

Abstract
Spinocerebellar ataxia type 7 (SCA7) is a neurodegenerative disease caused by expansion of a CAG repeat encoding a polyglutamine tract in ATXN7, a component of the SAGA histone acetyltransferase (HAT) complex. Previous studies provided conflicting evidence regarding the effects of polyQ–ATXN7 on the activity of Gcn5, the HAT catalytic subunit of SAGA. Here, we report that reducing Gcn5 expression accelerates both cerebellar and retinal degeneration in a mouse model of SCA7. Deletion of Gcn5 in Purkinje cells in mice expressing wild-type (wt) Atxn7, however, causes only mild ataxia and does not lead to the early lethality observed in SCA7 mice. Reduced Gcn5 expression strongly enhances retinopathy in SCA7 mice, but does not affect the known transcriptional targets of Atxn7, as expression of these genes is not further altered by Gcn5 depletion. These findings demonstrate that loss of Gcn5 functions can contribute to the time of onset and severity of SCA7 phenotypes, and suggest that non-transcriptional functions of SAGA may play a role in neurodegeneration in this disease.
INTRODUCTION
Spinocerebellar ataxia type 7 (SCA7) is one of 29 inherited SCAs, and features not only progressive spinocerebellar degeneration, but also a unique manifestation of retinal degeneration. Neurodegeneration in SCA7 patients leads to abnormal gait, uncoordinated movements, dysarthia, dysphagia and visual loss, usually followed by early lethality. SCA7 is dominantly inherited and is caused by expansion of a CAG repeat in the ATAXIN7 gene that is translated into a poly-glutamine (polyQ) tract in ATAXIN7 (ATXN7). SCA7 pathogenic expansion mutations range from 37 to more than 200 repeats (1–4). The length of the expanded CAG repeats correlates inversely with the onset of symptoms and directly with disease progression (2,5).
Genetic and biochemical studies have provided conflicting views as to the underlying causes of neurotoxicity associated with polyQ expansions in ATXN7 or other affected genes in spinocerebellar ataxias. Insertion of polyQ tracts into heterologous proteins or expression of a polyQ peptide alone in mice leads to neurological phenotypes and aggregation of the polyQ protein or peptide (6,7). These studies suggest that the expanded polyQ tract itself is toxic. However, more recent studies indicate that the native functions of the disease proteins are required for the development of neurological symptoms. For example, polyQ expansions in the androgen receptor (AR) are associated with spinobulbar muscular atrophy (SBMA) (8). Both ligand binding and nuclear translocation of polyQ–AR are required for manifestation of SBMA, and disrupting AR interactions with transcriptional coregulators suppresses SBMA neurodegeneration in a Drosophila model (9–11). These data indicate that the normal functions of AR are required for disease development, as further supported by the observation that over-expression of wt AR causes mild neurodegeneration, even in the absence of a polyQ expansion. Similarly, wt functions of the Ataxin-1 protein, particularly interactions with its native partners, are required for the development of SCA1 phenotypes in mice, and a SCA1-like phenotype can be induced by over-expression of Ataxin-1-lacking polyQ expansion (12–14).
SCA7 neuropathology has been proposed to be linked to the functions of ATXN7-interacting proteins within the multi-component SAGA complex (15) (also known as STAGA or TFTC in mammalian cells). SAGA plays an evolutionarily conserved role in transcriptional coactivation, mediated through its two histone-modifying activities—histone acetyltransferase (HAT) via Gcn5 (16), and deubiquitinase (DUB) via Usp22 (17,18). ATXN7 in itself has no enzymatic activity, but it resides within the DUB submodule of SAGA and may serve to anchor this module to the main complex (18,19). Loss of Gcn5 function displaces the DUB module from SAGA, compromising its enzymatic functions (20). However, the role of Atxn7 or the DUB module in SAGA during mammalian development has not yet been defined.
In a polyQ–Atxn7 knock-in mouse model, transcriptional downregulation was demonstrated to be an early event leading to photoreceptor dysfunction, retinal degeneration and visual impairment (21). These data are consistent with diminished transcriptional coactivator functions of SAGA in the presence of polyQ–Atxn7. Multiple studies indicate that polyQ–Atxn7 is incorporated into SAGA (22–24), and two of these studies indicate that it adversely affects the HAT activity of the complex (22,23). These studies point to a loss of function in Gcn5 and SAGA triggered by polyQ–Atxn7 in SCA7. In direct contrast to these results, Helmlinger et al. (25) reported that SAGA complexes isolated from SCA7 mouse models exhibited wt levels of HAT activity, and that polyQ–Atxn7 led to increased recruitment of SAGA to target promoters, resulting in hyperacetylation of histone H3. In this case, the wt functions of Atxn7 and SAGA would appear to be necessary for SCA7 disease, resembling the recent results reported for SCA1 and SBMA described above.
To gain further insights into the importance of Gcn5 and SAGA in SCA7, we examined the effects of Gcn5 mutations in mice bearing polyQ–Atxn7. Our previous work demonstrated that Gcn5 functions are essential for mouse embryo survival, as well as for multiple events during embryonic development, including neural tube closure and anterior–posterior patterning of the ribs (26–29). Here, we report that partial loss of Gcn5 functions accelerates neuronal dysfunction and pathology in an SCA7 mouse model. Interestingly, however, complete deletion of Gcn5 in Purkinje cells, the neuronal cells most affected in SCA7, is not sufficient to induce severe ataxia in the presence of wt Atxn7. Further, we show that in the SCA7 retina, this acceleration in pathogenesis is not accompanied by changes in the expression of known Atxn7 target genes, and so may involve non-transcriptional functions of SAGA. Together, these data demonstrate that Gcn5 loss-of-function can contribute to SCA7, further indicating that polyQ–Atxn7-interacting proteins within SAGA are important for development of this disease.
RESULTS
CAG repeat length inversely correlates with lifespan of polyQ–Atxn7 knock-in mice
To assess the role of Gcn5 in the neurodegeneration observed in a mouse model of SCA7, we introduced different mutant alleles of Gcn5 into polyQ–Atxn7 knock-in mice that express polyQ–Atxn7 from the endogenous Atxn7 locus. We predicted that if reduced Gcn5 function contributes to SCA7 pathogenesis, reducing Gcn5 expression would enhance disease progression. Although the previously reported polyQ–Atxn7 knock-in line, Atxn7266Q/5Q, nicely recapitulates the infantile form of SCA7 (21), disease progression in these mice occurs rapidly, making it difficult to observe an enhancement of the already severe SCA7 phenotypes. Therefore, to better characterize the role of Gcn5 loss-of-function, we utilized SCA7 mouse lines that have a more moderate disease progression.
