a less organized neural structure and, in addition, affected tail formation and movement. LRSAM1 is highly expressed in adult spinal cord motoneurons as well as in fetal spinal cord and muscle tissue. Recently, a homozygous mutation in LRSAM1 was proposed as a strong candidate for the disease in a family with recessive axonal polyneuropathy. Our data strongly support the hypothesis that LRSAM1 mutations can cause both dominant and recessive forms of CMT.
INTRODUCTION
Hereditary motor and sensory neuropathies (HMSN), also known as Charcot–Marie–Tooth (CMT) disease, consist of a group of common inherited peripheral neuropathies that are heterogeneous at the clinical and genetic levels. Clinically, they can be divided into demyelinating and axonal forms of the disease, primarily based on electrophysiological data. Demyelinating neuropathies show a marked decrease in median motor nerve conduction velocities (NCVs). The most frequent type of CMT with an autosomal dominant mode of inheritance, CMT1A, is most commonly caused by duplication of PMP22, although mutations and copy number variants in the vicinity of PMP22 are also described (1–6).
CMT2 is an axonal type of CMT with reduced compound motor axon potentials but initially no reduction in NCVs. However, the clinical features of patients of both types can be quite similar. X-linked forms of CMT also exist as well as subtypes with intermediate forms of NCVs and/or recessive modes of inheritance.
Many loci have been linked to CMT/HMSN and to date 44 genes have been implicated in the disease; mutations in 15 different genes have been identified to cause CMT2. Except for the PMP22 duplication that is responsible for CMT1A, mutations in other CMT genes occur at relatively low frequencies. Despite the high number of loci, the defect in many families remains unknown thereby precluding any genetic testing.
Most genes are associated with either demyelinating or axonal forms of subtypes of the disease, although GJB1 and MPZ have been associated with both (7–9). Severity of the disease varies between different individuals, even when an identical mutation is present in members of the same family. So far, only few clinical characteristics have been linked to a specific gene or mutation, thus presenting a diagnostic tool to determine which genes might be screened first. Here we report on a three-generation family with axonal neuropathy (CMT2) and a clear dominant mode of inheritance. Linkage analysis followed by next-generation sequencing of the linkage region on chromosome 9q33–34 and functional analysis led to the identification of the causative gene for this dominant type of CMT. We propose CMT2o for dominant LRSAM1-associated CMT2.
RESULTS
The clinical phenotype is associated with the chromosomal region 9q33–34
The index patient of Dutch descent presented at the age of 32 with progressive distal muscle weakness and mild sensory disturbances of both feet. Over decades, his symptoms were slowly progressive now also including weakness of hand muscles. He had relatively mild foot deformities with pes cavus and hammer toes and a marked atrophy of his lower legs with absent tendon reflexes. The vibration sense was mildly reduced, showing a proximal to distal gradient. His family history revealed various other family members with the same features (Fig. 1; Table 1). Nerve conduction studies and needle EMG showed a relatively severe axonal neuropathy with absent sural nerve sensory nerve action potentials, mildly reduced motor NCVs and severe distal neurogenic features on needle examination. Over two decades ago, a sural nerve biopsy was performed that showed severe axonal degeneration confirming the diagnosis of CMT2. The clinical phenotype can be summarized as a relatively mild, very slowly progressive axonal neuropathy with age of onset in the second or third decade of life. The pedigree of the family clearly showed an autosomal dominant inheritance of the disease.
Figure 1.

Pedigree of family with dominant axonal neuropathy. Filled symbols indicate disease, and open symbols represent unaffected persons. Grey symbols: not known if affected.
Table 1.
Characteristics of family members
| Clinical features | Affected family members |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Code in pedigree | 1.2 | 2.1 | 2.2 | 2.5 | 2.6 | 2.4 | 3.1 | 3.8 | 3.2 | 3.4 | 3.11 |
| Gender | F | F | M | M | F | M | M | M | F | M | F |
| Age at onset | 40 | Unknown | 30 | 30 | 37 | 40 | 27 | 20 | 28 | No complaints | No complaints |
| Age | died at the age of 72 | 70 | 68 | 64 | 62 | 63 | 34 | 32 | 32 | 26 | 25 |
| Distal muscle wasting/weakness (LL) | − | Unknown | + | + | + | + | + | + | + | + | − |
| Distal muscle wasting/weakness (UL) | − | Unknown | +(mild) | +(mild) | − | − | + | − | − | − | − |
| Upper limb reflexes | − | Unknown | N | − | Reduced | Reduced | Reduced | − | N | N | Reduced |
| Knee reflexes | − | Unknown | − | − | − | − | Reduced | Reduced | N | N | Reduced |
| Ankle reflexes | − | Unknown | − | − | − | − | − | − | − | − | − |
| Vibration sense | Unknown | Unknown | Reduced | Reduced | Reduced | Reduced | Reduced | Reduced | Reduced | ND | N |
| Pin-prick | Mildly reduced | Unknown | Mildly reduced | Mildly reduced | Reduced | Reduced | Reduced | Reduced | N | ND | N |
| Hearing loss | Unknown | Unknown | − | Mildly perceptive | − | − | − | − | − | − | − |
| Additional features | None | None | Parkinson | None | None | Parkinson | None | None | None | None | None |
| Nerve conduction studies | ND | + | + | + | + | + | + | + | + | + | + |
| Sural nerve response | − | − | − | − | − | − | − | − | − | − | |
| Peroneal nerve response | − | − | − | Strongly reduced | − | N | ND | Reduced | N | ND | |
| Needle EMG | ND | Neuro | Neuro | Neuro | Neuro | Neuro | ND | Neuro | Neuro | Neuro | |
−, absent; +, present; F, female; M, male; N, normal; ND, not done; Neuro, neurogenic.
Mutations in the coding regions of MFN2, MPZ, GJB1 and GARS, which are known to be associated with CMT2, were excluded. In view of the large number of CMT2-associated genes and the availability of many affected individuals in this family, we decided to locate the genetic defect by genome-wide linkage analysis using an Affymetrix 250K single nucleotide polymorphism (SNP) array. Due to the large number of family members, the family was split into two smaller pedigrees for fast multipoint parametric analysis using the ALLEGRO (10) program with the assumption of autosomal dominant inheritance and full penetrance. Analysis showed a single peak on chromosome 9 with a maximum LOD score of 4.52 (Fig. 2). When confining the analysis to markers on chromosome 9 using SimWalk2 (11), multipoint LOD scores could be calculated for the family as a whole resulting in a maximum LOD score of 5.12. Therefore, the single critical interval on 9q33–34, previously unknown as a CMT locus, of approximately 7 cm must harbor the causative mutation for the disease. Consequent construction of haplotypes with ALLEGRO revealed a specific haplotype that showed perfect co-segregation with the disease (Fig. 3). The borders of this haplotype shared between all affected individuals were defined by two limiting SNPs (rs10081624 and rs2039463) which are 4.4 Mb apart.
