

Abstract
Recently, researchers have created novel fluorescent proteins by harnessing the somatic hypermutation ability of B cells. In this study, we examined if this approach could be used to evolve a non-fluorescent protein, namely the anti-apoptosis protein Bcl-xL, using the Ramos B-cell line. After demonstrating that Ramos cells were capable of mutating a heterologous bcl-xL transgene, the cells were exposed to multiple rounds of the chemical apoptosis inducer staurosporine followed by rounds of recovery in fresh medium. The engineered B cells expressing Bcl-xL exhibited progressively lower increases in apoptosis activation as measured by caspase-3 activity after successive rounds of selective pressure with staurosporine treatment. Within the B-cell genome, a number of mutated bcl-xL transgene variants were identified after three rounds of evolution, including a mutation of Bcl-xL Asp29 to either Asn or His, in 8 out of 23 evaluated constructs that represented at least five distinct Ramos subpopulations. Subsequently, Chinese hamster ovary (CHO) cells engineered to overexpress the Bcl-xL Asp29Asn variant showed enhanced apoptosis resistance against an orthogonal apoptosis insult, Sindbis virus infection, when compared with cells expressing the wild-type Bcl-xL protein. These findings provide, to our knowledge, the first demonstration of evolution of a recombinant mammalian protein in a mammalian expression system.
Keywords: apoptosis, Bcl-xL, directed evolution, Ramos B cell, somatic hypermutation
Introduction
Directed evolution has become a powerful tool for biomolecular engineers interested in generating proteins with novel characteristics or enhanced function. Directed evolution involves diversification of a gene of interest through mutagenesis, expression of the mutant protein and selection based upon an easily identifiable and favorable trait. Such altered proteins may exhibit increased activity (Kauffmann and Schmidt-Dannert, 2001), specificity (Kuchner and Arnold, 1997; Liebeton et al., 2000) or properties including improved stability (Miyazaki et al., 2000; Lehmann and Wyss, 2001), novel fluorescent characteristics (Zacharias and Tsien, 2006) or ability to act as molecular switches (Guntas et al., 2005; Ostermeier, 2005).
Error-prone PCR has been the method of choice for generation of mutant libraries for directed evolution, and selection of mutant proteins of interest is typically carried out in bacteria or yeast. These organisms allow for high transformation efficiency, easy single-cell cloning and rapid molecular biology manipulation. However, the directed evolution of mammalian genes is not as well established for a number of reasons. Mammalian genes can be evolved in bacterial systems (Aharoni et al., 2004; Kumar et al., 2005), although the protein is not in its native physiological environment and its structure or function may be affected by the presence or absence of other proteins, ion concentrations and pH gradients. In addition, many mammalian proteins are transported to specific cellular compartments, perform specific cellular functions or undergo post-translational modifications, such as complex glycosylation, that are specific to mammalian cells. These differences between mammalian and bacterial proteins may significantly impact the protein structure and activity. A number of properties make mammalian cells less than ideal for expression and selection in traditional directed evolution experiments including the slow growth of mammalian cells, low efficiency of stable integration, tendency toward multiple gene insertions, highly variable expression levels and time-consuming molecular biology (Majors et al., 2009b).
Alternatively, nature's method for directed evolution in mammalian cells is most evident in B cells of the immune system. B cells, formed in the bone marrow, are responsible for generating the antibody diversity required for recognition of any number of antigens an organism may encounter. To generate antibodies with increasing specificity to antigens, B cells undergo random mutagenesis of their antibody-encoding genes, a process known as somatic hypermutation. Somatic hypermutation requires the protein activation-induced (cytidine) deaminase (AID), which preferentially targets transcriptionally active genes in the Ig variable locus (Bachl et al., 2001) and causes the deamination of cytosines to uracil. The mutagenesis is thought to occur during an error-prone repair process of the U:G base pairing, causing single-nucleotide changes and less often insertions and deletions due to double-stranded break repair activity (Perez-Duran et al., 2007). In B cells, the resulting mutations give rise to an antibody diversity that can bind new antigens or known antigens with increasing specificity (Cumbers et al., 2002).
Researchers have utilized this antibody diversification machinery to evolve both endogenous IgM from B cells (Cumbers et al., 2002) and exogenous genes introduced into the B cells (Bachl et al., 2001; Wang et al., 2004a,b; Kanayama et al., 2006; Wang and Tsien, 2006). The ability to evolve exogenous transgenes in B cells was first demonstrated using a novel red fluorescent protein (RFP). The scientists inserted the gene for RFP into the constitutively hypermutating B-cell line Ramos and selected cells containing fluorescent proteins with shifted emission wavelengths using fluorescence-activated cell sorting. They found that the hypermutation ability of the B cells created a collection of RFP protein variants (Wang et al., 2004b) including one RFP variant protein exhibiting an emission wavelength shift of 45 nm into the infrared. This protein is now being investigated for possible use in in vivo imaging. Additional studies utilizing the chicken DT40 B-cell line have shown the capacity to mutate fluorescent proteins by somatic hypermutation (Arakawa et al., 2008) and another endogenous B-cell gene diversification mechanism, gene conversion (Kanayama et al., 2006).
While using B cells for directed evolution has been demonstrated using fluorometric proteins, to our knowledge, no studies have been undertaken to demonstrate the potential of this technology for evolution of non-fluorescent recombinant proteins. One of the limitations is that a reliable method for selection of mutants is needed in order to be able to isolate proteins with improved function or altered activity. The goal of the current study was to use the induction of apoptosis as a method for evolving heterologous genes with improved anti-apoptotic function. In this study the target gene was bcl-xL, one of the principal anti-apoptotic Bcl-2 family members. Bcl-xL has been shown to inhibit apoptosis through direct maintenance of the mitochondrial membrane and through binding of pro-apoptosis proteins in the presence of apoptotic stimuli (Majors et al., 2007). In addition, expression of exogenous Bcl-xL has been employed in a biotechnology role to prevent cell death in mammalian cells used to produce recombinant proteins. Bcl-xL overexpression has been applied to enhance culture viability (Figueroa et al., 2003, 2004), increase viable cell density (Chiang and Sisk, 2005; Kim et al., 2009), and increase productivity (Meents et al., 2002; Chiang and Sisk, 2005; Majors et al., 2008) in mammalian cell lines engineered to express recombinant therapeutic proteins.
In this study, we employed the Ramos B-cell line as a mutagenesis and selection tool for evolution of a gain-of-function mutant of the bcl-xL gene. Cells expressing high levels of Bcl-xL were selected based on survival in the presence of apoptotic stimuli and the mutated transgenes characterized for novel gain of function. This study represents the first example of applying the B cell's mutation capabilities to evolve a mammalian protein in a mammalian host by taking advantage of the cell's natural programmed cell death pathway in order to select for altered properties of the protein.
Materials and methods
Maintenance of cell lines
Ramos B cells were a generous gift from the laboratory of Dr Chi Dang (Johns Hopkins Medical School) and were maintained in suspension in static culture in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented as instructed by American type culture collection for Ramos cells. Flp-In Chinese hamster ovary K1 (CHO-K1) cells (Invitrogen) were maintained in high glucose Dulbecco's modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), l-glutamine (Invitrogen) and non-essential amino acids (Invitrogen). CHO cell cultures were routinely detached using Trypsin–EDTA (Invitrogen) and passaged in fresh medium.