The length of the CAG repeat in the Atxn7 gene correlates inversely with the timing of disease-onset, and directly with severity, so we characterized the disease progression and the age of lethality in Atxn7 mutant mice bearing shorter polyQ tracts (2,4,5,30). Specifically, we compared the life span of mice bearing no expansion in Atxn7 (Atxn75Q/5Q) to those bearing 100 CAG repeats (Atxn7100Q/5Q) or 230 CAG repeats (Atxn7230Q/5Q) in one allele of Atxn7. Both Atxn7100Q/5Q and Atxn7230Q/5Q mice develop previously described symptoms of SCA7 mouse models including weight loss, kyphosis, ataxia, ptosis, tremors, gradual loss of mobility [data not shown; (21)]. We recorded the age at which these animals died or became moribund up to 24 months. Wt Atxn75Q/5Q mice (Fig. 1A) had a lifespan of more than 24 months, whereas Atxn7100Q/5Q mice had a significantly shorter lifespan (P < 0.001), averaging 18.7 months. Atxn7230Q/5Q mice died at a very early age, with a lifespan averaging only 3.5 months (Fig. 1A). The shorter lifespan of the animals bearing longer repeats confirms the correlation between repeat length and lethality in these mouse models of SCA7. Disease progression was accelerated by the presence of a second polyQ-expanded allele of Atxn7, as Atxn7100Q/100Q mice died at an earlier age than did the Atxn7100Q/5Q mice (P < 0.001), with a lifespan averaging 12.1 months (Fig. 1A and B).
Figure 1.

Gcn5 mutation reduces lifespan of 100Q-Atxn7 mice. (A) Survival rate of wt (thin black line), Atxn7230Q/5Q (blue line), Atxn7100Q/5Q (gray line) and Atxn7100Q/5Q; Gcn5Δ/+ (green line), until 24 months of age (wt, n = 10; Atxn7230Q/5Q, n = 10; Atxn7100Q/5Q, n = 34; Atxn7100Q/5Q; Gcn5Δ/+, n = 12; *P < 0.05 between Atxn7100Q/5Q and Atxn7100Q/5Q; Gcn5Δ/+, Kaplan–Meier analysis). Survival rate of Atxn7100Q/5Q; Gcn5fn/+ (dark red line) until 24 months of age is also shown (n = 31; *P < 0.05 between Atxn7100Q/5Q and Atxn7100Q/5Q; Gcn5fn/+, Kaplan–Meier analysis). (B) Survival rate of wt (thin black line), Atxn7100Q/100Q (thick black line) and Atxn7100Q/100Q; Gcn5Δ/+ (red line), until 24 months of age (wt, n = 10; Atxn7100Q/100Q, n = 18; Atxn7100Q/100Q; Gcn5Δ/+, n = 12; Kaplan–Meier analysis).
Reducing Gcn5 levels enhances the lethality of SCA7 mouse models
Gcn5 null mice are embryonic lethal (29). Therefore, to assess the role of partial loss-of-function of Gcn5 in SCA7 pathogenesis, we introduced a hypomorphic allele of Gcn5, which reduces the level of Gcn5 expression and activity, into Atxn7100Q/5Q mice. The hypomorphic Gcn5fn allele contains an intronic neomycin cassette insertion that lowers Gcn5 expression to ∼20% of wt levels in the homozygous state (27). Mice homozygous for this allele can survive to adulthood, albeit at 8% of expected frequency (28). We created Atxn7100Q/5Q; Gcn5fn/fn mice and found that the combination of these alleles resulted in lethality at postnatal day 1 (P1) (data not shown). This early lethality indicated that concomitant diminishment of both Gcn5 and Atxn7 functions is deleterious, but it also prohibited us from determining the effects of altered Gcn5 functions in mature neurons.
We therefore introduced single copies of either the Gcn5 hypomorphic allele (Gcn5fn/+), or a deletion allele (Gcn5Δ/+), resulting in a 50% reduction of Gcn5 expression, into Atxn7100Q/5Q or Atxn7100Q/100Q mice (28). In contrast to the P1 lethality of Atxn7100Q/5Q; Gcn5fn/fn animals, Atxn7100Q/5Q; Gcn5Δ/+ and Atxn7100Q/5Q; Gcn5fn/+ mice were born alive at expected Mendelian frequencies and had indistinguishable cage behaviors from those of wt littermates until the time of weaning, at 3 weeks of age.
The survival of Gcn5 mutant heterozygotes was no different from that of wt mice (Fig. 1 and data not shown), but the life span of Atxn7100Q/5Q; Gcn5Δ/+ and Atxn7100Q/5Q; Gcn5fn/+ mice were significantly reduced relative to Atxn7100Q/5Q mice (P < 0.01, Fig. 1A). Partial loss of Gcn5 function also reduced the survival of Atxn7100Q/100Q mice (average life span of 10.7 ± 7.5 months in Atxn7100Q/100Q; Gcn5Δ/+ mice when compared with 12.1 ± 2.9 months in Atxn7100Q/100Q mice). Interestingly, while 22% of the Atxn7100Q/100Q mice lived over 1.5 years, none of the Atxn7100Q/100Q; Gcn5Δ/+ mice survived more than 1 year (Fig. 1B). These data strongly suggest that decreased Gcn5 levels accelerate the early adult lethality of mice bearing Atxn7100Q alleles, supporting the hypothesis that Gcn5 loss-of-function may contribute to SCA7 pathogenesis.
Gcn5 partial loss-of-function hastens the onset of progressive ataxia in SCA7 mouse models
Since ataxia (uncoordinated locomotion) is one of the major neurological symptoms of SCA7 patients and mouse models (1,2,5,21,31), we next asked if reducing Gcn5 function contributes to the progression of ataxia in our SCA7 mouse model. We first examined mouse hind limb coordination using a tail suspension test (32). Wt and Gcn5Δ/+ mice were able to coordinate stretching of their hind limbs when suspended by their tails (to prepare for landing if dropped). By 7–8 months of age, 100% (12 out of 12) of Atxn7100Q/100Q mice also stretched their hind limbs normally upon tail suspension, while only 33.3% (two out of six) of Atxn7100Q/100Q; Gcn5Δ/+ mice were able to perform normally. Interestingly, one of the four uncoordinated Atxn7100Q/100Q; Gcn5Δ/+ mice developed a severe clasping phenotype (Fig. 2A), and the gait of this mouse deteriorated over time into a severe stagger, causing the mouse to frequently fall to one side (Supplementary Material, Movie S1) up until its death at 8 months of age. Conversely, 92% (11 out of 12) of Atxn7100Q/100Q mice were able to normally coordinate stretching of their hind limbs, and no clasping phenotype was observed in these 12 Atxn7100Q/100Q mice throughout their life.
Figure 2.

Reducing the level of Gcn5 enhances ataxia phenotypes of Atxn7100Q/100Q mice. (A) Clasping phenotype of Atxn7100Q/100Q; Gcn5Δ/+ but not Atxn7100Q/100Q or wt mice at 9 month of age during tail suspension. (B) Representative traces of the gait of wt, Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice recorded by footprint analysis, with red and blue footprints representing fore- and hind-paws, respectively. Uncoordinated gait of Atxn7100Q/100Q or Atxn7100Q/100Q; Gcn5Δ/+ mice at 9 months of age. Paired distance (PD) measures the distance between the prints of fore- and hind-paw on the same side. Hind-base width (HW) measures the distance between the prints of the two hind-paws. (C) Quantified HW or PD indicates Atxn7100Q/100Q; Gcn5Δ/+ mice have more uncoordinated walking gaits than Atxn7100Q/100Q mice at 9 months of age (wt, n = 3; Atxn7100Q/100Q, n = 4; Atxn7100Q/100Q; Gcn5Δ/+, n = 3; *P < 0.05 between wt and Atxn7100Q/100Q, wt and Atxn7100Q/100Q; Gcn5Δ/+, or Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+, Kruskal–Wallis test). Data are expressed as box plots (boxes, 25–75%; circles, < 10 or >90%; lines, median).
