

Abstract
Adiponectin is a protein hormone that can affect major metabolic processes including glucose regulation and fat metabolism. Our previous genome-wide association (GWA) study of circulating plasma adiponectin levels in Filipino women from the Cebu Longitudinal Health and Nutrition Survey (CLHNS) detected a 100 kb two-SNP haplotype at KNG1–ADIPOQ associated with reduced adiponectin (frequency = 0.050, P = 1.8 × 10−25). Subsequent genotyping of CLHNS young adult offspring detected an uncommon variant [minor allele frequency (MAF) = 0.025] located ∼800 kb from ADIPOQ that showed strong association with lower adiponectin levels (P = 2.7 × 10−15, n = 1695) and tagged a subset of KNG1–ADIPOQ haplotype carriers with even lower adiponectin levels. Sequencing of the ADIPOQ-coding region detected variant R221S (MAF = 0.015, P = 2.9 × 10−69), which explained 17.1% of the variance in adiponectin levels and largely accounted for the initial GWA signal in Filipinos. R221S was not present in 12 514 Europeans with previously sequenced exons. To explore the mechanism of this substitution, we re-measured adiponectin level in 20 R221S offspring carriers and 20 non-carriers using two alternative antibodies and determined that the presence of R221S resulted in artificially low quantification of adiponectin level using the original immunoassay. These data provide an example of an uncommon variant responsible for a GWA signal and demonstrate that genetic associations with phenotypes measured by antibody-based quantification methods can be affected by uncommon coding SNPs residing in the antibody target region.
INTRODUCTION
Secreted almost exclusively from adipocytes into the bloodstream, adiponectin is an abundant protein hormone that affects metabolic processes including glucose regulation and fat metabolism (1). Circulating plasma adiponectin level is substantially heritable (2–4), and low levels are associated with increased body mass index (BMI) and risk of cardiovascular disease, type 2 diabetes, atherosclerosis and metabolic syndrome (1,5–8). Understanding the genetic basis of adiponectin level may help unravel the etiology of these complex diseases.
The gene ADIPOQ, which encodes adiponectin, is an obvious candidate for influencing adiponectin level and its effects on target tissues. Candidate gene association studies (9–12) as well as genome-wide association (GWA) studies (13–16) of common SNPs [minor allele frequency (MAF) ≥ 0.05] have identified several potentially functional ADIPOQ variants. In vitro studies have shown that rare (MAF < 0.005) and uncommon (0.005 ≤ MAF < 0.05) missense variants can disrupt the multimerization necessary for adiponectin secretion (17) and that SNPs in the ADIPOQ promoter may alter its transcription (18). Nonetheless, the true causal variants responsible for the previously reported GWA signals at ADIPOQ have not yet been established.
We previously reported a 100 kb two-SNP haplotype spanning the KNG1–ADIPOQ gene region that is associated with reduced adiponectin level in 1776 Filipino women from the Cebu Longitudinal Health and Nutrition Survey (CLHNS) (19). Compared with the strongest single SNP associated with adiponectin in the GWA study (rs864265, MAF = 0.124, β = −0.123, P = 3.8 × 10−9), the C-T rs11924390-rs864265 haplotype showed substantially stronger evidence of association with a larger effect size (frequency = 0.050, β = −0.385, P = 1.8 × 10−25). Imputation of SNPs from the 1000 Genomes Pilot Project (June 2010) failed to identify any single SNP with association evidence at this level of significance, suggesting either that the true causal variant was poorly imputed, thus weakening its evidence of association, or that a lower frequency variant or more than one variant may be responsible for the GWA signal (19).
In the current study, we further characterized this KNG1–ADIPOQ haplotype association by identifying an uncommon missense variant responsible for much of the signal. In young adult offspring from the CLHNS, in whom the haplotype showed a similar effect (frequency = 0.052, β = −0.386, P = 8.7 × 10−32) (19), the observation of an additional putative association signal in the region led serendipitously, although indirectly, to an explanation for the original GWA signal at KNG1–ADIPOQ.
RESULTS
To study the genetic basis of circulating plasma adiponectin level and other metabolic traits, we genotyped using the MetaboChip a set of young adult offspring from the CLHNS. The offspring did not show evidence of population substructure (Supplementary Material, Fig. S1), and an assessment of global ancestry showed them clustering with other Asian individuals as expected (Supplementary Material, Fig. S2). Of 140 696 polymorphic MetaboChip SNPs tested for association with natural log-transformed adiponectin level in 1695 offspring, the two most strongly associated SNPs (P < 5.0 × 10−8) were located ∼800 kb apart on chromosome 3 (Fig. 1). Located upstream of the ADIPOQ gene, the first SNP, rs864265 (P = 1.0 × 10−13, Table 1), had been detected by our previous GWA study (19). The second SNP, rs117016164, showed stronger evidence of association (P = 2.7 × 10−15, Table 1), and is located downstream of the ETV5 gene. Originally identified by the 1000 Genomes Project as a singleton in a CHB HapMap sample (NA18593), rs117016164 had not been detected in CEU, JPT or YRI samples. This SNP was uncommon in the CLHNS offspring (MAF = 0.025), and exhibited very low linkage disequilibrium (LD) with all other SNPs within the densely typed ETV5 gene region (r2 < 0.1, chr3:187.2–187.4 Mb in Fig. 1). These data suggested a possible second and novel association signal 800 kb away from the original GWA signal.
Figure 1.

Two adiponectin association signals (P< 5.0 × 10−8) on chromosome 3 in 1695 CLHNS offspring. Regional plot of MetaboChip SNP associations with plasma adiponectin level on chromosome 3 (187.2–188.2 Mb). The previously associated 100 kb two-SNP KNG1–ADIPOQ haplotype of rs119243940 and rs864265 spans the region shaded gray. Plot was generated using LocusZoom (45).
Table 1.