Creation of Ramos B cells expressing yellow fluorescent protein (YFP) and YFP–Bcl-xL
The gene for enhanced YFP was PCR amplified from pEYFP-C1 (Clontech, Palo Alto, CA) and cloned into pLXSN retroviral plasmid (Clontech). Similarly, wild-type (WT) human bcl-xL was PCR amplified and cloned into pLXSN and the gene for YFP inserted in frame, 5′ of the bcl-xL gene. When expressed, the resulting YFP and bcl-xL genes are separated by a five amino acid linker. Expression of the YFP or YFP–bcl-xL gene is driven by the 5′ viral long terminal repeat which contains promoter/enhancer sequences. The retroviral vectors were transfected into the Amphopack 293 (Clontech) retroviral packaging cell line and a stable pool of packaging cells was selected in 500 µg/ml G418 (Invitrogen). Retrovirus-containing supernatants from the packaging cells were filtered through a 0.45 µM cellulose acetate filter (Nalgene, Rochester, NY) and used to infect actively dividing Ramos B cells by spin infection in 24-well plates (Becton Dickinson, Franklin Lakes, NJ) in the presence of 8 µg/ml polybrene (Sigma, St Louis, MO). Transfection efficiency was determined by fixing a portion of the cells and assaying for positive YFP fluorescence using a flow cytometer (Becton Dickinson, Mountain View, CA).
Western blot detection of Bcl-xL
Cells were lysed in RIPA buffer and the total protein concentration determined using a BCA Protein Assay Kit (Pierce, Rockford, IL). The indicated amount of total protein was run on a 4–20% Tris-glycine gel (Invitrogen) and subjected to western blot analysis according to Majors et al. (2008).
Measurement of caspase-3 activity
Caspase-3 activity of the B cells was measured using the EnzChek Caspase-3 Assay Kit (Invitrogen) according to the manufacturer's instructions.
Induction of apoptosis in B cells expressing YFP or YFP–Bcl-xL
Stable pools of B cells expressing YFP or YFP–Bcl-xL were seeded at 1 × 107 viable cells/ml in triplicate in either fresh medium or fresh medium containing 1 µM staurosporine (Cost et al., 2010) (Fisher Scientific, Fair Lawn, NJ). Three aliquots of 5 × 106 cells were removed at the indicated times for each condition. Cells were pelleted by centrifugation, washed once with PBS and frozen at −80°C until the level of caspase-3 activity was determined using the EnzChek Caspase-3 Assay Kit as stated above.
Protein evolution by staurosporine apoptosis induction
For rounds of selection by staurosporine-induced apoptosis, pools of Ramos B cells stably expressing YFP or YFP–Bcl-xL were expanded to ∼10 × 107 total viable cells. Staurosporine was added at 1 µM and the viable cell number determined using the trypan blue exclusion method on the indicated days. Once the total viable cell number decreased to less than 4 × 107 cells, the cells were pelleted by centrifugation, washed once in PBS and resuspended in fresh culture medium lacking staurosporine for recovery of the culture. During recovery, the culture medium was exchanged as necessary until the total number of viable cells again attained approximately 10 × 107, at which time an aliquot of the culture was frozen in cryovials prior to beginning the next round of staurosporine-induced apoptosis.
Characterization of mutations in B cells expressing YFP and YFP–Bcl-xL
Genomic DNA from 5 × 106 viable Ramos B cells expressing either YFP or YFP–Bcl-xL was isolated using the DNeasy Kit (Qiagen, Valencia, CA) and used as a template for high-fidelity PCR of the YFP or YFP–bcl-xL gene using primers specific to the pLXSN retroviral vector and VENT DNA polymerase (New England Biolabs, Ipswich, MA). The primers contained flanking sequences that allowed for cloning of the PCR product directly into the Gateway vector pDONR221 vector using the Gateway system enzymes (Invitrogen) and transformed into Subcloning Efficiency™ DH5α™ Competent Cells (Invitrogen). Plasmid DNA from individual bacterial colonies containing the bcl-xL gene was sequenced on both strands to confirm mutations.
Creation of stable Bcl-xL and mutant Bcl-xL CHO cell lines
The genes for Bcl-xL WT and the Bcl-xL D29N mutant were PCR amplified from the appropriate template and inserted into the pcDNA5/FRT vector (Invitrogen) using the EcoRV and XhoI sites. Constructs were cotransfected with the pOG44 vector (Invitrogen) into the CHO Flp-In cell line (Invitrogen) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Cells expressing the plasmid of interest were selected in 600 µg/ml hygromycin (Cellgro, Manassas, VA).
Sindbis virus infection
CHO cells were seeded in six-well plates at 1 × 106 viable cells/well and infected with Sindbis virus according to Figueroa et al. (2003) at a multiplicity of infection (MOI) of 10 the day after seeding. Cell viability was determined using trypan blue exclusion and caspase-3 activity was measured as described above.
Results
Generation of B cells expressing YFP and YFP–Bcl-xL
Previous studies applying B cells for directed evolution utilized retroviruses for insertion of the exogenous gene (Wang et al., 2004b). Retroviral delivery offers the capability to control gene copy number through adjustment of MOI, and retroviruses, by nature, provide for stable integration of the transgene into the target cell's genomic DNA. We chose to use a bcl-xL gene fused to that of YFP, which allows for easy analysis of transfection efficiency and expression levels. The sequence of the YFP–bcl-xL gene is shown in Fig. 1. To generate Ramos B cells expressing YFP or YFP–Bcl-xL, constructs containing the gene of interest and packaging signal domains were transfected into a retrovirus packaging cell line and viral supernatants were used to transduce Ramos cells. Ramos cells transduced with either YFP or YFP–bcl-xL showed transduction efficiencies of 5% or less (data not shown), as determined by YFP fluorescence using flow cytometry. Of the 5% population that was transduced, a Poisson distribution suggests 95% of these cells contain a single copy of the gene of interest (Wang et al., 2004a,b). Cell pools resistant to G418 selection were expanded, yielding populations exhibiting YFP fluorescence by fluorescence microscopy (data not shown). To determine whether the exogenous Bcl-xL protein was expressed, Ramos cells expressing YFP or YFP–Bcl-xL were subjected to western blot using an anti-Bcl-xL antibody. As shown in Fig. 2, a band corresponding to the 54 kDa size of YFP–Bcl-xL was detected only in the Ramos cells infected with retrovirus containing the YFP–Bcl-xL construct. To ensure that the western blot band intensities reflected the abundance of Bcl-xL protein, equal amounts of total cellular protein were loaded per lane.
Fig. 1.

YFP–Bcl-xL nucleotide and amino acid sequence. The region of interest is shown. The nucleotides within the 5′ UTR are shown in lower case, the nucleotides and amino acids corresponding to the YFP gene are denoted by the black arrow, and the nucleotides and amino acids corresponding to the bcl-xL gene are denoted by the gray arrow.