To further evaluate the progression of ataxic gaits, we used a footprint test to quantify changes in the gait of Atxn7100Q/100Q, Atxn7100Q/100Q; Gcn5Δ/+ and wt mice at 4 months and 8–9 months of age. At 4 months, we found no significant differences between the gaits of Atxn7100Q/100Q, Atxn7100Q/100Q; Gcn5Δ/+ and control mice (data not shown). However at 8–9 months of age, both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice walked with a significantly wider hind stance (wt, n = 5, Atxn7100Q/100Q; n = 5, Atxn7100Q/100Q; Gcn5Δ/+; n = 3, P < 0.05 for hindbase width) and dispersed fore- and hind-steps (P < 0.05, for paired distance) relative to wt mice (Fig. 2B–D). Moreover, this uncoordinated walking gait was significantly worse in Atxn7100Q/100Q; Gcn5Δ/+ and Atxn7100Q/5Q; Gcn5Δ/+ mice than in Atxn7100Q/100Q and Atxn7100Q/5Q mice, respectively (P < 0.05; Fig. 2B and C and Supplementary Material, Fig. S2). To assess motor balance, Atxn7100Q/100Q, Atxn7100Q/100Q; Gcn5Δ/+ and control wt mice were subjected to rotating rod, dowel and wire hang tests. Although Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice showed a trend of dysfunction, the differences observed were not statistically significant (Supplementary Material, Fig. S3). These results indicate that Atxn7100Q/5Q and Atxn7100Q/100Q mice progressively develop mild ataxia, and that Gcn5 partial loss-of-function accelerates the onset and progression of this ataxia in these SCA7 mouse models.
Gcn5 partial loss-of-function worsens cerebellar degeneration in SCA7
Since cerebellar degeneration occurs in human SCA7 patients at moderate/late stages of the disease and strongly correlates with uncoordinated movement, we next asked whether more severe cerebellar atrophy occurred in Atxn7100Q/100Q mice haploinsufficient for Gcn5. The cerebellar vermis showed mild cortical atrophy specifically in the molecular layer (ML) of lobules VI, VII and X in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice at 8–9 months of age; however, the atrophy was noticeably more severe in the double mutants (Fig. 3A and Supplementary Material, Fig. S4). Since the ML spans the dendrites or processes of Purkinje cells and Bergmann glia, and both of these cell types are affected in SCA7 patients and mouse models (31,33), we performed a more detailed analysis of these cells in the mutant mice. In particular, we focused on lobule VI of the cerebellar vermis, in which Purkinje and glial cell activities are implicated in motor coordination of the limb (34–36). For further comparison, we next stained Purkinje cells and Bergmann glia with cell-specific markers, Calbinin 1 and Glial fibrillary acidic protein (GFAP), respectively. Immunofluorescent staining revealed that Purkinje cells have weaker Calbindin 1 expression and significantly smaller soma size in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ relative to wt control mice (wt, 263 ± 42 μm2, n = 3; Atxn7100Q/100Q, 228 ± 34 μm2, n = 3; Atxn7100Q/100Q; Gcn5Δ/+, 226 ± 50 μm2, n = 2; P < 0.01; Fig. 3B and C). However, although no statistical difference was found for overall average soma size of the Purkinje cells between Atxn7100Q/100Q; Gcn5Δ/+ and Atxn7100Q/100Q mice, 9% of the Purkinje cells had uncommonly small soma in Atxn7100Q/100Q; Gcn5Δ/+ mice. We next asked whether there is a loss of Purkinje cells in these cerebella. The development of Purkinje cells has no significant defects in Atxn7100Q/100Q or Atxn7100Q/100Q; Gcn5Δ/+ cerebellum, since mice of all genotypes have similar number of Purkinje cells when 5 months old (data not shown). At 9 months, no significant loss of Purkinje cells occurred in Atxn7100Q/100Q (Purkinje cell count of all ten lobules per 6 μm sections in wt, 540 ± 34, n = 5; in Atxn7100Q/100Q, 514 ± 36, n = 4; P = 0.15). However, Atxn7100Q/100Q; Gcn5Δ/+ mice had significantly fewer Purkinje cells (489 ± 23, n = 3) when compared with wt mice (P < 0.05) at 9 months. Among all lobules, Purkinje cell loss was significantly worse in lobule X of Atxn7100Q/100Q; Gcn5Δ/+ mice compared with Atxn7100Q/100Q mice (Atxn7100Q/100Q; Gcn5Δ/+, 22 ± 2, n = 3; Atxn7100Q/100Q, 27 ± 2, n = 4; P < 0.05; Fig. 3D). In contrast, Bergmann glia exhibited similar elevated levels of GFAP in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice compared with control mice (Fig. 3B). Together, these data suggest that partial loss of Gcn5 function accelerates cerebellar and Purkinje cell degeneration, but does not further distort Bergmann glia in SCA7 mice.
Figure 3.

Cerebellar degeneration is more severe in Atxn7100Q/100Q animals in a background of Gcn5Δ/+. (A) Severe atrophy of molecular layer (ML) in lobule VI and X of mid-saggital cerebellar vermis of Atxn7100Q/100Q; Gcn5Δ/+. Representative images showing morphology of mid-sagittal cerebellar vermis of 9-month-old wt, Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice (n ≥ 3 mice of each genotype). Boxes of areas in lobule VI and X of 5- and 9-month-old mice were enlarged for detailed comparison. Thickness of ML was marked. (B) Anti-Calbindin 28K and Glial fibrillary acidic protein (GFAP) antibody immunofluorescent-labeled Purkinje cells and Bergmann glia, respectively, in cerebellar lobule VI of mice of indicated genotype. Areas containing soma of Purkinje cells were enlarged in the insets. (C) Area size frequency of Purkinje cell soma shows Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice have smaller soma (wt, n = 3 mice; Atxn7100Q/100Q, n = 3; Atxn7100Q/100Q; Gcn5Δ/+, n = 2; *P < 0.05 between wt and Atxn7100Q/100Q, or wt and Atxn7100Q/100Q; Gcn5Δ/+, Student's t-test). (D) Purkinje cell number is significantly less in lobule X of Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice (wt, n = 5 mice; Atxn7100Q/100Q, n = 4; Atxn7100Q/100Q; Gcn5Δ/+, n = 3; *P < 0.05 between wt and Atxn7100Q/100Q, or wt and Atxn7100Q/100Q; Gcn5Δ/+, or Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+, Student's t-test). Data are presented as mean ± SEM. PCL, Purkinje cell layer.