Association of three SNPs with plasma adiponectin level in CLHNS offspring and mothers
| SNP (gene) | Allele 1/2 | CLHNS offspring (n = 1695) |
CLHNS mothers (n = 1764) |
Combined samples |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | β (SE) | P-values | R2 | MAF | β (SE) | P-values | R2 | β (SE) | P-values | ||
| rs864265 (ADIPOQ) | T/G | 0.129 | −0.134 (0.018) | 1.0 × 10−13 | 0.032 | 0.124 | −0.124 (0.020) | 4.8 × 10−10 | 0.022 | −0.128 (0.014) | 4.2 × 10−20 |
| rs117016164 (ETV5) | T/C | 0.025 | −0.304 (0.038) | 2.7 × 10−15 | 0.036 | 0.030 | −0.198 (0.039) | 3.2 × 10−7 | 0.015 | −0.240 (0.028) | 7.0 × 10−17 |
| R221S (ADIPOQ) | A/C | 0.015 | −0.853 (0.046) | 2.9 × 10−69 | 0.171 | 0.015 | −0.811 (0.051) | 2.0 × 10−53 | 0.132 | −0.823 (0.035) | 4.3 × 10−99 |
Effect betas (β), standard errors (SE), P-values and squared type-II partial correlations (R2) are reported in terms of the minor allele (underlined) and for natural log-transformed values of adiponectin. MAFs are reported for all 1774 unrelated offspring and 1798 mothers, regardless of covariate completeness. In the analyses of offspring, models were adjusted for sex and BMI. In the analyses of mothers, models were adjusted for age, age2, household assets, natural log-transformed income, menopausal status and BMI. In the combined analyses, models accounting for sample relatedness were adjusted for sex, age, age2, household assets, natural log-transformed income and BMI. All covariates were from the 2005 survey.
To validate the evidence of association of rs117016164 with adiponectin in the CLHNS offspring, we re-genotyped the SNP and, observing concordant genotypes, then performed a permutation test, which generated strong and consistent evidence of association (Pperm < 10−9). In addition, the 1764 CLHNS mothers showed directionally consistent association (P = 3.2 × 10−7, Table 1). Using a general linear mixed model accounting for sample relatedness, the combined set of offspring and mothers strengthened the evidence of association of rs117016164 with adiponectin levels (Pcombined = 7.0 × 10−17).
We next examined the relationship between rs117016164 and the two SNPs from the previously identified adiponectin-associated KNG1–ADIPOQ haplotype (rs119243940 and rs864265). The SNP rs117016164 was in low LD with both rs119243940 (D′ = 0.38, r2 = 0.004) and rs864265 (D′ = 0.37, r2 = 0.024). Carrying the C-T haplotype at KNG1–ADIPOQ was associated with lower adiponectin levels in CLHNS offspring (P = 1.0 × 10−37, Fig. 2A). When rs117016164 was included to form three-SNP haplotypes, all carriers of the original two-SNP C-T haplotype still had significantly lower adiponectin levels than non-carriers; they carried haplotype H4 (C-C-T, P = 2.9 × 10−16) and/or haplotype H6 (T-C-T, P = 2.1 × 10−39) (Table 2). A post hoc comparison of these two haplotypes showed that their strengths of association were significantly different (P = 2.9 × 10−18), suggesting that the minor allele (T) of rs117016164 tagged a subset of KNG1-ADIPOQ haplotype carriers with even lower adiponectin levels. A consistent pattern of strong haplotype association was also observed in the mothers (Supplementary Material, Table S1). These data suggested that the H4 and H6 haplotypes together likely harbored two or more SNPs that might account for the observed evidence of association at the KNG1–ADIPOQ locus.
Figure 2.

Association of KNG1–ADIPOQ haplotype in CLHNS offspring is attenuated by ADIPOQ missense variant R221S. Adiponectin residuals plotted by number of copies of (A) the associated C-T KNG1–ADIPOQ haplotype, (B) the ADIPOQ missense variant R221S and (C) the KNG1–ADIPOQ haplotype when conditioned on R221S.
Table 2.
Association of three-SNP haplotypes with plasma adiponectin level in CLHNS offspring
| Haplotype | rs117016164 | rs11924390 | rs864265 | Haplotype frequency | Number of carriers | β | SE | P-value |
|---|---|---|---|---|---|---|---|---|
| H1 | C | C | G | 0.435 | 1196 | — | — | — |
| H2 | C | T | G | 0.421 | 1166 | 0.017 | 0.012 | 0.15 |
| H3 | C | T | T | 0.075 | 256 | 0.022 | 0.022 | 0.31 |
| H4 | C | C | T | 0.044 | 154 | −0.237 | 0.029 | 2.9 × 10−16 |
| H5 | T | C | G | 0.010 | 33 | −0.060 | 0.058 | 0.30 |
| H6 | T | C | T | 0.009 | 31 | −0.817 | 0.061 | 2.1 × 10−39 |
| H7 | T | T | G | 0.005 | 19 | 0.035 | 0.076 | 0.64 |
| H8 | T | T | T | 0.001 | 5 | −0.199 | 0.164 | 0.23 |
Effect betas (β), standard errors (SE) and P-values were calculated for each haplotype relative to the most common reference haplotype. Frequencies are reported for all 1774 unrelated offspring, regardless of covariate completeness. Significant associations (P< 0.05) are in boldface. Models were adjusted for sex and for BMI from the 2005 survey. A post hoc comparison of H4 and H6 showed that their strengths of association were significantly different (P = 2.9×10−18).
Three pieces of evidence led us to hypothesize that coding variants in ADIPOQ could explain the observed haplotype associations. First, although ETV5 was a plausible candidate gene based on a nearby SNP previously reported to be associated with BMI (20) and the inverse relationship between adiponectin level and BMI, rs117016164 was not located in a conserved region or a regulatory element predicted based on chromatin state. Second, the evidence of long haplotypes linking rs117016164 to the two adiponectin-associated SNPs in the KNG1-ADIPOQ region reinforced the possibility that causal variants could be located in other genes in the region; among these, ADIPOQ was the strongest biological candidate. Third, although decreased gene expression by a non-coding regulatory variant is a more likely scenario at many GWA loci, amino acid substitutions in ADIPOQ have been shown to disrupt the multimerization of the adiponectin protein necessary for its secretion into the bloodstream (17). We hypothesized then that one or more low-frequency-coding variants in ADIPOQ could have large effects on adiponectin levels consistent with the observed associations.