Figure 2.

Linkage analysis identifies 9q33–34 as the disease gene-containing region: LOD scores are plotted against the position (cm) on the chromosomes from 1–22, XX, XY and aligned from p-ter to q-ter.
Figure 3.

Haplotypes of affected and non-affected family members. Haplotypes were inferred for the grandparents. Different haplotypes were given a different color. All affected persons share the red haplotype.
Identification of heterozygous changes associated with the clinical phenotype
We designed a custom sequence capture array covering the entire linkage region (chr9: 127624486–132419012 hg18/NCBI36). Genomic DNA of an affected individual (subject 2.2) was enriched by sequence capture for this region, sequenced on a FLX Titanium sequencer (Roche) and mapped to the human consensus genome (hg18) with Newbler software. The captured region contains 78 annotated genes, 10 loci encoding predicted proteins and 2 microRNAs. Over 94% of all exons were sequenced with coverage of more than 10 times. Analysis yielded 5827 reliable nucleotide changes which is within the estimated range of nucleotide changes to be expected for a 5 Mb region. Since the mode of inheritance is autosomal dominant, the expected disease-causing mutation would be a heterozygous change. Within annotated genes, we found 330 nucleotide variations, 51 of these were heterozygous changes within exons or within 20 nucleotides of the exon–intron boundaries. The majority of sequence variants were already known as polymorphisms in the public databases. Five heterozygous changes were not known as polymorphisms and were located in the coding region of the corresponding genes, two of which were frameshift mutations, the other three being missense mutations (Table 2; Supplementary Material, Table S2). The sequence variants in GOLGA2, SLC27A4, WDR34 and LRSAM1 were all confirmed by Sanger sequencing, whereas the frameshift in ST6GALNAC6 represented a pyrosequencing artifact due to the presence of homopolymer stretches.
Table 2.
Found heterozygous changes
| GENE | Pos. chr9 (hg18) | Change | NT | Amino acid | Exon | Coverage | % mutant reads |
|---|---|---|---|---|---|---|---|
| GOLGA2 | 130059259 | G>A | c.2881C>T | p.Pro961Ser | 26 | 34 reads | 47 |
| SLC27A4 | 130155189 | A>G | c.1052A>G | p.Asn351Ser | 8 | 62 reads | 56 |
| WDR34 | 130435956 | G>A | c.1499G>A | p.Ala500Val | 9 | 50 reads | 50 |
| ST6GALNAC6 | 129692883–884 | CG>- | c.557_558delGC | p.Pro186Gln fsX38 | 5 | 63 reads | 27 |
| LRSAM1 | 129304950–951 | ->GC | c.2121_2122insGC | p.Leu708Arg fsX28 | 26 | 20 reads | 70 |
The position (pos.) on the chromosome is given, as well as the consequence for the gene involved at the nucleotide level (NT) and the protein level.
LRSAM1 is a candidate gene for CMT2
For segregation analysis, the presence of the four validated changes was determined by direct sequencing in seven additional family members (five affected and three non-affected persons). The missense mutation in WDR34 and the frameshift mutation in LRSAM1 both segregated with the disease. The other two missense mutations in GOLGA2 and SLC27A4 were variations located on the non-disease-associated allele. They were both found in two of three healthy individuals and, in addition, were absent in four of five affected individuals and should therefore be regarded as rare polymorphisms not related to the disease. Prediction programs (Polyphen, SIFT) indicated that the change in WDR34 was not expected to be harmful to the protein. Moreover, we identified two heterozygotes in 352 ethnicity-matched controls. In contrast, the 2 bp insertion in LRSAM1 was absent in 676 control chromosomes. Screening of all available family members, 11 affected and 8 non-affected individuals, showed complete co-segregation with the disease. The 2 bp insertion leads to a replacement of a leucine by an arginine residue near the predicted C-terminal RING finger domain and a concomitant frameshift with a stop codon 28 codons downstream (p.Leu708Argfx28). This would make the mutant protein slightly longer than the wild-type one, since normal translation would terminate only 16 amino acids downstream of this position. Thus, LRSAM1 is a very strong candidate gene for the neuropathy in this family. In addition, it was published recently that another mutation in LRSAM1 in a Canadian family was thought to be the cause for a recessive form of CMT2 (12). To determine if the found mutation of the Dutch family would possibly occur more frequently in Dutch CMT2 pedigrees, 11 patients as representatives of 11 additional CMT2 pedigrees in which no genetic aberration had been identified as well as 32 unrelated CMT1 patients without known mutations were screened for this specific insertion but we could not identify other Dutch families carrying the identical mutation.
LRSAM1 is highly expressed in the fetal and adult nervous system
We examined LRSAM1 expression by using locked nucleic acid (LNA) probes in RNA in situ experiments in neural tissue on sections of a 6–8-week-old human fetus and adult spinal cord. Motoneurons in the human adult spinal cord showed high LRSAM1 expression (Fig. 4) and a clear signal was also seen in some intermediate neurons (data not shown). Expression in a human fetus was found to be high in the fetal spinal cord, sensory ganglia and all developing muscles but was also observed in some other tissues such as the esophageal endothelium, epithelium, Meckel's cartilage and at a location that probably contains osteoblasts in the developing pharyngeal arches. Although expression is not restricted to neural tissue, a high LRSAM1 expression is present in all tissues that are affected by the disease (Fig. 4).
Figure 4.

LRSAM1 shows a high expression in neural and muscle tissue. RNA in situ hybridization using LNA probes on (A) adult spinal cord (10 × 10 magnification), (B) negative control on spinal cord; (C–H): cross-sections of human fetus 6–8 weeks, approximately at a level between the heart and the brain. (C–E) Overlapping pictures at 1.6× magnification, (F) negative control on the fetal spinal cord and (G and H) 4 × 10 magnification of specific parts of (E). Detection of RNA was visualized by the blue color, and counterstaining was performed with fast Nuclear red. M = muscle, V = developing vertebra, O = oesophagus, SC = spinal cord, SG = sensory ganglia
Interference with Lrsam1 in zebrafish embryos results in a neurodevelopmental phenotype
To study the role of LRSAM1 in neurodevelopment, we injected zebrafish embryos with morpholino oligonucleotides (MOs) to target the Lrsam1 gene (ENSDARG00000060683). We used MOs directed against the ATG to obtain a knockdown phenotype. We designed a splice site MO targeting the last exon, thereby mimicking the situation as found in our patients as closely as possible. MOs directed against splice sites usually lead to exon skipping but we expected that cryptic splice acceptor sites present at a short distance from the original site would be activated, resulting in a protein that is slightly different at the C-terminal end but not prone to nonsense-mediated decay similar to the situation in the patients. RT–PCR followed by sequence analysis on zebrafish embryos indeed showed that an alternative acceptor splice site at 39 nucleotides upstream of the normal acceptor site was used (data not shown) leading to a frameshift followed by a stopcodon after 13 codons, replacing the last 43 codons of the wild-type transcript.