Gcn5 loss-of-function in Purkinje cells is not sufficient to cause severe cerebellar ataxia
In the above experiments, Gcn5 expression was decreased uniformly in all cell types. However, in SCA7 patients, the most affected neurons in the cerebellum are Purkinje cells (37–40). Therefore, to determine whether Gcn5 loss in Purkinje cells is sufficient to cause an SCA7 phenotype, we generated Gcn5-conditional knockout mice using a transgenic Cre allele expressed specifically in Purkinje cells, Pcp2-cretg (41), and a ‘floxed’ allele of Gcn5 together with our deletion allele (Gcn5Δ/f). Cre-mediated recombination removes exons 3–18 in Gcn5, resulting in a null allele (42). The resulting Purkinje cell-specific Gcn5-conditional knock-out mice were present at Mendelian ratios at the time of weaning. Using RNA in situ hybridization, only minimal levels of Gcn5 transcripts were detected in Purkinje cells in these Gcn5-conditional mice (Fig. 4A). These mice exhibited normal hind limb extension in tail suspension assays, but by 11 months of age developed a mildly uncoordinated gait with wider paired distance (Fig. 4B and C). The cerebellar vermis in the conditional nulls appeared to be smaller than in wt mice (Fig. 4D). Since Gcn5 functions as a transcriptional coactivator, we also determined whether loss of Gcn5 affects Purkinje cell-specific transcripts. By quantitative realtime RT–PCR, we found that Calbindin 1 expression was mildly decreased in Purkinje cell-specific Gcn5 null cerebella, to 74.7 ± 5% of that in wt animals (n = 3, P < 0.05). These data suggest that loss of Gcn5 in Purkinje cells mildly affects Purkinje cell and cerebellar morphology, as well as cerebellar functions, but is not sufficient to induce a SCA7 phenotype. These data are consistent with the primary role of ATXN7 mutations in SCA7 disease and suggest that some other ATXN7-mediated interactions, beyond those mediated by GCN5, contribute to cerebellar pathology.
Figure 4.

Purkinje cell-specific loss of Gcn5 causes mild ataxia and cerebellar degeneration. (A) In situ hybridization for Gcn5 transcripts shows decreased Gcn5 expression in the Purkinje cell layer in Gcn5Δ/f;Pcp2-creTg mice compared with wt or Gcn5Δ/+ mice at postnatal day 21. (B) Coordinated hind limb stretching during tail suspension of wt, Gcn5Δ/+ and Gcn5Δ/f;Pcp2-creTg mice at 11 months of age. (C) Abnormal gait of Gcn5-conditional null mice at 11 months of age. Walking gait was recorded by footprint analysis with red and blue footprints representing fore- and hind-paws, respectively. Quantified footprints show Gcn5Δ/f;Pcp2-creTg mice have wider paired distance (n = 3 mice each genotype; *P < 0.05 between wt and Gcn5Δ/f;Pcp2-creTg, Kruskal–Wallis test). Data are expressed as box plots (boxes, 25–75%; circles, <10% or >90%; lines, median). (D) Representative H&E staining shows morphology of cerebellar vermis of 9-month-old control and Gcn5Δ/f;Pcp2-creTg mice.
Reducing Gcn5 accelerates progression of retinal degeneration in the SCA7 mouse
Since the retina is another major site of neural degeneration in SCA7 patients (1,2), we next asked whether reducing Gcn5 levels affects the progression of retinal degeneration in the SCA7 mouse model. We examined the morphology of the retinas of control and Atxn7100Q/100Q mice with or without Gcn5 deletion allele at postnatal day 14 (P14) at 1, 2, 4 or 8 months of age. At P14, when all cell types are formed in the retina, Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ had retinal morphologies similar to those of control mice (Fig. 5 and Supplementary Material, Fig. S5), indicating that normal retinal development had occurred. However, by the age of 2 months, the retina of Atxn7100Q/100Q; Gcn5Δ/+ mice had thinner inner (IS) and outer segment layers (OS) when compared with those of control or Atxn7100Q/100Q mice (Fig. 5 and Supplementary Material, Fig. S6), indicating photoreceptor degeneration. Further thinning of the outer nuclear layer (ONL) and inner nuclear layer (INL) was observed in Atxn7100Q/100Q; Gcn5Δ/+ retinas at 4 months of age, whereas only mild thinning of the ONL was observed in retinas of Atxn7100Q/100Q mice (Fig. 5). By the age of 8 months, retinal ONLs in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice were dramatically thinner when compared with those of the controls. The OS appeared to be completely gone in Atxn7100Q/100Q; Gcn5Δ/+ mice at this time (Fig. 5). In summary, both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice developed retinal atrophy. However, this retinal atrophy occurred earlier in Atxn7100Q/100Q; Gcn5Δ/+ mice (first observed at 1.5–2 months in Atxn7100Q/100Q; Gcn5Δ/+ mice compared with 4 months in Atxn7100Q/100Q mice), indicating that partial loss of function of Gcn5 enhances retinal degeneration.
Figure 5.