To identify rare or uncommon coding variants in ADIPOQ, we sequenced the translated exons of the gene in 47 CLHNS offspring, a sample set enriched for carriers of the H6 and H4 haplotypes. We observed three variants in the coding region, each in at least one sample: G15G (rs2241766), a common variant that previously showed modest association with adiponectin in the CLHNS mothers (P = 0.0077) (19); G48D, a novel missense variant in one individual with average adiponectin level (2.15 μg/ml); and R221S, a missense variant first reported in Japanese (21). To help predict their potential impact on protein structure and function, we performed bioinformatic annotation of the two observed missense variants. G48D was predicted by polymorphism phenotyping (PolyPhen) to be ‘likely damaging’ with a score of 0.997 and by sorting intolerant from tolerant (SIFT) to ‘affect protein function’ with a score of 0.00. R221S was predicted by PolyPhen to be ‘likely benign’ with a score of 0.000 and by SIFT to be ‘tolerated’ with a score of 0.41. The difference in the predicted effects of the two variants reflects that ADIPOQ amino acid 48 is more highly conserved across related protein sequences, and therefore more sensitive to substitution by an unlike residue. Eight other previously reported missense variants (9,10,21–24) were not found in these 47 offspring samples, including G45R, G84R, G90S, R92X, Y111H, R112C, I164T and H241P. However, as the I164T substitution was uncommon (MAF = 0.009) and strongly associated with lower adiponectin in Japanese (12), we included this SNP in follow-up genotyping.
We directly genotyped three candidate coding variants (G48D, I164T and R221S) in the complete sets of offspring and mothers, and tested each for association with plasma adiponectin level. No additional G48D carriers were identified, and I164T was monomorphic in both the mothers and offspring. In contrast, R221S was present in the offspring with MAF = 0.015, and carriers had significantly lower adiponectin levels than non-carriers (P = 2.9 × 10−69) (Fig. 2B, Table 1). The ADIPOQ R221S variant was in low LD with the ETV5 variant (rs117016164) (D′ = 0.54, r2 = 0.17). We observed similar evidence of R221S association in the mothers (P = 2.0 × 10−53) (Supplementary Material, Fig. S3, Table S1), and in the combined mothers and offspring (Pcombined = 4.3 × 10−99) (Table 1). R221S explained 17.1 and 13.2% of phenotypic variation in the offspring and mothers, respectively. Thus, only one of the three observed non-synonymous amino acid substitution variants in ADIPOQ (R221S) was strongly associated with adiponectin in Filipinos.
To determine the relationship between the uncommon coding variant R221S and the associated haplotypes, we performed conditional analyses. Conditioning on R221S, offspring carriers of the two-SNP C-T KNG1–ADIPOQ haplotype still had significantly lower adiponectin levels, but the association was substantially attenuated (P = 5.6 × 10−10, Fig. 2C). Carriers of the three-SNP H6 haplotype showed greatly attenuated association (P = 0.030), whereas carriers of H4 were still significantly associated with lower adiponectin levels (P = 1.3 × 10−8). The mothers showed similar attenuations (Supplementary Material, Fig. S3C and data not shown). We also re-phased haplotypes in the mothers after including the R221S variant and found that it segregated very infrequently (at most twice) on any haplotypes other than H4 and H6. These results suggested not only that the H6 haplotype association was largely explained by R221S, but also that one or more additional adiponectin-associated variants remained to be identified on the H4 haplotype, as its association was only partially explained by R221S.
In the CLHNS samples, adiponectin level shows modest but significant negative correlation (P < 0.0001) with multiple metabolic traits, including BMI, waist circumference, fasting glucose and insulin and homeostasis model assessment indices of insulin sensitivity and beta cell function (19). We tested R221S in both the mothers and offspring for association with these traits and lipid profiles and did not observe significant evidence of association (P = 0.13–0.95 in offspring and P = 0.20–0.98 in mothers, data not shown). In comparison, the ADIPOQ SNP rs864265 from the original GWA signal had shown nominal association (P < 0.05) in the offspring with several of these traits (19). With the comparatively smaller MAF of R221S, our study had <10% power to detect such associations with comparable effect sizes at P < 0.05. Given prior in vitro biochemical evidence that R221S does not affect adiponectin multimer formation or secretion (17), we hypothesized that the R221S substitution affected the original antibody detection of adiponectin, resulting in artificially low levels among carriers. The original enzyme-linked immunosorbent assay (ELISA) consisted of two monoclonal antibodies targeting amino acids 104–244 of ADIPOQ. We re-measured plasma adiponectin level in 20 R221S carriers and 20 sex- and BMI-matched non-carriers using two alternative polyclonal antibodies that recognize epitopes within amino acids 17–27 and 56–70, neither of which should be affected by amino acid 221. Adiponectin measurements were not consistent between the original ELISA and each of the alternative antibodies (Fig. 3 and data not shown). The subset of R221S carriers had lower adiponectin levels than non-carriers based on the original ELISA, but carriers and non-carriers showed similar distributions with the alternative antibodies. These results suggested that R221S changed the structure of adiponectin enough to alter the binding affinity of the monoclonal antibody in the original ELISA. These results confirmed that the very strong statistical association of R221S with lower adiponectin level was likely due to a methodological artifact of antibody specificity.
Figure 3.

Inconsistent plasma adiponectin measurements between assays are attributable to R221S. Plots of relative adiponectin levels of 20 R221S non-carriers (R/R: open circles) are shown compared with 20 R221S carriers (R/S: filled circles), as measured by two antibody-based assays for ADIPOQ [left: epitopes within amino acids (a.a.) 104–244, right: 56–70]. Within each assay, all adiponectin levels are natural log-transformed and zero-centered on the mean value of R221S non-carriers, and t-test P-values are reported for the difference in means between carriers and non-carriers.