Several injection experiments were performed to obtain the optimal concentration for the MO used. After 6h post-fertilization (hpf), a delay in development was readily observed for the ATG morphants and to a lesser extent for the splice MO-injected embryos, whereas it was hardly present if at all for the control MO-injected embryos (Fig. 5). After 24 hpf, death had occurred in a relatively high percentage and most of the surviving ATG and splice morphants were clearly affected. Only a low percentage of death and abnormalities was seen for the wild-type non-injected or control injected embryos. The phenotype at 2–3 days post-fertilization (dpf) varied from near-normal with a slightly smaller head, a slightly shorter body axis, slightly less pigmentation and bent tail tips to completely curled up and smaller embryos with bent tails with little pigmentation, smaller eyes, abnormal brain development and a less organized structure of the somites. The morphology of all MO-injected embryos at 54 hpf is given in Figure 5, typically affecting about 70–80% of the embryos with about one-third severely affected. The data for six independent experiments are given in Table 3. Despite the observed variation in the severity of the phenotype, the injections reproducibly gave similar results with a phenotype that was comparable between ATG and splice morphants. When allowed to develop for a longer time (experiment 1), the majority of morphants would not hatch. When excluding the most severely affected morphants which obviously would also show abnormal swimming behavior, we observed quite a high percentage with swimming abnormalities which was expected for the ones with curly tails but was also seen in the morphants that looked (near) normal. While wobbly swimming behaviour may partially be related to a developmental delay, some embryos showed no normal response at all when repeatedly touched using a tip of a pipette.
Figure 5.

Phenotype of MO-injected zebrafish embryos at 6 and 54 hpf. (A) Wt embryos at 6 hpf and embryos injected with a control MO (B), ATG MO (C) or splice MO (D). (E–H) Wt embryos at 54 hpf (e) and (F–H) embryos injected with a control MO (f), ATG MO (g) or splice MO (h) at 54 hpf. Magnification used was 0.8×. (I–L) A further magnification of the same ATG (I and J) and splice (K and L) MO-injected embryos where the black bar indicates the size of a wild-type embryo.
Table 3.
Injection experiments of zebrafish embryos.
| Exp. | MO | Conc. (µm) | # | %† 24 hpf | %† final | morphology and hatching | swimming |
|---|---|---|---|---|---|---|---|
| 2 | ATG | 750a | 47 | 44 | 65 | 2 NN, 9 ABN (5 SA) | 1/2 NN and all ABN swim abnormal |
| 1 | ATG | 600 | 31 | 48 | 58 | 6/13 non-hatched and SA | ND |
| 3 | ATG | 550 | 66 | 41 | 59 | 6 NN, 15 ABN (5 SA) | 2 N swimmers rest ABN |
| 4 | ATG | 550 | 60 | 20 | 20 | 13 NN, 35 ABN (10 SA) | 4 N swimmers |
| 2 | ATG | 500 | 62 | 6.5 | 17 | 30 NN, 6 ABN (2 SA) | 14 ABN swimmers |
| 5 | ATG | 450 | 75 | 13 | 17 | All N except 7 ABN 3 SA) | 10 ABN swimmers, rest N |
| 6 | ATG | 450 | 82 | 15 | 16 | All N except 10 weakly affected | 13 abnormal swimmers, rest normal |
| 1 | ATG | 300 | 32 | 15 | 15 | All N, 2/27 w abnormal tails | ND |
| 1 | splice | 800 | 41 | 100 | 100 | All dead | ND |
| 1 | splice | 400 | 46 | 39 | 60 | 1 N, 1 curly tail, rest ABN | ND |
| 2 | splice | 300 | 51 | 63 | 67 | 1 N, 11 ABN (5SA, 2 weak) | 1 N one swims abnormal |
| 3 | splice | 275 | 67 | 50 | 51 | 6 NN, 21 ABN (12 SA) | 2 N swimmers, rest ABN |
| 4 | splice | 275 | 51 | 47 | 49 | 4 NN 21 ABN (8 SA) | 3 N swimmers, rest ABN |
| 2 | splice | 250 | 44 | 38 | 65 | 1 N, 12 ABN | ABN ones swim ABN |
| 5 | splice | 200 | 138 | 30 | 33 | 30 NN, 54 ABN (15 SA) | 2–3 swim N, rest wobbly to ABN |
| 6 | splice | 200 | 86 | 14 | 18 | 44 NN, 24 ABN (3 SA) | 16 ABN swimmers, 3 N, rest wobbly |
| 1 | como | 800 | 39 | 0 | 0 | All N | ND |
| 3 | como | 550 | 39 | 5.1 | 9.1 | All N except one with curly tip of tail | ABN one swims ABN |
| 4 | como | 550 | 25 | 0 | 0 | All N except 1 | 5 ABN swimmers |
| 2 | como | 500 | 40 | 0 | 3.7 | All N except 2 with pigtails | 2 ABN ones move a bit do not swim |
| 5 | como | 450 | 42 | 19 | 2.1 | All N except 3 (1 curly, 1 curly tail tip) | ABN one swims ABN |
| 6 | como | 450 | 23 | 22 | 26 | All N except 1 with slightly curly tailtip | ABN one swims ABN |
| 1 | −(wt) | 33 | 60 | 6.0 | All N | ND | |
| 2 | −(wt) | 22 | 4.5 | 4.5 | All N | All swim N | |
| 2 | −(wt) | 40 | 2.5 | 4.1 | All N | All swim N | |
| 3 | −(wt) | 75 | 1 | 4.6 | All N except 1 with curly tail tip | Curly one swims ABN | |
| 4 | −(wt) | 41 | 2.5 | 2.5 | All N except 1with curly tail | 4 ABN swimmers | |
| 5 | −(wt) | 114 | 11 | 12 | All N except 3 curly (2 body, 1 tip) | 3–4 ABN swimmers | |
| 6 | −(wt) | 45 | 6.6 | 7.1 | All N except 1 | ABN one swims ABN |
Embryos in experiment (exp.) 1 were not dechorionated, but scored for the percentage of hatching and sacrificed at 4 dpf, embryos in exp. 2–4 were dechorionated at 48 hpf and sacrificed at 54 hpf, embryos in exp. 5 and 6 were dechorionated at 72 hpf if necessary and sacrificed at 78 hpf. For staining purposes, six embryos were removed at 30 hpf (splice), 6–16 (ATG), 6–12 embryos (como) and 10–16 (wt) (exp. 2 and 3). Removed embryos and embryos damaged by dechorionation were excluded from final counts. 750a: Volumes of 1 nl were injected except for exp. 2 in which the estimated injection size was 1.15 nl which equals 1 nl of a solution of 750 µm. ABN, abnormal; NN, near normal; A, clearly affected; SA, severely affected. †, Death.