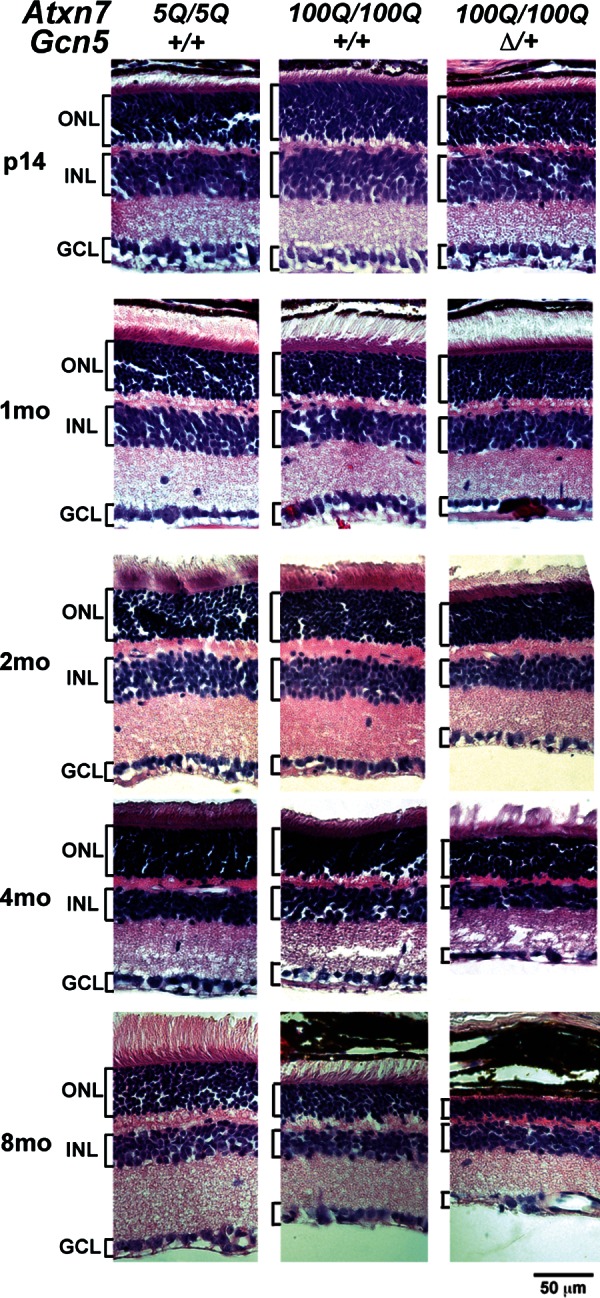
Reducing Gcn5 worsens retinal degeneration in Atxn7100Q/100Q mice. Representative images show progressively thinner retina in Atxn7100Q/100Q; Gcn5Δ/+ mice at 2, 4 and 8 months of age, whereas mice of all genotypes have similar retinal structures at P14 using H&E staining (n = 3 mice each genotype). OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Given that transcriptional deregulation is strongly associated with photoreceptor dysfunction and dystrophy in SCA7 mouse models (21,24,25), we next asked whether decreased Gcn5 expression induces transcriptional changes that accelerate retinal degeneration in SCA7 mice. We focused on eight transcripts (Opn1sw, Opn1mw, Rho, Gnat1, Rom1, Grk, Rbp3 and Cnga3) that were found previously to be decreased in early degenerating retinas of SCA7 mouse models, and one non-SCA7 target (Crx) as a control (21,24,25). RNAs were quantified after extraction from retinas of Atxn7100Q/100Q, Atxn7100Q/100Q; Gcn5Δ/+ and control mice at P14 at 1, 1.5 and 4 months of age. Consistent with our histological results, expression of all of the SCA7 targets examined was similar in all genotypes at P14; however, expression of these genes progressively decreased from 1.5 to 4 months in the SCA7 mice (Fig. 6). The pattern of transcriptional decrease was consistent with that previously reported for Atxn7266Q/5Q mice (21). While levels of Crx transcript remained steady, a gradual decrease of cone-specific transcripts (Opn1sw, Opn1mw), followed by rod-specific (Rho, Gnat1, Rom1) and cone–rod-shared (Grk, Rbp3) transcripts was detected in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ retinas. Transcript levels were decreased to a similar degree in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ retinas from 1 to 1.5 months. This trend continued to 4 months of age, except that the level of Gnat1 was lower in Atxn7100Q/100Q; Gcn5Δ/+ than in Atxn7100Q/100Q mice (Fig. 6). Although Atxn7100Q/100Q; Gcn5Δ/+ mice undergo accelerated retinal degeneration, the finding that transcript deregulation remains similar during early retinal degeneration in Atxn7100Q/100Q; Gcn5Δ/+ and Atxn7100Q/100Q mice suggests that reducing Gcn5 exacerbates retinal atrophy by mechanisms other than affecting the expression of known SCA7 target genes.
Figure 6.

Gradual decrease of SCA7 target transcripts in both Atxn7100Q/100Q and Atxn7100Q/100Q; Gcn5Δ/+ mice. Relative transcript levels to wt (dotted line) in the retinas of Gcn5Δ/+ (blue line), Atxn7100Q/100Q (red line) and Atxn7100Q/100Q; Gcn5Δ/+ (purple line) mice at P14, 1, 1.5 and 4 months of age (n = 3 mice of each genotype, *P < 0.05 between wt and Atxn7100Q/100Q or wt and Atxn7100Q/100Q; Gcn5Δ/+; Student's t-test). Data are presented as mean ± SD.
DISCUSSION
Previously, two independent studies using different SCA7 transgenic mouse models provided conflicting evidence regarding the effects of polyQ–Atxn7 on the functions of Gcn5 and SAGA complex (24,25). Specifically, one study pointed to a loss of function of Gcn5 and SAGA triggered by polyQ–Atxn7 (24), while the other showed that polyQ–Atxn7 led to increased recruitment of SAGA to target promoters, resulting in hyperacetylation of histone H3, arguing for enhanced SAGA function in pathogenesis (25). Data presented here demonstrate that decreased expression of Gcn5 worsens the severity of the two major neurological phenotypes of SCA7 mice (Figs 2–5), strongly indicating that loss of function of the SAGA complex contributes to polyQ–Atxn7 neurotoxicity.
Gcn5 itself appears to be necessary for normal Purkinje cell functions as Gcn5 depletion in these cells leads to mild ataxia (Fig. 4), which is similar to that observed in transgenic mice that express polyQ–Atxn7 only in Purkinje cells (33). To some degree, then, Gcn5 loss-of-function mimics the effects of the polyQ insult. The relatively mild phenotypes associated with Purkinje cell-specific polyQ–Atxn7 or Gcn5 deletion also suggest that other cell types may contribute to SCA7. Consistent with this idea, a non-cell autonomous effect from Bergmann glia has been linked previously to SCA7 neurotoxicity (31). Altered Gcn5 or SAGA functions in this cell type, then, might also contribute to SCA7 cerebellar degeneration. No further morphological distortion of Bergmann glia (Fig. 3B) was observed upon reduction of Gcn5 in Atxn7100Q/100Q mice beyond that caused by polyQ–Atxn7 alone, but it remains possible that the functions of Bergmann glia are altered upon Gcn5 depletion.
Another key finding reported here is that Gcn5 loss-of-function exacerbates retinal degeneration in SCA7 mice without affecting the transcription of several previously defined polyQ–Atxn7-dependent genes [Fig. 6 and (21)]. This finding suggests that lower levels of Gcn5 accelerate retinal degeneration in SCA7 mice either by affecting other, yet to be defined transcriptional targets or by affecting non-transcription-related SAGA functions. This point may provide further insight into the discordant results obtained by prior studies, which focused solely on the transcriptional role of Gcn5 and its alteration in SCA7 pathogenesis. Since Atxn7 is part of the SAGA DUB module (18,19), the more severe phenotype we observe upon depletion of Gcn5 in our Atxn7100Q/100Q; Gcn5Δ/+ mice may reflect combined diminishment of both the DUB and HAT modules of SAGA. As in Alzheimer's, Parkinson's and polyQ neurodegenerative diseases, abnormal accumulation of proteins is one feature of SCA7 neurons. One substrate of Usp22, ubiquitinated H2A, also accumulates in the nuclear inclusions of 100Q-Atxn7 neurons as indicated by immunostaining (Chen and Dent, unpublished data). Our previous work demonstrated that the DUB module in SAGA affects protein stability by regulating ubiquitination of proteins not involved in transcription (20), raising the possibility that accumulation of polyubiquitinated proteins upon crippling of the SAGA DUB module may directly contribute to SCA7. Future work with Usp22 mutant mice will test this idea.