We then re-evaluated the evidence of adiponectin association after conditioning on R221S. Overall, there were no substantial changes in any SNP associations at loci other than KNG1–ADIPOQ in the offspring (data not shown) or mothers (Supplementary Material, Fig. S4), including at CDH13, a previously reported GWA signal in the CLHNS (19); similar results were observed after removing R221S carriers from analysis (data not shown and Supplementary Material, Fig. S4). To examine the associations of additional variants in the 187.2–188.2 Mb region of chromosome 3, we re-imputed in the mothers SNPs from the November 2010 release of the 1000 Genomes Project (Supplementary Material, Fig. S5). Prior to conditioning on R221S, the most significantly associated SNP in the region was rs864265 (P = 2.2 × 10−10). After conditioning, this signal was highly attenuated (P = 0.076) and the most significantly associated SNP was rs58575091 (P = 3.7 × 10−5). We re-evaluated eight ADIPOQ SNPs that were identified as candidate functional variants in the literature (19), and of those, only rs266729 and rs182052 still showed nominal association (P < 0.05) after conditioning on R221S (Supplementary Material, Table S2). These associations remained significant after conditioning on both R221S and copies of the C-T haplotype (rs266729, P = 1.8 × 10−4 and rs182052, P = 4.6 × 10−4). When the two SNPs were included in the model together, rs266729 was modestly more significant than rs182052 (P = 0.067 versus P = 0.22). Comparable results were obtained when removing R221S carriers from analysis (data not shown). Thus, after conditioning on R221S, moderate evidence of association with adiponectin level remained. The conditional analyses suggested that two or more additional variants still play a role in trait variability in this study.
DISCUSSION
Follow-up of our previous GWA study in the CLHNS demonstrates that the two-SNP haplotype in the KNG1–ADIPOQ region associated with lower plasma adiponectin level can be attributed to a coding variant that affected antibody specificity. This uncommon non-synonymous amino acid substitution variant (R221S, MAF = 0.015) was responsible for most of the initial GWA signal, which was based on a survey of common variants. R221S explained 17.1 and 13.2% of adiponectin variation in the offspring and mothers, respectively. In comparison, the index SNP from the original GWA study (rs864265, MAF = 0.129) explained 3.2 and 2.2% of variation, respectively. We discovered R221S by detecting in the offspring a long three-SNP haplotype created by another uncommon SNP nearly 800 kb away from ADIPOQ. This SNP (rs117016164) was genotyped fortuitously, as it was included on the MetaboChip to fine-map the ETV5 gene region previously associated with BMI (20). The location of rs117016164 downstream of ETV5 and its lack of overlap with predicted regulatory elements in a relevant cell type, such as adipocytes, made ETV5 a less compelling candidate gene for regulating adiponectin level. Furthermore, rs117016164 was the only variant in the ETV5 region to show evidence of association comparable with that of rs864265 near ADIPOQ, suggesting the presence of a long haplotype spanning the entire region. Prior biochemical experiments have shown that ADIPOQ amino acid substitutions can disrupt the multimer aggregation required for adiponectin secretion (17). Targeted sequencing of ADIPOQ-coding regions in carriers of the associated haplotypes then revealed R221S, which segregated primarily on the two long haplotypes associated with lower adiponectin level in the entire cohort. Therefore, the causal variant was not only detectable 800 kb away, but was also nearly 10 times less frequent and explained a far greater proportion of phenotypic variance than the GWA index SNP, in line with a previously proposed association causation scenario (25). The proportion of reported GWAS signals explained by uncommon variants remains to be determined, but R221S is a clear example of one.
Because R221S has been reported previously only in Japanese (21), Korean (26,27) and Japanese–Brazilian (28) cohorts, the variant may be specific to Asian-ancestry populations. This R221S variant (CGT to AGT) was also observed to be monomorphic in 12 514 individuals of European ancestry with sequenced exons (M.R. Nelson et al., manuscript in preparation). A different ADIPOQ missense variant (I164T) with low frequency in Japanese (MAF = 0.009) has been shown to be associated with lower adiponectin levels (12), consistent with biochemical evidence that the I164T-containing adiponectin does not form trimers or high-molecular-weight multimers and impairs secretion from the adipocyte (17). However, we did not observe I164T or any other of seven known missense variants among the CLHNS samples.
The observed data did not support a biological role for R221S in lowering adiponectin level. Despite explaining ∼13–17% of variation in adiponectin levels in the CLHNS, we observed no evidence of association of R221S with metabolic traits or with waist circumference, which is a well-known predictor of adiponectin levels. This result could reflect limited power. R221S also did not show any deficiency in formation of adiponectin trimers, hexamers or high-molecular-weight multimers in vitro (17), and bioinformatic annotations that suggested R221S would not substantially alter the protein's conformation. Among the previous reports of R221S, adiponectin levels in carriers were significantly lower (28), indistinguishable from non-carriers (21), or not reported due to low MAF (26,27). We hypothesized that the R221S variant is located on the surface of the protein at a position that may alter the epitope recognized by the adiponectin antibody. Two previous reports support this scenario. The first describes an African-American-specific K29M amino acid substitution in the soluble circulating intracellular adhesion molecule-1 (sICAM-1) protein that resulted in the errant measurement of sICAM-1 by an ELISA using monoclonal antibodies to the region (29). The second describes a GWA study of European-ancestry samples in which a V32M substitution in the natriuretic peptide precursor A gene was strongly associated with mature plasma atrial natriuretic peptide only if the phenotype was measured by an immunoassay targeting the N-terminal epitope (30). Altered trait detection could be responsible for other associations of non-synonymous ADIPOQ variants with adiponectin or for any other genetic associations with protein traits assayed by monoclonal antibodies binding the region containing the mutation. Of further concern to the study of complex traits on a genome-wide level is that a study's power to detect associations at other loci could be affected by samples with noisy phenotypes, especially if they lie in the extreme tails of the distribution.