The abnormal phenotype in zebrafish is associated with abnormal motoneuron development
As CMT2 is an axonal neuropathy, the observed phenotype in zebrafish morphants would probably originate in neural abnormalities. To explore this possibility, we used an antibody, znp1, that detects caudal and middle motoneurons and Rohon Beard primary neurons. Immunohistochemical staining of embryos with a slightly to moderately affected phenotype of 30 hpf to 4 dpf showed several abnormalities. Severely affected embryos displayed a very weak staining due to a completely disorganized structure. In less affected embryos, staining seems reduced for both ATG and splice morphants and, in addition, showed a more irregular structure in contrast to the wild-type staining (Fig. 6). Motoneurons normally migrate halfway over the midline of the somite and then develop more extensive arborizations. This structure is less organized in the morphants. When examining older embryos at 4 dpf, a time point where the effects of the MOs are likely to have worn off, staining intensity increases to a level comparable to the controls, but the neural structure remains less well organized (Fig. 6, bottom panels). The observed neuronal aberrations probably explain at least a large part of the phenotype also affecting movement and swimming behavior.
Figure 6.
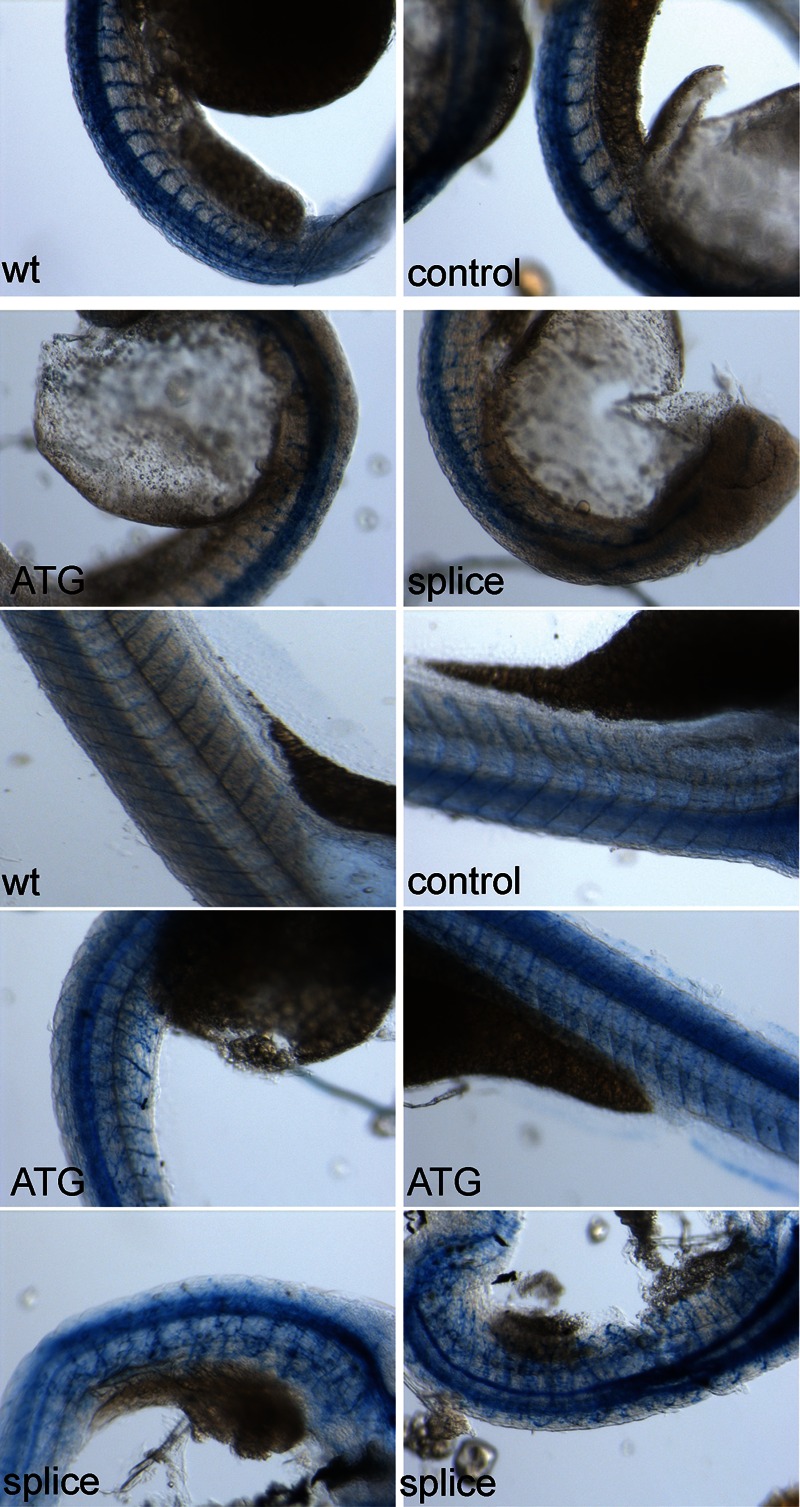
Znp-1 staining in MO-injected zebrafish embryos. Embryos were non-injected or injected as indicated with control MO, ATG MO or splice MO. The upper four pictures show 10× magnifications of zebrafish at 30 hpf (injection exp. 2), the bottom six panels show 10× magnifications of zebrafish at 4 dpf (injection exp.1).
The mutation in LRSAM1 affects its function
LRSAM1 encodes a multidomain protein and has been shown to possess E3-ubiquitin ligase activity owing to the C-terminal RING finger domain (13). E3-ubiquitin ligases constitute a large family of enzymes that can modify their target proteins through covalent attachment of ubiquitin molecules, which will promote their proteosomal degradation. The product of tumor suppressor gene 101, TSG101, is the only LRSAM1 target reported to date. Since the CMT2-associated mutation is located at the border of the C-terminal RING finger domain, it could perturb its E3 ligase activity. To test this hypothesis, HEK293T cells were co-transfected with LRSAM1 and TSG101 expression constructs. The introduction of a RING-inactivating mutation in LRSAM1 which completely abolishes its E3-ligase activity (p.C675A) (13) resulted in an increased abundance of exogenously introduced TSG101, despite equal transfection efficiencies (Fig. 7). Similarly, the introduction of the patient LRSAM1 mutation increased the abundance of the tagged TSG101 to a level even higher than that observed with the C675A mutant, thus supporting the notion that E3-ligase function of the CMT2-associated mutated LRSAM1 is affected.