Fig. 2.

Western blot analysis of YFP–Bcl-xL expression in B cells. Equal amounts of total cellular protein (25 µg) from stable pools of B cells expressing YFP or YFP–Bcl-xL were loaded in each lane and subjected to SDS–PAGE and western blot analysis with an anti-Bcl-xL antibody. The expected molecular mass of YFP–Bcl-xL is 54 kDa.
Effect of YFP or YFP–Bcl-xL overexpression on B cells exposed to an apoptotic stimulus
The next step was to determine whether exogenous expression of YFP or YFP–Bcl-xL could delay apoptosis in Ramos cells exposed to an apoptotic stimulus. As a model apoptosis insult, we chose to use the kinase inhibitor staurosporine. Staurosporine has been shown to cause the rapid onset of mitochondrial-induced apoptosis that is inhibited by the expression of Bcl-xL in a number of mammalian cell lines (Poppe et al., 2001; Takehara et al., 2001). To measure the apoptosis-inducing effects of staurosporine and the ability of Bcl-xL overexpression to delay apoptosis, we chose to monitor the activation of caspase-3 in the B cells. Caspase-3 is a member of the caspase family of proteases involved in apoptosis and is a crucial mediator of apoptotic cell death (Porter and Janicke, 1999). Therefore, the level of caspase-3 activation provides a measure of apoptosis activation in the B cells.
In the absence of staurosporine, G418-resistant Ramos B cells expressing either YFP or YFP–Bcl-xL did not exhibit any increase in apoptosis as measured by caspase-3 activity over the 6 h experiment (Fig. 3, no STS). In contrast, after just 2 h of exposure to 1 µM staurosporine, Ramos cells expressing YFP and YFP–Bcl-xL exhibited increases in the caspase-3 activity (Fig. 3, 1 µM STS) that progressively increased from 2 to 6 h. However, the increase in caspase-3 activity was lower in the YFP–Bcl-xL cells at the later time points (4–6 h). This reduced caspase-3 activity is consistent with the anti-apoptotic nature of the expressed bcl-xL gene.
Fig. 3.

Caspase-3 activity in B cells. Caspase-3 activity levels in stable pools of B cells expressing YFP or YFP–Bcl-xL in the presence or absence of 1 µM staurosporine (STS) were assessed over the time of exposure. The data represent averages from three independent experiments with two independent measurements per time point.
Mutation of exogenous YFP and YFP–Bcl-xL in B cells before selection in staurosporine
Directed evolution with B cells relies upon the effective mutation of exogenous genes incorporated into the cellular chromosomes. A schematic of the steps taken for the generation of the B-cell population expressing YFP and YFP–Bcl-xL during successive rounds of evolution is shown in Fig. 4. First, we examined whether the B cells could mutate exogenously inserted YFP or YFP–bcl-xL constructs prior to any direct apoptotic stimuli.
Fig. 4.

Workflow for generating B cells expressing YPF and YFP–Bcl-xL followed by rounds of selection in staurosporine and recovery.
Following G418 selection of stable pools expressing YFP and YFP–Bcl-xL (Fig. 4, round 0), aliquots of the cultures were harvested and the genomic DNA extracted from the population. To allow for efficient bacterial cloning, the integrated YFP–bcl-xL transgene was PCR amplified from the genomic DNA of the Ramos cells using a high-fidelity DNA polymerase, cloned into vectors, transformed into bacteria and purified for double-strand sequencing analysis.
Shown in Table I are the results of sequencing of bacterial isolates containing the recovered transgenes from stable pools of YFP–Bcl-xL-expressing B cells after round 0. Seventeen of 23 isolates had at least one nucleotide mutation within the sequenced region (Table I). Mutations in the coding sequence for the YFP–Bcl-xL construct included amino acid changes, silent mutations and premature stop sequences. A mutation of the Bcl-xL Trp169 codon occurred in three separate constructs, including one Bcl-xL Trp169Arg and two Bcl-xL Trp169stop mutations. Similarly, mutation of the Bcl-xL Gln207 codon occurred in four constructs, including two Gln207Leu and two Gln207stop changes, suggesting a slight increase in prevalence of cells harboring these mutations in culture. Sequencing of the B-cell culture expressing YFP alone that was transduced under identical conditions yielded a similar number of mutations after the initial selection in G418 (data not shown). These experiments demonstrated that native somatic hypermutation activity of the B cells introduced mutations into the exogenous genes even in the absence of selection for YFP or Bcl-xL activity.
Table I.
Mutations in the YFP–Bcl-xL gene region of transduced Ramos B cells at round 0
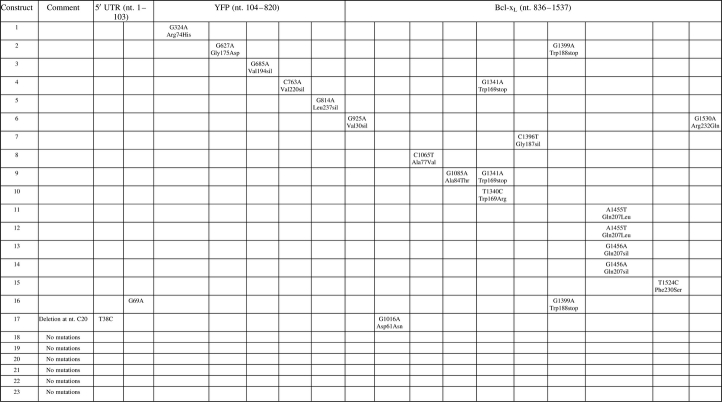 |
The YFP–Bcl-xL gene from genomic DNA of a pool of Ramos B cells from round 0 was PCR amplified, cloned and sequenced. The sequencing data for 23 different constructs isolated from bacterial colonies are shown. The location and nature of the nucleotide (nt.) mutation is shown as well as the resulting amino acid changes (amino acid location refers to residue within the YFP or Bcl-xL protein). Silent (sil) mutations are also shown.
Evaluation of Ramos B cells expressing YFP or YFP–Bcl-xL after multiple rounds of selection in staurosporine
B cells expressing YFP or YFP–Bcl-xL were then subjected to successive rounds of apoptosis induction by exposure to staurosporine followed by recovery in staurosporine-free media with the intent of generating cells with increased resistance to apoptosis. B cells were expanded to approximately 10 × 107 viable cells in culture and treated with 1 µM staurosporine to begin the first round of selection and recovery. When the number of viable cells declined to 4 × 107, the staurosporine was removed from this culture to limit further cell death. Cells were maintained in fresh medium until the number of viable cells again returned to 10 × 107 at which time the next round of selection by staurosporine would begin. This process of selection and recovery was repeated multiple times. During the third round of exposure to staurosporine the total number of viable cells declined for both YFP and YFP–Bcl-xL cells as shown in Fig. 5. For Ramos cells expressing the YFP construct, the total viable cell number began to decline rapidly after staurosporine treatment was initiated at day 0. After 4 days of staurosporine exposure, when the number of viable cells declined to 4 × 107, the staurosporine was removed. In contrast, Ramos cells expressing YFP–Bcl-xL showed a more gradual decline in viable cell number in response to staurosporine exposure, and the staurosporine was removed only after 6 days of exposure to allow for recovery of the pool. Similar death profiles were seen during the initial rounds (one and two) as well as in the following rounds of selection in staurosporine for these cell lines. The Ramos cells expressing YFP did not recover following round 3 of selection and thus only the Ramos cells expressing YFP–Bcl-xL were subsequently maintained and subjected to additional rounds of staurosporine selection followed by recovery.