The novel singleton missense variant (G48D) we identified in ADIPOQ is located in the conserved glycine repeat motif region. Other nearby glycine-to-aspartic acid substitutions in adiponectin (17) and the homologous mannose-binding protein (31) showed deficiency in the formation of high-molecular-weight multimers, suggesting that G48D may also disrupt multimer formation and reduce the stability of its collagen helix domain. However, as the single CLHNS G48D carrier had only slightly below average adiponectin level, the variant may have no effect.
We hypothesize that two or more variants of considerably smaller effect explain the remaining GWA signal at ADIPOQ after adjusting for R221S. The true causal variants in the remaining signal could be the same as those underlying ADIPOQ signals from GWA studies of European samples (13–16). The previously described candidate SNP rs266729 previously showed evidence of association in the CLHNS (19), and has been predicted to alter an SP1 transcription factor-binding site (32). It is also part of a three-SNP haplotype shown to have an in vitro effect on ADIPOQ promoter activity and on DNA-binding activity of nuclear proteins (18). Other putative regulatory regions near ADIPOQ (33–35) may also be promising targets for additional variants. Ultimately, both more comprehensive sequencing of the ADIPOQ gene region and improved imputation of SNPs from the 1000 Genomes Project may be needed to systematically identify additional candidate causal variants, especially of low frequency.
In summary, we characterized a GWA signal for lower plasma adiponectin level in a Filipino cohort and showed that a population-specific uncommon missense variant in the adiponectin-coding gene resulted in artificially low adiponectin measurements in a subset of samples and thus was largely responsible for the observed association signal detected with common variants. Our work highlights how genetic associations with phenotypes measured by antibody-based quantification methods can be affected by uncommon coding SNPs residing in the antibody target region. We also demonstrated the value of targeted sequencing for discovering a variant with potential functional relevance not yet present in public SNP databases. Although these findings as a whole do not elucidate the biology underlying the genetics of adiponectin level, they illustrate an unusual combination of association phenomena that may inform and caution other researchers performing large-scale genetic association analyses.
MATERIALS AND METHODS
Subjects
The CLHNS participants available for this study included 1798 mothers and 1779 male and female young adult offspring from a 1983–1984 Filipino birth cohort (36). Trained field staff conducted in-home interviews and collected quantitative anthropometric measurements, blood samples and comprehensive environmental data (available on-line at http://www.cpc.unc.edu/projects/cebu/). Outcome and covariate measures were taken from the 2005 survey. Informed consent was obtained from all CLHNS subjects, and the study protocol was approved by the University of North Carolina Institutional Review Board for the Protection of Human Subjects. Basic descriptive characteristics of the mothers and offspring are summarized in Supplementary Material, Table S3.
Genotyping and imputation
The 1779 CLHNS offspring were genotyped for 196 725 SNPs on the MetaboChip (Illumina, San Diego, CA, USA), a custom array of SNPs designed to replicate and fine-map loci associated with metabolic traits. Genotyping was performed by the Mammalian Genotyping Core at the University of North Carolina at Chapel Hill (UNC-CH) using the protocol recommended by the manufacturer. Individual sample success rates exceeded 98.6%. Due to poor genotype clustering, 8610 SNPs were removed from analysis. SNP quality control filtering was then performed using PLINK v1.07 (37). We removed 1652 SNPs for success rates ≤97%, 126 SNPs for deviation from Hardy–Weinberg equilibrium (P < 10−6), 228 SNPs for Mendelian inheritance errors (combined ≥3 discrepancies among 79 duplicate pairs and 4 HapMap CEPH trios genotyped on the MetaboChip), 21 SNPs for ≥3 genotype discrepancies with available HapMap genotypes for the 4 CEPH trios and 45 397 SNPs that were monomorphic. Five SNPs did not pass more than one of these filters, so the final set of high-quality MetaboChip data included 140 696 SNPs.
Using MACH version 1.0 (38), additional genotypes of SNPs in the 187.2–188.2 Mb region of chromosome 3 from the November 2010 release (23 November 2010) of the 1000 Genomes Project were imputed in the mothers based on reference haplotypes from CEU, CHB and JPT samples. MACH was also used to generate best-guess phased haplotypes (those with highest posterior probabilities) of 351 directly genotyped SNPs comprising the ETV5-KNG1-ADIPOQ gene region in the mothers (19) and of 1148 SNPs in the offspring. To infer high-quality haplotypes, we specified 200 rounds of Markov sampling considering 500 haplotype states when updating each individual.
Additional SNPs were genotyped using TaqMan allelic discrimination (Applied Biosystems, Foster City, CA, USA), including ADIPOQ missense SNPs G48D (ss469105329), I164T (ss469105330) and R221S (ss469105331), and MetaboChip ETV5 SNP rs117016164 (chr3:187 239 040, hg18). Primer sequences are provided in Supplementary Material, Table S4. The two KNG1-ADIPOQ SNPs rs119243940 and rs864265 were previously genotyped (19). Success rates were >95% and all SNP genotypes were consistent with Hardy–Weinberg equilibrium (P> 0.05). The non-missing TaqMan genotypes of rs117016164 had 99.8% concordance with the original MetaboChip genotypes. Bioinformatic annotations were assigned to missense variants in the adiponectin protein (Uniprot ID: Q15848) using PolyPhen (version 2) (39) and SIFT (40), each using default parameters.
Sequencing
We sequenced the two translated exons of ADIPOQ in 47 offspring samples by PCR amplification and Sanger sequencing. We chose all samples with adiponectin residuals >2.5 standard deviations above or below the population mean. Primer sequences were previously published (21). Sequencing was performed by the UNC-CH Genome Analysis Facility on a 3730xl DNA Analyzer (Applied Biosystems).