Clinical features of adult-onset SCA7 include progressive spinocerebellar and retinal degeneration, and these neurological symptoms are recapitulated in Atxn7100Q/100Q mice after the age of weaning (>3 weeks). In SCA7 patients, cortical cerebellar atrophy, Purkinje cell loss and increased gliosis are frequently reported (37,39,40,43). Our Atxn7100Q/100Q mice also exhibited these features of adult-onset SCA7, in distinction to Atxn7266Q/5Q mice, which have phenotypes that more resemble those of infantile SCA7 disease (21). The inverse correlation between repeat length and disease-onset, and direct relationship with the progression in SCA7 patients suggests differential neurotoxicity from polyQ–ATXN7 proteins with different polyQ lengths (5,30). Consistent with these observations, we found that the length of the polyQ in Atxn7 inversely correlates to lifespan in Atxn7 knock-in mice, which provides a read-out of both disease-onset and progression.
Although our data clearly support a role for the loss of SAGA functions in SCA7, it is important to note that we have not ruled out the possibility that the polyQ–Atxn7 causes a gain of function in some SAGA components. Recent studies of SBMA and SCA1 indicate that the polyQ expansion can have differential effects on the functions of the affected protein, and in some cases can cause both gain- and loss-of-function, depending on post-translational modifications and interacting proteins (9,12,13,44–50). Additional studies examining the functions of SAGA components in SCA7 models are needed to address this question.
In conclusion, our data highlight the potential importance of considering the non-transcriptional functions of SAGA, particularly the SAGA DUB module, in disease development, as we demonstrate that polyQ–Atxn7 has additional toxic effects that are probably mediated by other proteins, independent of Gcn5. It will be crucial to identify these other functions as we work towards developing ways to intervene in this devastating disease.
MATERIALS AND METHODS
Generation of Gcn5;Atxn7 and Purkinje cell-specific Gcn5 mutant mouse models
Atxn7 230Q and Atxn7 100Q alleles were naturally occurring contracted mutations of the 266Q tract in Atxn7 obtained when maintaining Atxn7266Q/5Q mouse line (21). Atxn7100Q/5Q; Gcn5fn/+ or Atxn7100Q/5Q; Gcn5Δ/+ mice were generated by crossing Gcn5flox(neo)/+ or Gcn5Δ3−18/+ mice (42) with Atxn7100Q/5Q mice. Atxn7100Q/5Q; Gcn5fn/fn mice were generated by crossing Gcn5fn/+ and Atxn7100Q/5Q; Gcn5fn/+ mice. Atxn7100Q/100Q; Gcn5Δ/+ mice were generated by crossing Atxn7100Q/5Q; Gcn5Δ/+ and Atxn7100Q/5Q mice.
Purkinje cell-specific Gcn5-conditional nulls were obtained from crossing Gcn5Δ/+ mice carries Pcp2-cretg (41,51) with Gcn5flox/flox mice. Genotyping for Gcn5 and Atxn7 alleles was performed using PCR primers as previously described (21,27). Pcp2-cre genotype was assessed following the protocol for generic Cre by JAX laboratory. All procedures were performed in accordance with the approved IACUC protocols at MDAnderson Cancer Center.
Survival analysis
PolyQ–Atxn7 mutant mice were housed with control littermates and given ad libitum supply of moist food upon ataxia as diagnosed. Moribund was defined by losing body weight of more than 20%, or loss of mobility. Statistical significant differences were determined by Kaplan–Meier test.
Histological analyses
Deeply anesthetized animals were transcardially perfused with phosphate-buffered saline followed by 4% paraformaldehyde. Isolated organs were fixed in 4% paraformaldehyde overnight. Fixed tissues were then preserved in paraffin or cryo-blocks. For hematoxylin and eosin Y staining (H&E), 6 μm sections of paraffin-embedded mid-sagittal brain or eye were stained with H&E staining protocol (Fisher). Pictures were taken using Olympus SZX12 or Leica DM4000 microscope. For immunofluorescent staining, 20 μm sections of cryo-preserved tissues were used. Briefly, sections were boiled in 10 mm sodium citrate to retrieve antigen. PBST 0.2% was used for permeablizing and washing prior to incubation in anti-Calbindin (1:1000, Sigma) and anti-GFAP antibody (1:600, Dako). Signals were visualized using Alexa 555-conjugated donkey anti-mouse or Alexa 488-conjugated donkey–anti rabbit antibody (all 1:750, Invitrogen). DAPI (Roche) counterstains the nucleus. All pictures were obtained using FV1000 Olympus confocal microscope.
Purkinje cell counts were done by counting nuclei of Purkinje cells in pictures of H&E-stained 6 μm mid-sagittal cerebellar sections (three sections per mouse) from three to four mice of each genotype using Image J software (National Institutes of Health). Samples were renumbered to avoid bias. Statistical significance was determined using Student's t-tests.
Quantification of Purkinje cell soma size. Purkinje cell perikaryons were manually outlined from 12 μm z-stacked images of Calbindin1-labeled mid-sagittal cerebellums (three to four images of each mouse) using Image J software (National Institutes of Health). A total of 135 cells in lobule VI from two to three animals were analyzed for each genotype.
Footprint analyses
Behavior tests were performed between 2 and 6 pm with animals maintained on a C57BL/6J:129/SvEv mixed background at F3 or F4 backcrossing to C57BL/6J.
Footprint analysis was done as previously described (52). Briefly, fore- and hind-paws of mouse were painted with non-toxic red and blue paints. Then the mouse was allowed to walk through a tunnel (17 in.-long, 3 in.-wide, 3 in.-height) lined with a fresh paper at the bottom of each test. Statistical difference was first tested using one-way ANOVA between males and females. Data were only pooled together when there is no difference between genders. Statistical significant difference was determined by Kruskal–Wallis test.
Quantitative RT–PCR and primers
All tissues were dissected and snap-frozen in liquid nitrogen at 5–8 pm after overdose anesthesia. Total RNA was prepared using TRIZOL Reagent (Invitrogen) according to the instructions of manufacturer followed by on-column DNase I (Qiagen) treatment. cDNA were generated using 200 ng of total RNA by SuperScript III reverse transcriptase (Invitrogen) with poly dT primers following the instructions of manufacturer. cDNA were quantified using specific primers and SYBR Green PCR Master Mix (Applied Biosystems). Triplicates were performed. β-actin cDNA was quantified as normalizing control. The mean value for three animals was used for the level of each genotype. Statistical difference was calculated using Student's t-test. Primers used: Gcn5-f: 5′-CAGTGGTGG AGGGGTCTCTA-3′; Gcn5-r: 5′-AAACATTGTCTGGCGCTCTC-3′; Actb-f: 5′-GGCTGTATTCCCCTCCATCG-3′; Actb-r: 5′-CCAGTTGGTAACAATGCCATGT-3′; S-opsin-f: 5′-CAGCCTTCATG GGATTTGTCT-3′ ; S-opsin-r: 5′-CAAAGAGGAAGTATCCGTGACAG-3′; M-opsin-f: 5′-ATGGCCCAAAGGCTTACAGG-3′; M-opsin-r: 5′-CCACAAGAATCATCCAGGTGC-3′; rhodopsin-f: 5′-CCCTTCTCCAACGTCACAGG-3′; rhodopsin-r: 5′-TGAGGAAGTTGATGGG GAAGC-3′; Grk-f: 5′-CGGGGCAGTTTTGACGGAA-3′; Grk-r: 5′-AGCTGAGGTTGTCACGGAGA-3′; Rom1-f: 5′-CTCCAACCCCGT ATCCGTTTG-3′; Rom1-r: 5′-GAGCAGGGAATGAACAAGAGG-3′; Cnga3-f: 5′-TCGACCAC GTAGAGAACGG-3′; Cnga3-r: 5′-TGGAGGGGTCCACCACAAT-3′; Rbp3-f: 5′-ATGAGAGAATGGGTCCTGGTT-3′; Rbp3-r: 5′-GCCCAGAATCTCGTGACTC TTC-3′; Gnat1-f: 5′-GATGCCCGCACTGTGAAAC-3′; Gnat1-r: 5′-CCAG CGAATACCCGTCCTG-3′; Crx-f: 5′-GTTCAAGAATCGTAGGGCGAA-3′; Crx-r: 5′-TGAGATGCCCAAAGGATCTGT-3′.