Figure 7.

Mutation of the RING domain of LRSAM1 affects the abundance of its target TSG101. HEK293T cells were transfected with the indicated expression plasmids for FLAG-LRSAM1 (450 ng) in combination with TSG101-HA (450 ng) and GFP (100 ng). Total cell lysates were analyzed by immunoblotting as indicated. WT, wild-type; C675, RING mutant; PAT, patient mutation; -, no construct.
DISCUSSION
We identified a mutation in LRSAM1, c.2121_2122insGC, p.Leu708ArgfsX28, causative for CMT in a large three-generation family with axonal neuropathy. The 2 bp insertion in the last exon of this gene results in the exchange of the last leucine by an arginine at the border of the C-terminal RING finger motif (amino acids 674–713). In addition, it leads to a frameshift predicted to result in a protein that is 12 amino acids longer than the normal protein. Since the mutation is located in the last exon, the mutant mRNA would not be prone to degradation by nonsense-mediated decay, which may explain the dominant nature of the phenotype. As expected, both mRNAs were found to be present at comparable levels in patient fibroblasts, as determined by RT–PCR followed by the sequence analysis of cDNA fragments containing the mutation site (Supplementary Material, Fig. S1). In contrast, the mutation identified in a Canadian family as the probable cause for CMT2 was a homozygous mutation in the consensus splice acceptor AG dinucleotide of the penultimate exon into an AA dinucleotide which leads to degradation by nonsense-mediated decay of the mRNA and the absence of the protein which was confirmed in EBV-transformed B-lymphocytes of one of their patients. The described clinical phenotype of the Canadian family was that of a relatively mild autosomal recessive CMT2 quite similar to the phenotype found in our family, albeit the mode of inheritance is different. This phenomenon has also been reported for other CMT-associated genes. Mutations in HSP27/HSPB1 can cause both dominant and recessive forms of CMT2 (14). Likewise, most mutations in GDAP that have been reported (15,16) are associated with autosomal recessive CMT disease (CMT4A) but some cause a dominant form of the disease (17).
The phenotype we observed in zebrafish was also similar when comparing two different antisense morpholinos directed against Lrsam1, one blocking the transcription start site resembling a null mutation and the other one blocking splicing at the 3′ end of the messenger, thereby mimicking the patient mutation described here. When screening the sequence of the used morpholinos against other zebrafish transcripts, no other mRNA was remotely similar when allowing up to four mismatches. With the concentrations used, it seems unlikely that the morphological effects and consequences on zebrafish and their ability to respond and move with underlying deficient and less organized motoneurons would be due to a block of other unrelated messengers, coincidentally in both cases leading to the same neurodevelopmental phenotype. Recently, another CMT2 model was described in zebrafish using mitofusin morpholinos (18) with a phenotype quite strikingly similar to Lrsam1 morphants.
LRSAM1 (leucine-rich repeat and sterile alpha motif-containing protein 1) was reported independently by two groups who named the gene TAL, for TSG101-associated protein, and RIFLE, which stands for RING finger and leucin repeat-rich protein, respectively (13,19). The protein was initially implicated in PC12 cells in E-cadherin-mediated cell adhesion and WNT signaling. Subsequent studies showed that it controls sorting of ubiquitinylated cargo and viral budding, which is attributed to ubiquitinylation of TSG101, a component of the ESCRT-I (endosomal-sorting complexes required for transport) sorting machinery (20). Ubiquitinylation by LRSAM1 or Mahogunin RING finger 1 (MGRN1), another RING finger E3-ubiquitin ligase (21), is an important post-translational determinant for expression of TSG101. The found CMT2-associated mutation that is located in the C-terminal RING domain of LRSAM1 required for ubiquitinylation indeed affects TSG101 abundance in co-transfected cells akin to what we observed when using the RING finger dead LRSAM1 mutant C675A (13) which acts as a non-functional allele in this assay. Although it is currently unknown if the readout for LRSAM1 function used here is the (most) relevant aspect ultimately leading to the disease, we can conclude from Figure 7 that the introduction of a high expression of wild-type LRSAM1 does not significantly change the level of exogenously introduced TSG101 when comparing it with the sample in which no exogenous LRSAM1 was introduced, whereas mutant LRSAM1, similar to the C675A loss-of-function mutant, does affect these levels. We propose that the found mutation is a loss-of-function mutant that exerts a downstream dominant effect, and therefore should be regarded as a dominant negative mutation. However, we cannot exclude that mutant LRSAM1 could have acquired some additional characteristic of a dominant nature which we cannot measure using this assay. Based on these results and the expression of both mutant and wild-type mRNAs in patient fibroblasts, haploinsufficiency can be excluded as the mechanism leading to the disease as is the case for the carriers of the Canadian recessive mutation, who have the wild-type allele in addition to a null allele and are not affected. Due to the presence of a sterile alpha motif domain that attributes to oligomerization processes (22), LRSAM1 may act as a homodimer, like other RING finger E3-ubiquitin ligases, or be part of a multimeric ligase complex, but be defective in evoking the normal response, probably by a change in ubiquitinylation signaling. Consequent downstream effects such as dysregulation of expression levels of TSG101, (wild-type) LRSAM1 or other targets or changes in dimerization capacity and/or binding affinity may explain the dominant outcome of the mutation with clinical features similar to having no LRSAM1 protein at all.
Recently, LRSAM1 was also reported as a novel candidate modifier for Huntington's disease, with a role in the regulation of localization and clearance of the protein (23). The upregulation of LRSAM1 led to the amelioration of the Htt phenotype in mice and prevented the loss of NeuN-positive cell bodies in primary striatum cultures, concomitant with a reduction in nuclear aggregates. Conversely, the loss of normal LRSAM1 expression or function in the case of a homozygous recessive mutation or a dominant negative mutation would result in the loss of this neuroprotective effect. Except for TSG101 and Huntington protein, more proteins may be regulated by LRSAM1 activity and affect CNS neurons as well. In this respect, it is remarkable that two of our CMT2 patients with the LRSAM1 mutation also developed Parkinson's disease. Another report described LRSAM1 as a potential component of the antibacterial autophagic response. This would be regulated by its leucine-rich repeats which are typically involved in the detection of pathogen-associated molecular patterns by the innate immune system (24) that is known to be relevant for neural regeneration (25). Clearly, LRSAM1 may affect several pathways and the mechanism how this will lead to a neuropathy will be the subject for further study.