Fig. 5.

Total viable cell number (TVCN) profiles from Ramos B cells expressing YFP or YFP–Bcl-xL during and after exposure to staurosporine in round 3. The TVCN was plotted against the duration of the culture. Staurosporine (1 µM) was added to the cell cultures on day 0 and removed as indicated by the arrows when the cultures declined to 4 × 107 total viable cells.
Next, we wanted to assess the apoptosis resistance of B cells expressing YFP–Bcl-xL generated from successive rounds of apoptosis induction by staurosporine followed by recovery in fresh medium. To do so, we compared the level of caspase-3 activity in response to staurosporine exposure in B cells pools generated from 0, 2, 4 and 6 successive rounds of selection and recovery (Fig. 6). Aliquots of cells from the end of each round of recovery were frozen and later cultured and tested in parallel for this experiment. In this experiment, the level of caspase-3 activity was measured after subtracting the basal level of caspase-3 signal in cells not exposed to staurosporine. For B cells expressing YFP from round 0, the caspase-3 activity level increased after 2 h of exposure to staurosporine. When the B cells expressing YFP–Bcl-xL obtained from rounds of selection and recovery were exposed to staurosporine in this experiment, all cultures exhibited lower increases in caspase-3 activity compared to the YFP cells from round 0. Furthermore, Ramos cells expressing YFP–Bcl-xL from round 2 showed lower caspase-3 activity increases compared to cells from round 0 and cells from round 4 showed lower caspase-3 activity levels compared to cells from round 2. In fact, the Ramos B expressing YFP–Bcl-xL from rounds 4 and 6 showed almost no increase in caspase-3 activity after staurosporine exposure compared to the same cells in the absence of staurosporine.
Fig. 6.

Caspase-3 activation in response to staurosporine in recovered Ramos B cells expressing YFP (round 0) and YFP–Bcl-xL after rounds of staurosporine exposure and recovery. Recovered cells from the indicated round of selection (R) were exposed to 1 µM staurosporine for 2 h and the caspase-3 activity was measured. The basal caspase-3 activity for cells in the absence of staurosporine was subtracted from the caspase-3 activity levels for cells in the presence of staurosporine.
One possible explanation for the increased survival during repeated rounds of staurosporine selection may be that subsequent rounds selected Ramos cells with higher levels of Bcl-xL expression. To examine the relative level of Bcl-xL expression in the Ramos B cells over an increasing number of selection rounds, the expression level of YFP–Bcl-xL in lysates from Ramos cells from rounds 0, 2, 4 and 6 was determined by western blot (Fig. 7). There was no detection of endogenous Bcl-xL in the Ramos cells expressing YFP only. Ramos cells from various rounds of treatment expressing the YFP–Bcl-xL gene showed detectable levels of Bcl-xL, and the relative levels of Bcl-xL protein in the Ramos B-cell population increased from round 0 to round 2 and round 2 to round 4, suggesting that the selection process enriched for cells exhibiting increased expression of Bcl-xL. To ensure that the western blot band intensities reflected the abundance of Bcl-xL protein, equal amounts of total cellular protein were loaded per lane.
Fig. 7.

Western blot expression of Bcl-xL in Ramos B cells expressing the YFP or YFP–Bcl-xL protein. Equal amounts of total cellular protein (50 µg) from Ramos B cells from the indicated round (R) of the selection process were loaded in each lane to ensure that band intensities reflect actual relative expression levels in the cells. Samples were subjected to SDS–PAGE and western blot analysis with an anti-Bcl-xL antibody.
Mutation of exogenous YFP–Bcl-xL in B cells following rounds of selection in staurosporine
Another possible reason for the increased survival and reduced caspase-3 activity was the presence of an altered Bcl-xL protein. The sequence of the YFP–bcl-xL construct was analyzed from the lysates of cells following rounds 3 and 6 of the selection and recovery experiment. The results of the sequence analysis for Ramos cells harvested following round 3 of selection and recovery are shown in Table II. These constructs exhibited multiple mutations in both the YFP and Bcl-xL coding sequence and in multiple colonies (Table II). If a gain-of-function mutant of the Bcl-xL protein were to evolve within the culture, the construct would be present in the pool of genomic DNA from the selected B cells and may exhibit an increased predominance afforded by the survival phenotype. Interestingly, in the Bcl-xL coding sequence, 8 of 23 constructs had a conserved amino acid change at the Bcl-xL Asp29 residue (Table II). This amino acid was changed to Asn in seven bacterial isolates (as a result of a G→A transition) and to His in one instance (as a result of a G→C transversion). A variety of other mutations in both the YFP coding sequence and Bcl-xL sequence were also found in cells harboring the Bcl-xL Asp29 mutation. Therefore, at least five of the Bcl-xL Asn29 variants were separate isolates because five out of the eight included a variety of different mutations at other points in the sequence. Aside from the Bcl-xL Asp29 change, one other mutation, Bcl-xL Gly117Glu, appeared in two separate Bcl-xL constructs. However, this mutation was found in only 2 of 23 isolates and was thus much less frequently observed than the Bcl-xL Asp29 change. In addition, the Asp29 amino acid was the only residue that exhibited a transformation to two different amino acids as observed in the sequenced isolates. From round 0 (Table I) to round 3 (Table II), two silent mutations and one nonsense mutation were maintained in the YFP–Bcl-xL coding region. Of the 23 colonies with sequencing data, 5 did not show any detectable mutations in the entire YFP–Bcl-xL coding sequence (including UTR regions) and 3 had mutations in the YFP coding region only. Other genomic mutations outside the sequenced region may have allowed these cells to survive three rounds of selection and recovery. Surprisingly, sequencing of the pooled genomic DNA constructs from round 6 of the selection and recovery experiment yielded no colonies with the Asp29 mutation (data not shown). Additionally, 12 of 19 isolates from round 6 were identified with no mutations in the bcl-xL coding sequence.
Table II.