Statistical analyses
Adiponectin levels were natural log-transformed to approximate normality and SNPs were tested for association using multivariable linear regression models in PLINK. We assumed an additive mode of inheritance reporting β coefficients representing the estimated change in mean transformed trait value due to each additional copy of the minor allele. In the offspring, 1 member of each of 5 twin pairs were removed and 79 additional samples were excluded because they lacked non-pregnant measures of BMI in 2005. The final sample sets consisted of 1695 non-sibling offspring and 1764 mothers with complete sets of outcomes and covariates. Sex and BMI covariates were significantly associated (P < 0.05) with plasma adiponectin level in the offspring (Supplementary Material, Table S5). In the mothers, age, median-centered age2, household assets, natural log-transformed household income and menopausal status during the 2005 survey were included as covariates, as previously described (19). For combined analysis, age, mean-centered age2, household assets, natural log-transformed household income and BMI during the 2005 survey were used as covariates. Combined analysis of selected SNPs in the offspring and mothers together was performed in SAS version 9.3 (SAS, Inc., Cary, NC, USA) using a general linear mixed model that accounted for the relatedness between mother–child pairs. Conditional analyses of directly genotyped SNPs in the offspring and mothers were performed in SAS using linear regression. Conditional analyses of imputed SNPs in the mothers adjusting for R221S were performed using MACH2QTL (38). Haplotype association analyses in the offspring and mothers were performed in SAS. All conditional and haplotype analyses used the corresponding outcome and covariates from the main association analyses.
To capture population substructure in the offspring, we constructed 10 principal components (PCs) using the software EIGENSOFT (41,42). We first identified a set of 40 239 independent MetaboChip SNPs (estimated r2 <0.001 between all pairs of SNPs within 1 Mb windows) with observed MAF >0.05. PCs were then constructed in 1670 CHLNS offspring who had pair-wise identity-by-descent (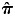 ) <0.1 with all other samples, as estimated using PLINK. The remaining 104 offspring who had
) <0.1 with all other samples, as estimated using PLINK. The remaining 104 offspring who had 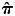 ≥ 0.1 with any other sample(s) were then projected onto these PCs. Supplementary Material, Figure S1 shows the first two PCs in all 1774 offspring samples. We also performed a complementary analysis of global ancestry in these samples (Supplementary Material, Fig. S2) by re-generating PCs using a subset of 24 033 SNPs shared with 165 CEU, 84 CHB, 86 JPT and 167 YRI HapMap Phase III samples (genotypes available at ftp://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/hapmap3/plink_format/draft_2/). The construction of PCs in the CLHNS mothers has been described previously (43). No PCs were associated with adiponectin level in either the offspring (Supplementary Material, Table S5) or mothers (19).
≥ 0.1 with any other sample(s) were then projected onto these PCs. Supplementary Material, Figure S1 shows the first two PCs in all 1774 offspring samples. We also performed a complementary analysis of global ancestry in these samples (Supplementary Material, Fig. S2) by re-generating PCs using a subset of 24 033 SNPs shared with 165 CEU, 84 CHB, 86 JPT and 167 YRI HapMap Phase III samples (genotypes available at ftp://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/hapmap3/plink_format/draft_2/). The construction of PCs in the CLHNS mothers has been described previously (43). No PCs were associated with adiponectin level in either the offspring (Supplementary Material, Table S5) or mothers (19).
Quantification of adiponectin level
Adiponectin level in plasma was originally measured in the offspring and mothers with a commercially available ELISA kit (R&D Systems, Minneapolis, MN, USA, #DY1065) consisting of two monoclonal antibodies that recognize epitopes within amino acids 104–244. Selected offspring samples (n = 40) re-measured by western blot included 20 R221S carriers and 20 sex- and BMI-matched non-carriers. The distributions of adiponectin and BMI were comparable between these two subsets and the respective groups from which they were drawn (data not shown). Plasma proteins were resolved by polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Invitrogen, Carlsbad, CA, USA). Duplicate membranes were separately probed with rabbit polyclonal antisera generated by peptide antigens DQETTTQGPGV (44) and DGRDGTPGEKGEKGD (Sigma, St Louis, MO, USA), corresponding to amino acids 17–27 and 56–70, respectively. Alexa Fluor 680-conjugated goat anti-rabbit IgG was then used as a secondary antibody (Invitrogen). Proteins were detected using an Odyssey imaging system (LI-COR, Lincoln, NE, USA) and the adiponectin bands (30 kDa) were quantified by densitometry using the LI-COR software. The adiponectin standards used for each assay did not allow direct comparison of the original and alternative measurements. Thus, all densitometry readings using a given antibody were natural log-transformed and zero-centered on the mean value of R221S non-carriers from that group.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by National Institutes of Health grants (DK078150, TW05596, HL085144, K01DK075573 and P30ES10126), pilot funds (RR20649, ES10126 and DK56350) and a training grant (T32 GM007092).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the research and data collection teams from the Office of Population Studies Foundation as well as the study participants who generously provided their time. We also thank Matthew Nelson, Margaret Ehm, Liling Warren and Dawn Waterworth for sharing pre-publication sequencing data, as well as Amanda Floyd Beaty and Michael Andre of the UNC Mammalian Genotyping Core for their genotyping assistance.