Conflict of Interest statement: None declared.
FUNDING
This work was supported by the National Institutes of Health (NS049065 to P.A.G. with a subcontract to S.Y.R.D., NS27699 to H.Y.Z.); and the Howard Hughes Medical Institute to H.Y.Z.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Richard Behringer, John Wilson, Howard Gutstein and Stephanie Watowich for critical discussions; Marek Napierala, Marenda Wilson-Pham and Cheng-Chiu Huang for comments on the manuscript; Howard Gutstein, Juliette Kahle and Eric Kandel provided advice with the behavioral tests; and Institutional Core Grant No. CA16672 for High-Resolution Electron Microscopy Facility, UTMDACC.
REFERENCES
- 1.David G., Abbas N., Stevanin G., Durr A., Yvert G., Cancel G., Weber C., Imbert G., Saudou F., Antoniou E., et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 2.David G., Durr A., Stevanin G., Cancel G., Abbas N., Benomar A., Belal S., Lebre A.-S., Abada-Bendib M., Grid D., et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum. Mol. Genet. 1998;7:165–170. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 3.Del-Favero J., Krols L., Michalik A., Theuns J., Löfgren A., Goossens D., Wehnert A., Van den Bossche D., Van Zand K., Backhovens H., et al. Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA Type II) caused by CAG triplet repeat expansion. Hum. Mol. Genet. 1998;7:177–186. doi: 10.1093/hmg/7.2.177. [DOI] [PubMed] [Google Scholar]
- 4.Johansson J., Forsgren L., Sandgren O., Brice A., Holmgren G., Holmberg M. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum. Mol. Genet. 1998;7:171–176. doi: 10.1093/hmg/7.2.171. [DOI] [PubMed] [Google Scholar]
- 5.Giunti P., Stevanin G., Worth P.F., David G., Brice A., Wood N.W. Molecular and clinical study of 18 families with ADCA type ii: evidence for genetic heterogeneity and de novo mutation. Am. J. Hum. Genet. 1999;64:1594–1603. doi: 10.1086/302406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marsh J.L., Walker H., Theisen H., Zhu Y.-Z., Fielder T., Purcell J., Thompson L.M. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum. Mol. Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- 7.Ordway J.M., Tallaksen-Greene S., Gutekunst C.-A., Bernstein E.M., Cearley J.A., Wiener H.W., Dure Iv L.S., Lindsey R., Hersch S.M., Jope R.S., et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 8.Spada A.R.L., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 9.Katsuno M., Adachi H., Kume A., Li M., Nakagomi Y., Niwa H., Sang C., Kobayashi Y., Doyu M., Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 10.Nedelsky N.B., Pennuto M., Smith R.B., Palazzolo I., Moore J., Nie Z., Neale G., Taylor J.P. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeyama K.-i., Ito S., Yamamoto A., Tanimoto H., Furutani T., Kanuka H., Miura M., Tabata T., Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- 12.Duvick L., Barnes J., Ebner B., Agrawal S., Andresen M., Lim J., Giesler G.J., Zoghbi H.Y., Orr H.T. SCA1-like disease in mice expressing wild-type Ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A.B., Richman R., Hill D.E., Orr H.T., Zoghbi H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Ann. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 15.Helmlinger D., Hardy S., Sasorith S., Klein F., Robert F., Weber C., Miguet L., Potier N.l., Van-Dorsselaer A., Wurtz J.-M., et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 16.Martinez E., Palhan V.B., Tjernberg A., Lymar E.S., Gamper A.M., Kundu T.K., Chait B.T., Roeder R.G. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol. 2001;21:6782–6795. doi: 10.1128/MCB.21.20.6782-6795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X.-Y., Varthi M., Sykes S.M., Phillips C., Warzecha C., Zhu W., Wyce A., Thorne A.W., Berger S.L., McMahon S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell. 2008;29:102–111. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y., Lang G., Ito S., Bonnet J., Metzger E., Sawatsubashi S., Suzuki E., Le Guezennec X., Stunnenberg H.G., Krasnov A., et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol. Cell. 2008;29:92–101. doi: 10.1016/j.molcel.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 19.Kohler A., Schneider M., Cabal G.G., Nehrbass U., Hurt E. Yeast Ataxin-7 links histone deubiquitination with gene gating and mRNA export. Nat. Cell. Biol. 2008;10:707–715. doi: 10.1038/ncb1733. [DOI] [PubMed] [Google Scholar]
- 20.Atanassov B.S., Evrard Y.A., Multani A.S., Zhang Z., Tora L., Devys D., Chang S., Dent S.Y. Gcn5 and SAGA regulate sheltering protein turnover and telomere maintenance. Mol. Cell. 2009;35:352–364. doi: 10.1016/j.molcel.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoo S.-Y., Pennesi M.E., Weeber E.J., Xu B., Atkinson R., Chen S., Armstrong D.L., Wu S.M., Sweatt J.D., Zoghbi H.Y. SCA7 knock-in mice model human SCA7 and reveal gradual accumulation of mutant Ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/s0896-6273(02)01190-x. [DOI] [PubMed] [Google Scholar]
- 22.Helmlinger D., Hardy S., Eberlin A., Devys D., Tora L. Both normal and polyglutamine-expanded ataxin-7 are components of TFTC-type GCN5 histone acetyltransferase-containing complexes. Biochem. Soc. Symposia. 2006;73:155–163. doi: 10.1042/bss0730155. [DOI] [PubMed] [Google Scholar]
- 23.McMahon S.J., Pray-Grant M.G., Schieltz D., Yates J.R., Grant P.A. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc. Natl Acad. Sci. USA. 2005;102:8478–8482. doi: 10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palhan V.B., Chen S., Peng G.-H., Tjernberg A., Gamper A.M., Fan Y., Chait B.T., La Spada A.R., Roeder R.G. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl Acad. Sci. USA. 2005;102:8472–8477. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helmlinger D., Hardy S., Abou-Sleymane G., Eberlin A., Bowman A.B., Gansmüller A., Picaud S., Zoghbi H.Y., Trottier Y., Tora L., et al. Glutamine-expanded Ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bu P., Evrard Y.A., Lozano G., Dent S.Y. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol. Cell. Biol. 2007;27:3405–3416. doi: 10.1128/MCB.00066-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin W., Zhang Z., Chen C.H., Behringer R.R., Dent S.Y. Proper Gcn5 histone acetyltransferase expression is required for normal anteroposterior patterning of the mouse skeleton. Dev. Growth Diff. 2008;50:321–330. doi: 10.1111/j.1440-169X.2008.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin W., Zhang Z., Srajer G., Chen Y.C., Huang M., Phan H.M., Dent S.Y. Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev. Dyn. 2008;237:928–940. doi: 10.1002/dvdy.21479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu W., Edmondson D.G., Evrard Y.A., Wakamiya M., Behringer R.R., Roth S.Y. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 2000;26:229–232. doi: 10.1038/79973. [DOI] [PubMed] [Google Scholar]