Mutations in the YFP–Bcl-xL gene region of transduced Ramos B cells following round 3
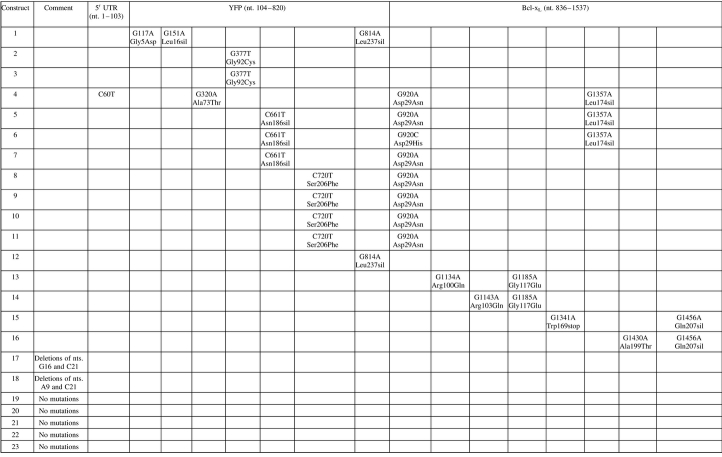 |
The YFP–Bcl-xL gene from genomic DNA of a pool of Ramos B cells was PCR amplified, cloned and sequenced. The sequencing data for 23 different constructs isolated from bacterial colonies are shown. The location and nature of the nucleotide (nt.) mutation is shown as well as the resulting amino acid changes (amino acid location refers to residue within the YFP or Bcl-xL protein). Silent (sil) mutations are also shown.
Effect of Bcl-xL mutant on cell survival for a model Sindbis apoptotic insult
To assess the extent that the predominant Bcl-xL Asp29Asn (D29N) mutation altered the properties of the protein, mammalian cells lines expressing the Bcl-xL WT protein, the Bcl-xL D29N mutant protein or the Null vector were created. Previously, expression of recombinant WT anti-apoptosis genes including Bcl-xL has been shown to inhibit cell death in CHO cells (Figueroa et al., 2003; Chiang and Sisk, 2005). Thus, CHO cells in combination with the Flp-In integration system, which provides for integration into a single, identical chromosomal site, were used in this study. The use of the Flp-In system helps to eliminate genomic location effects to ensure that stable selection of the Bcl-xL WT and Bcl-xL D29N cells resulted in comparable levels of the Bcl-xL protein. We determined the relative level of Bcl-xL expression by western blot (Fig. 8). While a low level of endogenous Bcl-xL was detected in the CHO cells expressing the Null vector, Bcl-xL was detected at much higher levels in CHO cells transfected with Bcl-xL or Bcl-xL D29N. Within the limits of quantification possible using western blot, both Bcl-xL-expressing cell lines showed the presence of similar, high levels of stable Bcl-xL protein. Equal amounts of total cellular protein were loaded per lane during the western blot so that relative band intensities reflected actual protein expression levels.
Fig. 8.

Western blot detection of Bcl-xL from Flp-In CHO cells. Flp-In CHO cells were transfected with either the Bcl-xL WT gene, the Bcl-xL D29N gene or the null vector and lysates from stable pools were subjected to SDS–PAGE and western blot analysis with an anti-Bcl-xL antibody. Equal amounts of total cellular protein (50 µg) were loaded per lane to ensure that band intensities reflected the relative expression levels of Bcl-xL. The expected molecular weight of Bcl-xL (WT and D29N) is 25 kDa.
To determine the effects of Bcl-xL or Bcl-xL mutant overexpression on apoptosis in CHO cells, we next exposed the CHO cells to a staurosporine apoptosis insult. While staurosporine was able to induce apoptosis rapidly in Ramos cells expressing YFP–Bcl-xL (Fig. 3) and cell death in CHO cells containing the null vector, CHO cells expressing Bcl-xL WT and CHO Bcl-xL D29N exhibited a much reduced rate of cell death in the presence of staurosporine (data not shown). Therefore Sindbis virus infection was considered as a more potent apoptosis inducer as it has been shown to cause rapid apoptosis (Mastrangelo et al., 1999; Figueroa et al., 2001). Furthermore, Sindbis virus infection is also known to cause apoptosis through a mitochondrial (Moriishi et al., 2002) and caspase-dependent (Nava et al., 1998) pathway.
Therefore, CHO cells expressing the Null, Bcl-xL WT and Bcl-xL D29N constructs were seeded at identical concentrations in six-well plates and exposed to Sindbis virus at a MOI of 10 to ensure that all cells were infected. After 24 h of Sindbis virus infection, the viabilities of the cultures were determined by the trypan blue exclusion method (Fig. 9). The null cell line showed less than 15% viability 24 h after infection, whereas CHO cells expressing either Bcl-xL construct showed much higher viabilities. Specifically, CHO Bcl-xL WT cells were 40% viable after 24 h of infection. However, CHO cells expressing the Bcl-xL D29N protein showed average viabilities that were even higher (at 55%). These experiments were repeated multiple times, with the CHO Bcl-xL D29N cell line exhibiting statistically-significant increases in viability relative to both Null and CHO Bcl-xL WT according to the Student's t-test criterion in all cases.
Fig. 9.

Viability of CHO cells following exposure to Sindbis virus. Flp-In CHO cells stably expressing Bcl-xL WT, Bcl-xL D29N or the null vector were exposed to Sindbis virus at an MOI of 10. The data represent averages of two independent experiments with two counts per experiment at 24-h post-infection. The higher viability of the Bcl-xL D29N cells was determined to be significantly higher than that of the Bcl-xL WT cells (Student's t-test P < 0.001).
Discussion
Directed evolution has already proven to be a powerful tool for the generation of novel polypeptides in a variety of applications. Such experiments typically employ bacterial or yeast systems to evolve proteins with easily identifiable traits. Ideally, it would be useful to have a mammalian system to evolve mammalian proteins, especially those in which modifications or functional selection depends on the presence of a mammalian host. Emerging higher eukaryotic directed evolution systems that employ self-mutating B-cell lines such as Ramos, 18–81 (Wang et al., 2004a) and DT-40 (Arakawa et al., 2008; Arakawa and Buerstedde, 2009) have recently demonstrated their capacity to evolve protein variants (Blagodatski and Katanaev, 2011). Yet, demonstration of this technology has been limited primarily to evolution of fluorescent proteins for ease of screening. Here, we describe, to our knowledge, the first successful directed evolution experiment of an exogenously inserted but naturally occurring mammalian protein in a mammalian protein evolution system. This work shows that the ability of using hypermutating B-cell lines for mammalian protein evolution is possible for proteins using screening criteria other than fluorescence. Such a study should provide the impetus to use mammalian evolution systems in other biotechnology and medical applications.
We first generated Ramos B-cell populations expressing either YFP or YFP–Bcl-xL. Unlike previous mammalian evolution experiments (Wang et al., 2004b; Arakawa et al., 2008), our fluorescent proteins were simply used to determine the transfection efficiency in our system and not used for selection. The efficiency of retrovirus transduction of Ramos cells was 5% for the constructs in this study, suggesting the difficult nature of gene delivery to B cells as seen in previous reports (Wang et al., 2004b). Viral gene delivery is reported to follow a Poisson distribution, meaning that at 5% transduction efficiency, one can assume that 95% of the cells transduced are the result of a single virion introducing a single copy of the gene of interest (Wang et al., 2004a). This is important in our study, since a cell harboring multiple genes of interest may lead to higher expression levels and multiple, different mutant proteins in a single cell. Such effects would likely compromise the cell-versus-cell competitive nature necessary for success in these evolution experiments.