REFERENCES
- 1.Diez J.J., Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol. 2003;148:293–300. doi: 10.1530/eje.0.1480293. [DOI] [PubMed] [Google Scholar]
- 2.Comuzzie A.G., Funahashi T., Sonnenberg G., Martin L.J., Jacob H.J., Black A.E., Maas D., Takahashi M., Kihara S., Tanaka S., et al. The genetic basis of plasma variation in adiponectin, a global endophenotype for obesity and the metabolic syndrome. J. Clin. Endocrinol. Metab. 2001;86:4321–4325. doi: 10.1210/jcem.86.9.7878. [DOI] [PubMed] [Google Scholar]
- 3.Chuang L.M., Chiu Y.F., Sheu W.H., Hung Y.J., Ho L.T., Grove J., Rodriguez B., Quertermous T., Chen Y.D., Hsiung C.A., et al. Biethnic comparisons of autosomal genomic scan for loci linked to plasma adiponectin in populations of Chinese and Japanese origin. J. Clin. Endocrinol. Metab. 2004;89:5772–5778. doi: 10.1210/jc.2004-0640. [DOI] [PubMed] [Google Scholar]
- 4.Lindsay R.S., Funahashi T., Krakoff J., Matsuzawa Y., Tanaka S., Kobes S., Bennett P.H., Tataranni P.A., Knowler W.C., Hanson R.L. Genome-wide linkage analysis of serum adiponectin in the Pima Indian population. Diabetes. 2003;52:2419–2425. doi: 10.2337/diabetes.52.9.2419. [DOI] [PubMed] [Google Scholar]
- 5.Renaldi O., Pramono B., Sinorita H., Purnomo L.B., Asdie R.H., Asdie A.H. Hypoadiponectinemia: a risk factor for metabolic syndrome. Acta Med. Indones. 2009;41:20–24. [PubMed] [Google Scholar]
- 6.Gable D.R., Hurel S.J., Humphries S.E. Adiponectin and its gene variants as risk factors for insulin resistance, the metabolic syndrome and cardiovascular disease. Atherosclerosis. 2006;188:231–244. doi: 10.1016/j.atherosclerosis.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Weyer C., Funahashi T., Tanaka S., Hotta K., Matsuzawa Y., Pratley R.E., Tataranni P.A. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001;86:1930–1935. doi: 10.1210/jcem.86.5.7463. [DOI] [PubMed] [Google Scholar]
- 8.Hotta K., Funahashi T., Arita Y., Takahashi M., Matsuda M., Okamoto Y., Iwahashi H., Kuriyama H., Ouchi N., Maeda K., et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi M., Arita Y., Yamagata K., Matsukawa Y., Okutomi K., Horie M., Shimomura I., Hotta K., Kuriyama H., Kihara S., et al. Genomic structure and mutations in adipose-specific gene, adiponectin. Int. J. Obes. Relat. Metab. Disord. 2000;24:861–868. doi: 10.1038/sj.ijo.0801244. [DOI] [PubMed] [Google Scholar]
- 10.Vasseur F., Helbecque N., Dina C., Lobbens S., Delannoy V., Gaget S., Boutin P., Vaxillaire M., Lepretre F., Dupont S., et al. Single-nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of the APM1 gene modulate adipocyte-secreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians. Hum. Mol. Genet. 2002;11:2607–2614. doi: 10.1093/hmg/11.21.2607. [DOI] [PubMed] [Google Scholar]
- 11.Sutton B.S., Weinert S., Langefeld C.D., Williams A.H., Campbell J.K., Saad M.F., Haffner S.M., Norris J.M., Bowden D.W. Genetic analysis of adiponectin and obesity in Hispanic families: the IRAS Family Study. Hum. Genet. 2005;117:107–118. doi: 10.1007/s00439-005-1260-9. [DOI] [PubMed] [Google Scholar]
- 12.Tanimura D., Shibata R., Izawa H., Hirashiki A., Asano H., Murase Y., Miyata S., Nakatochi M., Ouchi N., Ichihara S., et al. Relation of a common variant of the adiponectin gene to serum adiponectin concentration and metabolic traits in an aged Japanese population. Eur. J. Hum. Genet. 2010;19:262–269. doi: 10.1038/ejhg.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heid I.M., Henneman P., Hicks A., Coassin S., Winkler T., Aulchenko Y.S., Fuchsberger C., Song K., Hivert M.F., Waterworth D.M., et al. Clear detection of ADIPOQ locus as the major gene for plasma adiponectin: results of genome-wide association analyses including 4659 European individuals. Atherosclerosis. 2010;208:412–420. doi: 10.1016/j.atherosclerosis.2009.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jee S.H., Sull J.W., Lee J.E., Shin C., Park J., Kimm H., Cho E.Y., Shin E.S., Yun J.E., Park J.W., et al. Adiponectin concentrations: a genome-wide association study. Am. J. Hum. Genet. 2010;87:545–552. doi: 10.1016/j.ajhg.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards J.B., Waterworth D., O'Rahilly S., Hivert M.F., Loos R.J., Perry J.R., Tanaka T., Timpson N.J., Semple R.K., Soranzo N., et al. A genome-wide association study reveals variants in ARL15 that influence adiponectin levels. PLoS Genet. 2009;5:e1000768. doi: 10.1371/journal.pgen.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ling H., Waterworth D.M., Stirnadel H.A., Pollin T.I., Barter P.J., Kesaniemi Y.A., Mahley R.W., McPherson R., Waeber G., Bersot T.P., et al. Genome-wide linkage and association analyses to identify genes influencing adiponectin levels: the GEMS Study. Obesity. 2009;17:737–744. doi: 10.1038/oby.2008.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waki H., Yamauchi T., Kamon J., Ito Y., Uchida S., Kita S., Hara K., Hada Y., Vasseur F., Froguel P., et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J. Biol. Chem. 2003;278:40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- 18.Laumen H., Saningong A.D., Heid I.M., Hess J., Herder C., Claussnitzer M., Baumert J., Lamina C., Rathmann W., Sedlmeier E.M., et al. Functional characterization of promoter variants of the adiponectin gene complemented by epidemiological data. Diabetes. 2009;58:984–991. doi: 10.2337/db07-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y., Li Y., Lange E.M., Croteau-Chonka D.C., Kuzawa C.W., McDade T.W., Qin L., Curocichin G., Borja J.B., Lange L.A., et al. Genome-wide association study for adiponectin levels in Filipino women identifies CDH13 and a novel uncommon haplotype at KNG1-ADIPOQ. Hum. Mol. Genet. 2010;19:4955–4964. doi: 10.1093/hmg/ddq423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorleifsson G., Walters G.B., Gudbjartsson D.F., Steinthorsdottir V., Sulem P., Helgadottir A., Styrkarsdottir U., Gretarsdottir S., Thorlacius S., Jonsdottir I., et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 21.Kondo H., Shimomura I., Matsukawa Y., Kumada M., Takahashi M., Matsuda M., Ouchi N., Kihara S., Kawamoto T., Sumitsuji S., et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes. 2002;51:2325–2328. doi: 10.2337/diabetes.51.7.2325. [DOI] [PubMed] [Google Scholar]