Based on the number of expressed sequence tags and other information in publicly available databases, LRSAM1 appears to be widely expressed, although it was also reported to be higher in tissues with a high energy demand such as the brain, heart, skeletal muscle, liver and kidney. Although we did find a high expression in the tissues that are affected in CMT, it was not restricted to these tissues. Many CMT-associated genes are widely expressed and have been implied in the protein machinery, ranging from amino-acyl transferases to transport. As such, a mutation in LRSAM1 with a potential role in protein clearance would fit into this picture. It is currently unclear whether the large distances in and energy requirement of neural cells make these cells more vulnerable to these mutations or whether the proteins involved in CMT have an additional neural-specific function, as for the case of LRSAM1 the outcome of the phenotype in the zebrafish model could suggest. Our data indicate that LRSAM1 function is relevant for neural development and functioning, and although not neural-specific, it is highly expressed in tissues relevant for CMT both at the fetal level and at the adult level. The identified mutation in the C-terminal part of the protein affects its function to such an extent that the consequent effect is of a dominant nature. In summary, we can conclude that the LRSAM1 mutation we identified in this family is causative for a dominant form of CMT2 enabling further genetic testing for next generations in this family. The absence of LRSAM1 in zebrafish which would represent a null mutation results in a phenotype comparable to the mimicked patient mutation in this model. This provides evidence that the homozygous recessive LRSAM1 mutation that was reported is indeed the causative mutation. We have shown that LRSAM1 mutations can cause both dominant and recessive forms of CMT2 and have also provided a new CMT2 model for inherited neuropathies.
MATERIALS AND METHODS
Patients and DNA
Patients were all seen by an experienced neurologist and subjected to detailed examination. The index person of the family was a Dutch male presenting with the disease at 32 years of age (subject 2.5). There was no medical history of note. Detailed clinical information for all affected family members is given in Table 1. In addition, two brothers of the index patient developed Parkinson's disease at the age of 64 (subject 2.2) and 52 (subject 2.4), respectively. Both showed slowly progressive extrapyramidal features with an asymmetrical onset, a relatively good response to levodopa and a slowly progressive course. Extrapyramidal features were not observed in the younger family members. Clinical data and blood samples were collected as part of the diagnostic procedure for hereditary neuropathies for which all patients gave their consent. Genomic DNA was isolated from blood by proteinase K/SDS treatment followed by phenol/chloroform extractions and ethanol precipitation. Tissue samples for expression analysis were obtained from the department of pathology.
Linkage analysis
DNA samples from 11 affected and 8 unaffected members of the family were genotyped using the Affymetrix GeneChip® Human Mapping 250K Sty Array as described before (26). The gender of samples was verified by counting heterozygous SNPs on the X chromosome. Relationship errors were evaluated with the help of the program graphical representation of relationships (27). Linkage analysis was performed assuming autosomal dominant inheritance, full penetrance and a disease gene frequency of 0.0001. Multipoint LOD scores were calculated using the programs ALLEGRO (10) and SimWalk2 (11). The latter program is much slower but allows for the analysis of large families without splitting them. Haplotypes were constructed with ALLEGRO and presented graphically with Haplopainter (28). All data handling was performed using the graphical interface ALOHOMORA (29).
Next-generation and Sanger sequencing
Sequence capture was performed using a custom design 385K capture array (chr9: 127624486–132419012 hg18/NCBI36) according to the protocol supplied by the manufacturer (Nimblgen, Roche). Basically, a representing library was made of the patient's DNA sheared to an average length of 500–800 bp by cavitation (Covaris). After hybridization, DNA enriched for target sequences was eluted, amplified, quantified (Qubit) and analyzed on a BioAnalyzer DNA 7500 chip (Agilent) followed by pyrosequencing on an FLX titanium sequencer. Analysis was performed with the Newbler program (v2.3, Roche). About 1 × 106 reads were obtained from 1.4 × 106 key passed sequences. Of the 6.5 × 105 filter passed reads, 96% could be mapped, and 50% of the mapped reads were uniquely mapped to the target region and were scanned for differences with the human consensus sequence using the Roche software. All high-quality differences were analyzed for their heterozygous or homozygous state, position within coding exons or distance of at most 20 nucleotides from exon–intron boundaries, known occurrence as polymorphisms and potential effect on the protein.
Direct Sanger sequencing was performed on amplified exons containing the heterozygous nucleotide changes. Amplification was performed with exon-specific M13-tagged primers using 20 ng of genomic DNA, 4 mm MgCl2, Hotfire polymerase (Solys Biodyne) and a touchdown PCR program. Primer sequences are provided in Supplementary Material, Table S1. The resulting PCR products were treated with shrimp alkaline phosphatase and exonuclease I prior to sequencing using the ABI Big Dye Terminator cycle sequencing kit and an ABI3730xl sequencer (Applied Biosystems).
LNA in situ hybridizations
Fluorescein-labeled LNA/2-O-Me probes (locked nucleic acid/2′-O-methyl RNA) were synthesized by Ribotask ApS (Odense, Denmark) and hybridized according to Budde et al. (26) followed by anti-fluorescein-labeled alkaline phosphatase-coupled Fab fragment (Roche, 1:200) incubation as a secondary step. After several washes with a decreasing salt concentration to 0.2× SSC, a final wash was done at ambient temperature and the SK5300 alkaline phosphatase detection kit (Vector Laboratories, Burlingham, CA, USA) was used for visualization. Pretreatment conditions were slightly different for adult and fetal sections. Fetal tissue sections were pretreated using 20 μg/ml protK, followed by post-fixation in 4% paraformaldehyde (PFA), and denaturation of proteins in adult sections was achieved by a 10min incubation in a microwave in 0.1 m citric acid/0.1 m trisodium citrate (pH = 6). Probe concentrations were 0.25–1 µm. Secondary antibody fragment dilutions were 1:200 and 1:1000, respectively. Counterstaining was performed using fast Nuclear red. Pictures were taken on an Olympus BX41 microscope using CellD software. As a control, samples were taken where the probe was omitted from the hybridization mixture. The sequence of the LNA probe used was: 5′-FAM-UcuGgcUccUgaUcuGccG-3′ where capital letters represent LNA, normal letters are 2-O-methyl oligos.