After selection of Ramos B cells harboring the YFP or YFP–Bcl-xL genes, each was sequenced prior to applying apoptotic selective pressure. In the case of YFP–Bcl-xL, many mutations were found in the coding sequence, including three that were found in multiple constructs, even prior to staurosporine exposure when there was no selective pressure other than G418. Thus, these mutations are most likely the result of random mutagenesis by the hypermutation machinery. Most importantly, it shows that the Ramos B cells are able to introduce mutations in the exogenous genes during the culture process.
The transduced B cells stably expressing the YFP–Bcl-xL gene showed enhanced tolerance to the kinase inhibitor and apoptosis inducer staurosporine compared to cells expressing YFP only. This was expected since Bcl-xL expression has previously been shown to inhibit apoptosis in a Ramos B cell (Alam et al., 1997). Furthermore, our results confirm that the YFP fusion did not inhibit Bcl-xL function (Chu et al., 2004). After staurosporine exposure, the number of viable Ramos cells expressing only the YFP protein declined precipitously compared to YFP–Bcl-xL-expressing Ramos cells, which showed a slower decline. The cells continued to decrease in total viable number even after staurosporine was removed from the culture which may indicate either incomplete removal of the chemical or that the apoptosis cascade had already been activated in some cells.
After three rounds of staurosporine selection and cell recovery, the genomic DNA was harvested from YFP and YFP–Bcl-xL cells, PCR amplified with high-fidelity polymerase and multiple clones were sequenced. Numerous mutations were observed throughout the sequenced construct. The mutations in the Bcl-xL coding sequence are of particular interest. It is quite interesting that 8 of 23 sequenced constructs at round 3 had a mutation at Asp29. This can arise from either multiple independent mutations of the codon for Asp29 in multiple cells or from the survival or growth advantage of a subset of cells containing the mutant gene overtaking the culture. Interestingly, seven of the eight constructs showing the Asp29 mutation were a result of a G to A transition resulting in an Asn residue, whereas one of the eight constructs had an atypical G to C transversion that resulted in a His residue. A silent Leu174 mutation in the Bcl-xL coding sequence occurred in three Asp29-containing constructs, a Ser206Phe mutation was present in the YFP coding sequence for four Asp29-containing constructs, and another variant included an Ala73Thr mutation in the YFP coding region. These changes indicate that the Asp29 mutation arose multiple times in culture leading to perhaps five separate populations.
Subsequently, we tested the anti-apoptotic potential of the predominant mutation, Bcl-xL Asp29Asn (or D29N), found after three rounds in our studies. We chose to test the effects of Bcl-xL and Bcl-xL D29N expression in a CHO cell system that enables recombinant protein expression from a single, common genomic locus. CHO cells overexpressing the Bcl-xL D29N mutation showed higher viabilities in multiple experiments following Sindbis virus infection. Thus, we confirmed, in a non-hypermutating and commercially relevant cell line, the improved anti-apoptotic effects of the Bcl-xL mutant.
The reason that the D29N mutation is able to impart improved anti-apoptosis protection may be multifaceted. Most strikingly, aspartic acid residues are the known substrate for caspase-dependent cleavage during apoptosis signaling. Furthermore, the Asp29 residue is located within the unstructured loop domain of Bcl-xL, a region where two other well-characterized caspase cleavage residues, Asp61 and Asp76, are located (Clem et al., 1998). Interestingly, Bcl-2, an anti-apoptotic member of the Bcl-2 family of apoptosis-related proteins, exhibits significant homology to Bcl-xL and has a caspase cleavage residue in the vicinity of the Bcl-xL Asp29 location. Specifically, Bcl-2 has been shown to be cleaved by caspases at Asp34, and mutagenesis of this cleavage site abolished caspase cleavage and increased protection to apoptosis in cells expressing the mutant protein (Cheng et al., 1997). Surprisingly, the mutations found in the Bcl-xL gene failed to change amino acids at the other known Bcl-xL cleavage sites Asp61 and Asp76. When there are multiple known cleavage locations in a protein, the cleavage site acted upon by caspases will vary based on the apoptotic insult (Marie Hardwick, personal communication) and possibly the particular cell lines used.
It is possible that the D29N mutation in Bcl-xL may result in other modifications or interactions that alter Bcl-xL function. Bcl-xL interacts with the mitochondria (Vander Heiden et al., 1997; Shimizu et al., 2000), endoplasmic reticulum (White et al., 2005; Jeong et al., 2004) and with a multitude of other proteins both with direct apoptosis and non-apoptosis functions in the different cellular compartments.
It was also observed that the expression of Bcl-xL increased at later rounds of selection. Indeed, it has been shown that anti-apoptosis proteins exhibit dose-dependent behavior in which their anti-apoptotic properties increase with the levels of these proteins within the cells (Majors et al., 2009a). The increased stability brought about by removing a caspase cleavage site such as D29 may increase the levels of the full length protein. In addition to the D29N mutation, a number of additional mutations were also detected in the Bcl-xL, YFP and 5′ and 3′ UTR domains, including a Gly177Glu mutation in two separate isolates. These mutations as well as others outside the sequenced region may serve to increase either the stability or expression of Bcl-xL. Modifications that increase expression levels or protein stability will act in concert with those modifications that increase the function of the Bcl-xL protein and represent two complimentary evolving traits in the hypermutating B cells.
Ramos cell populations expressing YFP–Bcl-xL harvested after six rounds of staurosporine exposure and recovery no longer showed the widespread mutation of the Asp29 residue. Indeed, a majority of the cells even at round 3 of selection did not contain the Asp29 mutation. Prolonged stress using a B-cell type capable of hypermutating its own genomic DNA (Parsa et al., 2007) may result in a number of outcomes that can overcome staurosporine exposure, such as upregulation of pumps capable of removing the toxin or mutation of the kinases affected by staurosporine treatment. Furthermore, the apoptotic machinery of these cells may be rendered non-functional by other mutations, thus conferring apoptosis resistance superior to any mutation in the Bcl-xL protein. These particular changes may be superior to the increased protection afforded by the widespread Asp29 mutation in round 3. What the data suggest is that distinct subpopulations of Ramos can evolve even over a few rounds, and fewer rounds of B-cell evolution may perhaps be more suitable for identification of novel, non-fluorescent proteins while the phenotype imparted by overexpression of the transgene is most prominent. Extended culture or selection in hypermutating cells may allow for non-identifiable mutations that circumvent the overexpressed transgene. For these cases, systems that allow for inducible hypermutation ability (Faili et al., 2002) may provide a more flexible alternative to the constitutive hypermutating systems used in this study.
This study has demonstrated the ability of the Ramos B-cell line to induce mutations in exogenous non-fluorescent genes. Using rounds of selection and recovery in the presence of an apoptotic insult, we were able to isolate a mutant of Bcl-xL with improved anti-apoptotic ability. This Ramos B-cell mutagenesis system relies upon the mutagenesis properties of AID which may have preferential DNA targets and limited nucleotide changes (Seki et al., 2005). Future studies could employ the use of other hypermutating systems to generate greater diversity. For example, the DT40 chicken B-cell line can introduce genetic mutations by both somatic hypermutation and gene conversion. Furthermore, as more information is gained about the mutation capacity of B-cell lines, vectors targeting a region of increased hypermutation (such as the IgV locus in Ramos cells) can be utilized for faster mutagenesis. Regardless, the main factor to consider in all directed evolution experiments is identifying a selectable phenotype based on transgene overexpression. Mammalian proteins that can link functional changes to an appropriate selectable readout will likely provide excellent opportunities for directed evolution in mammalian cells in the coming years.