- 30.Martin J., Van Regemorter N., Del-Favero J., Lofgren A., Van Broeckhoven C. Spinocerebellar ataxia type 7 (SCA7): correlations between phenotype and genotype in one large Belgian family. J. Neurol. Sci. 1999;168:37–46. doi: 10.1016/s0022-510x(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 31.Custer S.K., Garden G.A., Gill N., Rueb U., Libby R.T., Schultz C., Guyenet S.J., Deller T., Westrum L.E., Sopher B.L., et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat. Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 32.Lalonde R. Motor abnormalities in staggerer mutant mice. Exp. Brain Res. 1987;68:417–420. doi: 10.1007/BF00248807. [DOI] [PubMed] [Google Scholar]
- 33.Yvert G., Lindenberg K.S., Picaud S., Landwehrmeyer G.B., Sahel J.-A., Mandel J.-L. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum. Mol. Genet. 2000;9:2491–2506. doi: 10.1093/hmg/9.17.2491. [DOI] [PubMed] [Google Scholar]
- 34.de Solages C., Szapiro G., Brunel N., Hakim V., Isope P., Buisseret P., Rousseau C., Barbour B., Léna C. High-frequency organization and synchrony of activity in the purkinje cell layer of the cerebellum. Neuron. 2008;58:775–788. doi: 10.1016/j.neuron.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 35.Harvey R.J., Porter R., Rawson J.A. The natural discharges of Purkinje cells in paravermal regions of lobules V and VI of the monkey's cerebellum. J. Physiol. 1977;271:515–536. doi: 10.1113/jphysiol.1977.sp012012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nimmerjahn A., Mukamel E.A., Schnitzer M.J. Motor behavior activates bergmann glial networks. Neuron. 2009;62:400–412. doi: 10.1016/j.neuron.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carpenter C.S., Schumacher G.A. Familial infantile cerebellar atrophy associated with retinal degeneration. Arch. Neurol. 1966;14:82–94. doi: 10.1001/archneur.1966.00470070086010. [DOI] [PubMed] [Google Scholar]
- 38.Gouw L.G., Digre K.B., Harris C.P., Haines J.H., Ptacek L.J. Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology. 1994;44:1441–1447. doi: 10.1212/wnl.44.8.1441. [DOI] [PubMed] [Google Scholar]
- 39.Martin J., Krols L., Ceuterick C., Van Broeckhoven C., Van Regemorter N., Hayer-Delatte F., Brucher J., de Barsy T., Szliwowski H., Evrard P., et al. On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol. 1994;88:277–286. doi: 10.1007/BF00310370. [DOI] [PubMed] [Google Scholar]
- 40.Rub U., Brunt E.R., Gierga K., Seidel K., Schultz C., Schols L., Auburger G., Heinsen H., Ippel P.F., Glimmerveen W.F., et al. Spinocerebellar ataxia type 7 (SCA7): first report of a systematic neuropathological study of the brain of a patient with a very short expanded CAG-repeat. Brain Pathol. 2005;15:287–295. doi: 10.1111/j.1750-3639.2005.tb00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barski J.J., Mörl K., Meyer M. Conditional inactivation of the calbindin D-28k (Calb1) gene by Cre/loxP-mediated recombination. Genesis. 2002;32:165–168. doi: 10.1002/gene.10045. [DOI] [PubMed] [Google Scholar]
- 42.Lin W., Srajer G., Evrard Y.A., Phan H.M., Furuta Y., Dent S.Y. Developmental potential of Gcn5(-/-) embryonic stem cells in vivo and in vitro. Dev. Dyn. 2007;236:1547–1557. doi: 10.1002/dvdy.21160. [DOI] [PubMed] [Google Scholar]
- 43.Jobsis G.J., Weber J.W., Barth P.G., Keizers H., Baas F., van Schooneveld M.J., van Hilten J.J., Troost D., Geesink H.H., Bolhuis P.A. Autosomal dominant cerebellar ataxia with retinal degeneration (ADCA II): clinical and neuropathological findings in two pedigrees and genetic linkage to 3p12-p21.1. J. Neurol., Neurosurgery Psychiatry. 1997;62:367–371. doi: 10.1136/jnnp.62.4.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chevalier-Larsen E.S., O'Brien C.J., Wang H., Jenkins S.C., Holder L., Lieberman A.P., Merry D.E. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J. Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crespo-Barreto J., Fryer J.D., Shaw C.A., Orr H.T., Zoghbi H.Y. Partial loss of Ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010;6:e1001021. doi: 10.1371/journal.pgen.1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gehrking K.M., Andresen J.M., Duvick L., Lough J., Zoghbi H.Y., Orr H.T. Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum. Mol. Genet. 2011;20:2204–2212. doi: 10.1093/hmg/ddr108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goold R., Hubank M., Hunt A., Holton J., Menon R.P., Revesz T., Pandolfo M., Matilla-Dueñas A. Down-regulation of the dopamine receptor D2 in mice lacking ataxin 1. Hum. Mol. Genet. 2007;16:2122–2134. doi: 10.1093/hmg/ddm162. [DOI] [PubMed] [Google Scholar]
- 48.Lam Y.C., Bowman A.B., Jafar-Nejad P., Lim J., Richman R., Fryer J.D., Hyun E.D., Duvick L.A., Orr H.T., Botas J., et al. ATAXIN-1 interacts with the Repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 49.Mizutani A., Wang L., Rajan H., Vig P.J.S., Alaynick W.A., Thaler J.P., Tsai C.-C. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1. EMBO J. 2005;24:3339–3351. doi: 10.1038/sj.emboj.7600785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuda H., Jafar-Nejad H., Patel A.J., Sun Y., Chen H.-K., Rose M.F., Venken K.J.T., Botas J., Orr H.T., Bellen H.J., et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 51.Barski J.J., Hartmann J., Rose C.R., Hoebeek F., Mörl K., Noll-Hussong M., De Zeeuw C.I., Konnerth A., Meyer M. Calbindin in cerebellar purkinje cells is a critical determinant of the precision of motor coordination. J. Neurosci. 2003;23:3469–3477. doi: 10.1523/JNEUROSCI.23-08-03469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carter R.J., Lione L.A., Humby T., Mangiarini L., Mahal A., Bates G.P., Dunnett S.B., Morton A.J. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J. Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.