Morpholino experiments in zebrafish
All zebrafish (Danio rerio) procedures complied with standard animal care guidelines according to Dutch Animal Ethics Committee guidelines. Breeding and maintenance of all zebrafish stocks were performed as described by others (30). Wild-type TL zebrafish outbred strains were crossed to generate embryos for MO injections. MO antisense 25mers were designed against the sequences surrounding the ATG and the last splice acceptor site by the manufacturing company (Genetools; ATG MO: GAACAAAAACATCGTGAGGCTTAGC; splice MO: TGAGACTACACAAAGCAGAACAAGA). A range of 1.7–6.4 ng of MO (1 nl of a 200–750 μm MO solution) was injected into the yolk sac of one to two cell-stage embryos using fine glass needles and a microinjector (World Precision Instruments). Standard control MOs (GeneTools) and non-injected wild-types were taken as controls (31). The developing embryos were allowed to recover in E3 medium and incubated at 28°C. Morphology was monitored over time and images were taken using a Leica MZFLIII equipped with a Retiga 2000R Fast1394 Q imaging camera and the QCapture Pro program. Zebrafish were dechorionated in all experiments at 2–3 dpf, at least 6 h before final pictures were taken except for one experiment where the percentage of hatching was examined. Swimming behavior was examined by the response upon repeatedly touching with a pipette tip. Surviving zebrafish were fixed in 4% PFA after which they were kept in PBS prior to immunohistochemistry (IHC) or stored at −20°C in methanol.
Wholemount IHC zebrafish
Zebrafish older than 30 hpf were bleached for 3–4min using a bleaching solution containing 5% formamide, 0.5× SSC and 9% H2O2. After several washes in PBST, the embryos were permeabilized with 2.5 mg/ml trypsin in PBST for 3–4 min on ice, followed by three PBST washes and incubation with the primary antibody in a 1:400–1:2000 dilution in blocking buffer at 4°C for 16–48 h under gentle agitation. Similarly, secondary antibody detections were performed overnight at 4°C in a 1:1000–2000 dilution (Southern Biotechnology, AP-coupled goat anti-mouse IgG) followed by extensive washing prior to detection using the SK5300 (Vector Laboratories) alkaline phosphatase detection kit. Images were captured using a Leica M125 stereomicroscope equipped with a DFC425 camera and LASv3.7 software.
Transfections and western blot analysis
HEK 293T cells were obtained from the American Tissue Culture Collection and maintained in DMEM (Invitrogen) supplemented with 10% FBS at 37°C and 5% CO2. Cells were transfected with Lipofectamine 2000 (Invitrogen). Full-length wild-type LRSAM1 cDNA was amplified by PCR from IMAGE clone ID 5852; cat#OHS1770-9382089 (Thermo Scientific) using oligonucleotides flanking the coding part of LRSAM1 and recombined into the gateway entry plasmid pDONR221 (Invitrogen). Site-directed mutagenesis was used to introduce the C675A mutation in the RING domain of LRSAM1 with the QuickChange site mutagenesis kit (Stratagene). Full-length LRSAM1 carrying the c.2121_2122duplGC mutation was cloned with a two-step PCR strategy. Briefly, genomic DNA from fibroblasts of an individual carrying this mutation was used to amplify exon 26 of LRSAM1. Subsequently, the obtained PCR fragment was used to amplify the full-length LRSAM1 cDNA in combination with the forward primer that was used to clone full-length wt LRSAM1. The pCAGGS-hTSG101-HA vector was a kind gift from Dr R. Harty (University of Pennsylvania) (32). To generate mammalian expression plasmids for LRSAM1, we used LR recombination between LRSAM1-containing pDONR221 and pDEST-FLAG. Transfection efficiency was monitored by co-transfection of an expression plasmid for GFP and was consistently >90%.
Total cell lysates were prepared in RIPA buffer (150 mm NaCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 100 mm Tris–HCl, pH 7.4) supplemented with protease inhibitors (Roche Molecular Biochemicals). Lysates were cleared by centrifugation at 4°C for 10 min at 10 000g. Protein concentration was determined using the Bradford assay (Biorad) with BSA as reference. Samples (10–40 µg) were separated on NuPAGE Bis–Tris gels (Invitrogen) and transferred to nitrocellulose. Membranes were probed with the following antibodies: HA (Covance, clone HA.11, 1:4000–10000), tubulin (Calbiochem, clone CP06, 1:10000), FLAG-HRP (Sigma, clone M2, 1:8000), GFP (affinity purified rabbit polyclonal anti-GFP was a gift from Dr Mireille Riedinger, UCLA, 1:10000). Secondary HRP-conjugated antibodies (Zymed) were used and visualized with chemiluminescence on a Fuji LAS4000 (GE Healthcare). The immunoblots are representative of at least three independent experiments.
WEB RESOURCES
CMT mutation databases: http://www.molgen.ua.ac.be, http://www.neuromuscular.wustl.edu.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
Znp-1 was obtained from the Zebrafish International Resource Center which is supported by grant P40 RR012546 from the NIH-NCRR.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Professor Dr M. Kamermans (NIN, Amsterdam), Professor Dr D. Troost (Department of Pathology, AMC Amsterdam), Dr P. van Tijn (NIN, Amsterdam) and Dr K. de Jong (Department of Anatomy, Embryology and Physiology, AMC Amsterdam) for use of facilities and valuable advice and Z. Qahar for technical support.