Acknowledgments
The authors would like to thank the contributions of Christopher Tonkin from the Biogen Idec sequencing group, Marie Hardwick from the Johns Hopkins School of Public Health and George Oyler. M.J.B.'s contribution was supported in part from grant 1RO1GM095685 from NIGMS.
References
- Aharoni A., Gaidukov L., Yagur S., Toker L., Silman I., Tawfik D.S. Proc. Natl Acad. Sci. USA. 2004;101:482–487. doi: 10.1073/pnas.2536901100. First published on 2003/12/30, doi: 10.1073/pnas.2536901100; 2536901100 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M.K., Davison S., Siddiqui N., Norton J.D., Murphy J.J. Eur. J. Immunol. 1997;27:3485–3491. doi: 10.1002/eji.1830271249. First published on 1998/02/17, doi: 10.1002/eji.1830271249. [DOI] [PubMed] [Google Scholar]
- Arakawa H., Buerstedde J.M. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:639–644. doi: 10.1098/rstb.2008.0202. First published on 2008/11/15, doi: 95780M5276381811 [pii]; 10.1098/rstb.2008.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H., Kudo H., Batrak V., Caldwell R.B., Rieger M.A., Ellwart J.W., Buerstedde J.M. Nucleic Acids Res. 2008;36:e1. doi: 10.1093/nar/gkm616. First published on 2007/12/13, doi: gkm616 [pii]; 10.1093/nar/gkm616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachl J., Carlson C., Gray-Schopfer V., Dessing M., Olsson C. J. Immunol. 2001;166:5051–5057. doi: 10.4049/jimmunol.166.8.5051. First published on 2001/04/06. [DOI] [PubMed] [Google Scholar]
- Blagodatski A., Katanaev V.L. Cell Mol. Life Sci. 2011;68:1207–1214. doi: 10.1007/s00018-010-0610-5. First published on 2010/12/31, doi: 10.1007/s00018-010-0610-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng E.H., Kirsch D.G., Clem R.J., Ravi R., Kastan M.B., Bedi A., Ueno K., Hardwick J.M. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. First published on 1998/01/07. [DOI] [PubMed] [Google Scholar]
- Chiang G.G., Sisk W.P. Biotechnol. Bioeng. 2005;91:779–792. doi: 10.1002/bit.20551. First published on 2005/06/30, doi: 10.1002/bit.20551. [DOI] [PubMed] [Google Scholar]
- Chu F., Borthakur A., Sun X., Barkinge J., Gudi R., Hawkins S., Prasad K.V. Apoptosis. 2004;9:83–95. doi: 10.1023/B:APPT.0000012125.01799.4c. First published on 2004/01/24, doi: 10.1023/B:APPT.0000012125.01799.4c; 5256690 [pii] [DOI] [PubMed] [Google Scholar]
- Clem R.J., Cheng E.H., Karp C.L., Kirsch D.G., Ueno K., Takahashi A., Kastan M.B., Griffin D.E., Earnshaw W.C., Veliuona M.A., et al. Proc. Natl. Acad. Sci. USA. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. First published on 1998/01/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost G.J., Freyvert Y., Vafiadis A., Santiago Y., Miller J.C., Rebar E., Collingwood T.N., Snowden A., Gregory P.D. Biotechnol. Bioeng. 2010;105:330–340. doi: 10.1002/bit.22541. First published on 2009/09/25, doi: 10.1002/bit.22541. [DOI] [PubMed] [Google Scholar]
- Cumbers S.J., Williams G.T., Davies S.L., Grenfell R.L., Takeda S., Batista F.D., Sale J.E., Neuberger M.S. Nat. Biotechnol. 2002;20:1129–1134. doi: 10.1038/nbt752. First published on 2002/10/16, doi: 10.1038/nbt752; nbt752 [pii] [DOI] [PubMed] [Google Scholar]
- Faili A., Aoufouchi S., Flatter E., Gueranger Q., Reynaud C.A., Weill J.C. Nature. 2002;419:944–947. doi: 10.1038/nature01117. First published on 2002/11/01, doi: 10.1038/nature01117; nature01117 [pii] [DOI] [PubMed] [Google Scholar]
- Figueroa B., Jr, Sauerwald T.M., Mastrangelo A.J., Hardwick J.M., Betenbaugh M.J. Biotechnol. Bioeng. 2001;73:211–222. doi: 10.1002/bit.1053. First published on 2001/03/21, doi: 10.1002/bit.1053 [pii] [DOI] [PubMed] [Google Scholar]
- Figueroa B., Jr, Sauerwald T.M., Oyler G.A., Hardwick J.M., Betenbaugh M.J. Metab. Eng. 2003;5:230–245. doi: 10.1016/s1096-7176(03)00044-2. First published on 2003/12/04, doi: S1096717603000442 [pii] [DOI] [PubMed] [Google Scholar]
- Figueroa B., Jr, Chen S., Oyler G.A., Hardwick J.M., Betenbaugh M.J. Biotechnol. Bioeng. 2004;85:589–600. doi: 10.1002/bit.10913. First published on. [DOI] [PubMed] [Google Scholar]
- Guntas G., Mansell T.J., Kim J.R., Ostermeier M. Proc. Natl Acad. Sci. USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. First published on 2005/08/03, doi: 0502673102 [pii]; 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S.Y., Gaume B., Lee Y.J., Hsu Y.T., Ryu S.W., Yoon S.H., Youle R.J. EMBO J. 2004;23:2146–2155. doi: 10.1038/sj.emboj.7600225. First published on 2004/05/08, doi: 10.1038/sj.emboj.7600225; 7600225 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama N., Todo K., Takahashi S., Magari M., Ohmori H. Nucleic Acids Res. 2006;34:e10. doi: 10.1093/nar/gnj013. First published on 2006/01/20, doi: 34/2/e10 [pii]; 10.1093/nar/gnj013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.G., Kim J.Y., Mohan C., Lee G.M. Biotechnol. Bioeng. 2009;103:757–766. doi: 10.1002/bit.22298. First published on 2009/03/07, doi: 10.1002/bit.22298. [DOI] [PubMed] [Google Scholar]
- Kauffmann I., Schmidt-Dannert C. Pro. Eng. 2001;14:919–928. doi: 10.1093/protein/14.11.919. doi:10.1093/protein/14.11.919. [DOI] [PubMed] [Google Scholar]
- Kuchner O., Arnold F.H. Trends Biotechnol. 1997;15:523–530. doi: 10.1016/S0167-7799(97)01138-4. First published on 1998/01/07, doi: S0167-7799(97)01138-4 [pii]; 10.1016/S0167-7799(97)01138-4. [DOI] [PubMed] [Google Scholar]