- 22.Bowden D.W., An S.S., Palmer N.D., Brown W.M., Norris J.M., Haffner S.M., Hawkins G.A., Guo X., Rotter J.I., Chen Y.D., et al. Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum. Mol. Genet. 2010;19:4112–4120. doi: 10.1093/hmg/ddq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hara K., Boutin P., Mori Y., Tobe K., Dina C., Yasuda K., Yamauchi T., Otabe S., Okada T., Eto K., et al. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes. 2002;51:536–540. doi: 10.2337/diabetes.51.2.536. [DOI] [PubMed] [Google Scholar]
- 24.Schaffler A., Barth N., Palitzsch K.D., Drobnik W., Scholmerich J., Schmitz G. Mutation analysis of the human adipocyte-specific apM-1 gene. Eur. J. Clin. Invest. 2000;30:879–887. doi: 10.1046/j.1365-2362.2000.00722.x. [DOI] [PubMed] [Google Scholar]
- 25.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang Y., Lee J.H., Kim O.Y., Koh S.J., Chae J.S., Woo J.H., Cho H., Lee J.E., Ordovas J.M. The SNP276G>T polymorphism in the adiponectin (ACDC) gene is more strongly associated with insulin resistance and cardiovascular disease risk than SNP45T>G in nonobese/nondiabetic Korean men independent of abdominal adiposity and circulating plasma adiponectin. Metabolism. 2006;55:59–66. doi: 10.1016/j.metabol.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Jang Y., Chae J.S., Koh S.J., Hyun Y.J., Kim J.Y., Jeong Y.J., Park S., Ahn C.M., Lee J.H. The influence of the adiponectin gene on adiponectin concentrations and parameters of metabolic syndrome in non-diabetic Korean women. Clin. Chim. Acta. 2008;391:85–90. doi: 10.1016/j.cca.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 28.Vendramini M.F., Pereira A.C., Ferreira S.R., Kasamatsu T.S., Moises R.S. Association of genetic variants in the adiponectin encoding gene (ADIPOQ) with type 2 diabetes in Japanese Brazilians. J. Diabetes Complication. 2010;24:115–120. doi: 10.1016/j.jdiacomp.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Register T.C., Burdon K.P., Lenchik L., Bowden D.W., Hawkins G.A., Nicklas B.J., Lohman K., Hsu F.C., Langefeld C.D., Carr J.J. Variability of serum soluble intercellular adhesion molecule-1 measurements attributable to a common polymorphism. Clin. Chem. 2004;50:2185–2187. doi: 10.1373/clinchem.2004.036806. [DOI] [PubMed] [Google Scholar]
- 30.Newton-Cheh C., Larson M.G., Vasan R.S., Levy D., Bloch K.D., Surti A., Guiducci C., Kathiresan S., Benjamin E.J., Struck J., et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 2009;41:348–353. doi: 10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallis R., Cheng J.Y. Molecular defects in variant forms of mannose-binding protein associated with immunodeficiency. J. Immunol. 1999;163:4953–4959. [PubMed] [Google Scholar]
- 32.Zhang D., Ma J., Brismar K., Efendic S., Gu H.F. A single nucleotide polymorphism alters the sequence of SP1 binding site in the adiponectin promoter region and is associated with diabetic nephropathy among type 1 diabetic patients in the Genetics of Kidneys in Diabetes Study. J. Diabetes Complications. 2009;23:265–272. doi: 10.1016/j.jdiacomp.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Kita A., Yamasaki H., Kuwahara H., Moriuchi A., Fukushima K., Kobayashi M., Fukushima T., Takahashi R., Abiru N., Uotani S., et al. Identification of the promoter region required for human adiponectin gene transcription: association with CCAAT/enhancer binding protein-beta and tumor necrosis factor-alpha. Biochem. Biophys. Res. Commun. 2005;331:484–490. doi: 10.1016/j.bbrc.2005.03.205. [DOI] [PubMed] [Google Scholar]
- 34.Qiao L., Maclean P.S., Schaack J., Orlicky D.J., Darimont C., Pagliassotti M., Friedman J.E., Shao J. C/EBPalpha regulates human adiponectin gene transcription through an intronic enhancer. Diabetes. 2005;54:1744–1754. doi: 10.2337/diabetes.54.6.1744. [DOI] [PubMed] [Google Scholar]
- 35.Segawa K., Matsuda M., Fukuhara A., Morita K., Okuno Y., Komuro R., Shimomura I. Identification of a novel distal enhancer in human adiponectin gene. J. Endocrinol. 2009;200:107–116. doi: 10.1677/JOE-08-0376. [DOI] [PubMed] [Google Scholar]
- 36.Adair L.S., Popkin B.M., Akin J.S., Guilkey D.K., Gultiano S., Borja J., Perez L., Kuzawa C.W., McDade T., Hindin M.J. Cohort profile: the Cebu Longitudinal Health and Nutrition Survey. Int. J. Epidemiol. 2011;40:619–625. doi: 10.1093/ije/dyq085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 43.Lange L.A., Croteau-Chonka D.C., Marvelle A.F., Qin L., Gaulton K.J., Kuzawa C.W., McDade T.W., Wang Y., Li Y., Levy S., et al. Genome-wide association study of homocysteine levels in Filipinos provides evidence for CPS1 in women and a stronger MTHFR effect in young adults. Hum. Mol. Genet. 2010;19:2050–2058. doi: 10.1093/hmg/ddq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Combs T.P., Wagner J.A., Berger J., Doebber T., Wang W.J., Zhang B.B., Tanen M., Berg A.H., O'Rahilly S., Savage D.B., et al. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology. 2002;143:998–1007. doi: 10.1210/endo.143.3.8662. [DOI] [PubMed] [Google Scholar]
- 45.Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R., Willer C.J. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.