REFERENCES
- 1.Matsunami N., Smith B., Ballard L., Lensch M.W., Robertson M., Albertsen H., Hanemann C.O., Muller H.W., Bird T.D., White R. Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot–Marie–Tooth 1A. Nat. Genet. 1992;1:176–179. doi: 10.1038/ng0692-176. [DOI] [PubMed] [Google Scholar]
- 2.Patel P.I., Roa B.B., Welcher A.A., Schoener-Scott R., Trask B.J., Pentao L., Snipes G.J., Garcia C.A., Francke U., Shooter E.M., et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot–Marie–Tooth disease type 1A. Nat. Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 3.Timmerman V., Nelis E., van Hal W., Nieuwenhuijsen B.W., Chen K.L., Wang S., Ben O.K., Cullen B., Leach R.J., Hanemann C.O. The peripheral myelin protein gene PMP-22 is contained within the Charcot–Marie–Tooth disease type 1A duplication. Nat. Genet. 1992;1:171–175. doi: 10.1038/ng0692-171. [DOI] [PubMed] [Google Scholar]
- 4.Valentijn L.J., Baas F., Wolterman R.A., Hoogendijk J.E., van den Bosch N.H., Zorn I., Gabreels-Festen A.W., de Visser M., Bolhuis P.A. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot–Marie–Tooth disease type 1A. Nat. Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 5.Zhang F., Seeman P., Liu P., Weterman M.A., Gonzaga-Jauregui C., Towne C.F., Batish S.D., De Vriendt E., De Jonghe P, Rautenstrauss B., et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weterman M.A., van Ruissen F., de Wissel M., Bordewijk L., Samijn J.P., van der Pol W.L., Meggouh F., Baas F. Copy number variation upstream of PMP22 in Charcot–Marie–Tooth disease. Eur. J. Hum. Genet. 2010;18:421–428. doi: 10.1038/ejhg.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuesta A., Pedrola L., Sevilla T., Garcia-Planells J., Chumillas M.J., Mayordomo F., LeGuern E., Marin I., Vilchez J.J., Palau F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot–Marie–Tooth type 4A disease. Nat. Genet. 2002;30:22–25. doi: 10.1038/ng798. [DOI] [PubMed] [Google Scholar]
- 8.Baxter R.V., Ben O.K., Rochelle J.M., Stajich J.E., Hulette C., Dew-Knight S., Hentati F., Ben H.M., Bel S., Stenger J.E., et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot–Marie–Tooth disease type 4A/8q21. Nat. Genet. 2002;30:21–22. doi: 10.1038/ng796. [DOI] [PubMed] [Google Scholar]
- 9.Shy M.E., Jani A., Krajewski K., Grandis M., Lewis R.A., Li J., Shy R.R., Balsamo J., Lilien J., Garbern J.Y., Kamholz J. Phenotypic clustering in MPZ mutations. Brain. 2004;127:371–384. doi: 10.1093/brain/awh048. [DOI] [PubMed] [Google Scholar]
- 10.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 11.Sobel E., Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 12.Guernsey D.L., Jiang H., Bedard K., Evans S.C., Ferguson M., Matsuoka M., Macgillivray C., Nightingale M., Perry S., Rideout A.L., et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot–Marie–Tooth disease. PLoS Genet. 2010;6:e.1001081. doi: 10.1371/journal.pgen.1001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amit I., Yakir L., Katz M., Zwang Y., Marmor M.D., Citri A., Shtiegman K., Alroy I., Tuvia S., Reiss Y., et al. Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004;18:1737–1752. doi: 10.1101/gad.294904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houlden H., Laura M., Wavrant-de Vrieze F., Blake J., Wood N., Reilly M.M. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71:1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]
- 15.Bouhouche A., Birouk N., Azzedine H., Benomar A., Durosier G., Ente D., Muriel M.P., Ruberg M., Slassi I., Yahyaoui M., et al. Autosomal recessive axonal Charcot–Marie–Tooth disease (ARCMT2): phenotype-genotype correlations in 13 Moroccan families. Brain. 2007;130:1062–1075. doi: 10.1093/brain/awm014. [DOI] [PubMed] [Google Scholar]
- 16.Georgiou D.M., Nicolaou P., Chitayat D., Koutsou P., Babul-Hirji R., Vajsar J., Murphy J., Christodoulou K. A novel GDAP1 mutation 439delA is associated with autosomal recessive CMT disease. Can. J. Neurol. Sci. 2006;33:311–316. doi: 10.1017/s0317167100005199. [DOI] [PubMed] [Google Scholar]
- 17.Claramunt R., Pedrola L., Sevilla T., Lopez de M.A., Berciano J., Cuesta A., Sanchez-Navarro B., Millan J.M., Saifi G.M., Lupski J.R., et al. Genetics of Charcot–Marie–Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet. 2005;42:358–365. doi: 10.1136/jmg.2004.022178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vettori A., Bergamin G., Moro E., Vazza G., Polo G., Tiso N., Argenton F., Mostacciuolo M.L. Developmental defects and neuromuscular alterations due to mitofusin 2 gene (MFN2) silencing in zebrafish: a new model for Charcot–Marie–Tooth type 2A neuropathy. Neuromuscul. Disord. 2011;21:58–67. doi: 10.1016/j.nmd.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Ammar N., Nelis E., Merlini L., Barisic N., Amouri R., Ceuterick C., Martin J.J., Timmerman V., Hentati F., de Jonghe P. Identification of novel GDAP1 mutations causing autosomal recessive Charcot–Marie–Tooth disease. Neuromuscul. Disord. 2003;13:720–728. doi: 10.1016/s0960-8966(03)00093-2. [DOI] [PubMed] [Google Scholar]
- 20.McDonald B., Martin-Serrano J. Regulation of Tsg101 expression by the steadiness box: a role of Tsg101-associated ligase. Mol. Biol. Cell. 2008;19:754–763. doi: 10.1091/mbc.E07-09-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim B.Y., Olzmann J.A., Barsh G.S., Chin L.S., Li L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol. Biol. Cell. 2007;18:1129–1142. doi: 10.1091/mbc.E06-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchler-Bauer A., Lu S., Anderson J.B., Chitsaz F., Derbyshire M.K., DeWeese-Scott C., Fong J.H., Geer L.Y., Geer R.C., Gonzales N.R., et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang B., Seredenina T., Coppola G., Kuhn A., Geschwind D.H., Luthi-Carter R., Thomas E.A. Gene expression profiling of R6/2 transgenic mice with different CAG repeat lengths reveals genes associated with disease onset and progression in Huntington's disease. Neurobiol. Dis. 2011;42:459–467. doi: 10.1016/j.nbd.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng A.C., Eisenberg J.M., Heath R.J., Huett A., Robinson C.M., Nau G.J., Xavier R.J. Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl Acad. Sci. USA. 2011;108(Suppl 1):4631–4638. doi: 10.1073/pnas.1000093107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramaglia V., Tannemaat M.R., de Koning M., Wolterman R., Vigar M.A., King R.H., Morgan B.P., Baas F. Complement inhibition accelerates regeneration in a model of peripheral nerve injury. Mol. Immunol. 2009;47:302–309. doi: 10.1016/j.molimm.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 26.Budde B.S., Namavar Y., Barth P.G., Poll-The BT, Nurnberg G., Becker C., van Ruissen F., Weterman M.A., Fluiter K., te Beek E.T., et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 2008;40:1113–1118. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- 27.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 28.Thiele H., Nuernberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 29.Rueschendorf F., Nuernberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 30.Klooster J., Yazulla S., Kamermans M. Ultrastructural analysis of the glutamatergic system in the outer plexiform layer of zebrafish retina. J. Chem. Neuroanat. 2009;37:254–265. doi: 10.1016/j.jchemneu.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Kasher P.R., Namavar Y., van Tijn P., Fluiter K., Sizarov A., Kamermans M., Grierson A.J., Zivkovic D., Baas F. Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum. Mol. Genet. 2011;20:1574–1584. doi: 10.1093/hmg/ddr034. [DOI] [PubMed] [Google Scholar]
- 32.Irie T., Licata J.M., McGettigan J.P., Schnell M.J., Harty R.N. Budding of PPxY-containing rhabdoviruses is not dependent on host proteins TGS101 and VPS4A. J. Virol. 2004;78:2657–2665. doi: 10.1128/JVI.78.6.2657-2665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.