- Kumar S., Chen C.S., Waxman D.J., Halpert J.R. J. Biol. Chem. 2005;280:19569–19575. doi: 10.1074/jbc.M500158200. First published on 2005/03/19, doi: M500158200 [pii]; 10.1074/jbc.M500158200. [DOI] [PubMed] [Google Scholar]
- Lehmann M., Wyss M. Curr. Opin. Biotechnol. 2001;12:371–375. doi: 10.1016/s0958-1669(00)00229-9. First published on 2001/09/12, doi: S0958-1669(00)00229-9 [pii] [DOI] [PubMed] [Google Scholar]
- Liebeton K., Zonta A., Schimossek K., Nardini M., Lang D., Dijkstra B.W., Reetz M.T., Jaeger K.E. Chem. Biol. 2000;7:709–718. doi: 10.1016/s1074-5521(00)00015-6. First published on 2000/09/12, doi: S1074-5521(00)00015-6 [pii] [DOI] [PubMed] [Google Scholar]
- Majors B.S., Betenbaugh M.J., Chiang G.G. Metab. Eng. 2007;9:317–326. doi: 10.1016/j.ymben.2007.05.003. First published on 2007/07/06, doi: S1096-7176(07)00030-4 [pii]; 10.1016/j.ymben.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Majors B.S., Betenbaugh M.J., Pederson N.E., Chiang G.G. Biotechnol. Bioeng. 2008;101:567–578. doi: 10.1002/bit.21917. First published on 2008/08/30, doi: 10.1002/bit.21917. [DOI] [PubMed] [Google Scholar]
- Majors B.S., Betenbaugh M.J., Pederson N.E., Chiang G.G. Biotechnol. Prog. 2009a;25:1161–1168. doi: 10.1002/btpr.192. First published on 2009/06/25, doi: 10.1002/btpr.192. [DOI] [PubMed] [Google Scholar]
- Majors B.S., Chiang G.G., Betenbaugh M.J. Mol. Biotechnol. 2009b;42:216–223. doi: 10.1007/s12033-009-9156-x. First published on 2009/04/16, doi: 10.1007/s12033-009-9156-x. [DOI] [PubMed] [Google Scholar]
- Mastrangelo A.J., Zou S., Hardwick J.M., Betenbaugh M.J. Biotechnol. Bioeng. 1999;65:298–305. First published on 1999/09/15, doi: 10.1002/(SICI)1097-0290(19991105)65:3<298::AID-BIT7>3.0.CO;2-S [pii] [PubMed] [Google Scholar]
- Meents H., Enenkel B., Eppenberger H.M., Werner R.G., Fussenegger M. Biotechnol. Bioeng. 2002;80:706–716. doi: 10.1002/bit.10449. First published on 2002/10/16, doi: 10.1002/bit.10449. [DOI] [PubMed] [Google Scholar]
- Miyazaki K., Wintrode P.L., Grayling R.A., Rubingh D.N., Arnold F.H. J. Mol. Biol. 2000;297:1015–1026. doi: 10.1006/jmbi.2000.3612. First published on 2000/03/29, doi: 10.1006/jmbi.2000.3612; S0022-2836(00)93612-X [pii] [DOI] [PubMed] [Google Scholar]
- Moriishi K., Koura M., Matsuura Y. Virology. 2002;292:258–271. doi: 10.1006/viro.2001.1206. First published on 2002/03/07, doi: 10.1006/viro.2001.1206; S0042682 201912061 [pii] [DOI] [PubMed] [Google Scholar]
- Nava V.E., Rosen A., Veliuona M.A., Clem R.J., Levine B., Hardwick J.M. J. Virol. 1998;72:452–459. doi: 10.1128/jvi.72.1.452-459.1998. First published on 1998/01/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier M. Protein Eng. Des. Sel. 2005;18:359–364. doi: 10.1093/protein/gzi048. First published on 2005/07/27, doi: gzi048 [pii]; 10.1093/protein/gzi048. [DOI] [PubMed] [Google Scholar]
- Parsa J.Y., Basit W., Wang C.L., Gommerman J.L., Carlyle J.R., Martin A. Mol. Immunol. 2007;44:567–575. doi: 10.1016/j.molimm.2006.02.003. First published on 2006/03/18, doi: S0161-5890(06)00031-9 [pii]; 10.1016/j.molimm.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Perez-Duran P., de Yebenes V.G., Ramiro A.R. Carcinogenesis. 2007;28:2427–2433. doi: 10.1093/carcin/bgm201. First published on 2007/09/07, doi: bgm201 [pii]; 10.1093/carcin/bgm201. [DOI] [PubMed] [Google Scholar]
- Poppe M., Reimertz C., Dussmann H., Krohn A.J., Luetjens C.M., Bockelmann D., Nieminen A.L., Kogel D., Prehn J.H. J. Neurosci. 2001;21:4551–4563. doi: 10.1523/JNEUROSCI.21-13-04551.2001. First published on 2001/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A.G., Janicke R.U. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. First published on 1999/04/14, doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Seki M., Gearhart P.J., Wood R.D. EMBO Rep. 2005;6:1143–1148. doi: 10.1038/sj.embor.7400582. First published on 2005/12/02, doi: 7400582 [pii]; 10.1038/sj.embor.7400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S., Shinohara Y., Tsujimoto Y. Oncogene. 2000;19:4309–4318. doi: 10.1038/sj.onc.1203788. First published on 2000/09/12, doi: 10.1038/sj.onc.1203788. [DOI] [PubMed] [Google Scholar]
- Takehara T., Liu X., Fujimoto J., Friedman S.L., Takahashi H. Hepatology. 2001;34:55–61. doi: 10.1053/jhep.2001.25387. First published on 2001/06/30, doi: S0270-9139(01)57355-3 [pii]; 10.1053/jhep.2001.25387. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. First published on 1997/12/11, doi: S0092-8674(00)80450-X [pii] [DOI] [PubMed] [Google Scholar]
- Wang C.L., Yang D.C., Wabl M. Protein Eng. Des. Sel. 2004a;17:659–664. doi: 10.1093/protein/gzh080. First published on 2004/11/02, doi: gzh080 [pii]; 10.1093/protein/gzh080. [DOI] [PubMed] [Google Scholar]
- Wang L., Jackson W.C., Steinbach P.A., Tsien R.Y. Proc. Natl Acad. Sci. USA. 2004b;101:16745–16749. doi: 10.1073/pnas.0407752101. First published on 2004/11/24, doi: 0407752101 [pii]; 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Tsien R.Y. Nat. Protoc. 2006;1:1346–1350. doi: 10.1038/nprot.2006.243. First published on 2007/04/05, doi: nprot.2006.243 [pii]; 10.1038/nprot.2006.243. [DOI] [PubMed] [Google Scholar]
- White C., Li C., Yang J., Petrenko N.B., Madesh M., Thompson C.B., Foskett J.K. Nat. Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. First published on 2005/09/24, doi: ncb1302 [pii]; 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias D.A., Tsien R.Y. Methods Biochem. Anal. 2006;47:83–120. First published on 2005/12/13. [PubMed] [Google Scholar]