

Abstract
Sox2, which encodes an SRY-like HMG box transcription factor, is critical for vertebrate development. Sox2 mediates its transcriptional effects through the formation of complexes with specific co-factors, many of which are unknown. In this report, we identify Oct-1, encoded by the Pou2f1 gene, as a co-factor for Sox2 in the context of mouse lens and nasal placode induction. Oct-1, Sox2, and Pax6 are co-expressed during lens and nasal placode induction and during subsequent developmental stages. Genetic combination of Sox2 and Pou2f1 mutant alleles results in impaired induction of the lens placode, an ocular phenotype that includes anophthalmia, and a complete failure of nasal placode induction. These ocular and nasal phenotypes closely resemble those observed in Pax6 null embryos. Moreover, we identify DNA binding sites that support the cooperative formation of a complex between Sox2 and Oct-1 and mediate Sox2/Oct-1-dependent transactivation of the Pax6 lens ectoderm enhancer in vitro. We demonstrate that the same Sox and Octamer binding sites are essential for Pax6 enhancer activity in the lens placode and its derivatives in transgenic mouse embryos. Collectively, these results indicate that Pou2f1, Sox2 and Pax6 are interdependent components of a molecular pathway utilized in both lens and nasal placode induction.
Keywords: Sox2, Pax6, Pou2f1, Oct-1, lens placode, nasal placode, pre-placode, E-cadherin, cataract
INTRODUCTION
Vertebrate sensory structures, including the lens, ear and nose, develop through multistep inductive processes that involve the formation of a neural plate stage pre-placodal region (PPR) and the subsequent formation of discrete ectodermal thickenings called placodes (reviewed in Streit, 2004, Bhattacharyya and Bronner-Fraser, 2004; Brugmann and Moody, 2005; Schlosser, 2006). Induction of the PPR in chick and Xenopus is dependent upon Fgf signaling and attenuation of Wnt and Bmp signaling (Litsiou et al., 2005; Ahrens and Schlosser 2005). Similarly, Fgf signals are required for the induction of the individual sensory placodes (Faber et al., 2001; Phillips et al., 2004; Martin and Groves, 2005; Ladher et al., 2005; Bailey et al., 2006), while Wnt signaling is required for appropriate placode placement (Smith et al., 2005; Ohyama et al., 2006). In contrast to PPR induction, Bmp signals play a positive role in the induction of the mouse lens placode (Furuta and Hogan, 1998; Wawersik et al., 1999). Several transcription factor families including Six, Eya, Pax, Pitx, Msx and Sox are expressed in and important for the formation of all placodes (reviewed in Streit, 2004, Bhattacharyya and Bronner-Fraser, 2004; Brugmann and Moody, 2005; Schlosser, 2006). Thus, while many molecules critical for sensory placode induction have been identified, detailed molecular mechanisms that control different stages of placode induction, particularly in mammals, are still not fully understood.
Sox2, a Group B1 SRY-related HMG box containing transcription factor, has emerged as an important factor in vertebrate sensory development (Dong et al., 2002; Fantes et al., 2003; Kiernan et al., 2005; Ragge et al., 2005; Hagstrom et al., 2005; Taranova et al., 2006). While Sox2 is not expressed in the early PPR (Wood and Episkopou, 1999), it is expressed in the distinct lens, nasal, and otic placodes (Kamachi et al., 1998; Wood and Episkopou, 1999). Disruption of Sox2 expression in the mouse otocyst disrupts formation of the ear prosensory domain and severely disrupts inner ear development (Kiernan et al., 2005). In the chick lens, Sox2 cooperates with Pax6 to regulate crystallin gene expression (Kamachi et al., 2001), and mutation of SOX2 in humans results in severe eye phenotypes ranging from microphthalmia to anophthalmia (Fantes et al., 2003; Ragge et al., 2005; Zenteno et al., 2005; Hagstrom et al., 2005).
As with other Sox family members, Sox2 requires co-factors to mediate transcriptional control over its targets (reviewed in Kamachi et al., 2000). Two Sox2 co-factor families have been identified, the paired-type homeodomain containing family (Pax), and members of the POU family (Ambrosetti et al., 1997; Botquin et al., 1998; Nishimoto et al., 1999; Ma et al., 2000; Kamachi et al., 2001; DiRocco et al., 2001; Aota et al., 2003; Tanaka et al., 2004; Rodda et al., 2005; Okumura-Nakanishi et al., 2005). In the context of placode formation, however, the cofactors that interact with Sox2 are unknown.
Here, we identify Oct-1 as a Sox2 co-factor in the context of murine lens and nasal placode induction. We demonstrate that Oct-1, Sox2 and Pax6 are co-expressed in cells of the nasal and lens placodes, and that compound mutations in Pou2f1 and Sox2 disrupt lens and nasal morphogenesis while isolated mutation of Pou2f1 or Sox2 does not. We provide in vitro biochemical evidence that Sox2 and Oct-1 synergistically activate expression of Pax6 by cooperatively binding to the Pax6 lens ectoderm enhancer (EE). Furthermore, we show that the same Sox and Octamer DNA binding sites are required for Pax6 lens enhancer activity during embryogenesis in transgenic mouse models. Thus, compromised Pax6 expression likely accounts for the failure of lens and nasal placode induction in Oct-1−/−, Sox2+/− embryos. These data are consistent with common molecular mechanisms controlling Pax6 expression during lens and nasal placode induction.
MATERIALS AND METHODS
Cell culture and RT PCR
The rabbit lens epithelial cell lines B3, LEP2, and N/N1003A were cultured in DMEM supplemented with 10% rabbit serum and penicillin/streptomycin. Cells were harvested and total RNA isolated utilizing Qiagen RNeasy® Kits according to the manufacturer’s instructions. RNA from E13.5 eyes was screened for Pou2 genes using the degenerate primers POU2FF 5′-AGTTYGCYMRSACYTTCAARCA and POU2FR 5′-GTCRTTKCCATABARYTTBCCC. Specific primers for Sox2 (SOX2LRT 5′-CACAACTCGGAGATCAGCAA and SOX2RRT 5′-CTCCGGGAAGCGTGTACTTA); Pax6 (PAX6LRT 5′-GCAGATGCAAAAGTCCAGGT and PAX6RRT 5′-TTCCCAAGCAAAGATGGAAG; Pou2f1/Oct-1 (POU2F1 5′-GCTCTTGCTTCTAGTGGCTCT and POU2R1 5′-TGAAACTCTTCTCTAAGGCC); Pou2f2/Oct-2 (OCT2F 5′-CAAGCCTACCCAGCCCAAAC and OCT2R 5′-GAAGCGGACATTCGTCTCGA); and Pou5f1/Oct-3 (POU5FF 5′-GAGTCCCAGGACATGAAAGC and POU5FR 5′-AGATGGTGGTCTGGCTGAAC) were utilized for RT-PCR on RNA from lens epithelial cell cultures. RT-PCR was performed using Superscript™ One-Step RT-PCR with Platinum® Taq according to the manufacturer’s instructions (Invitrogen).
Mouse lens epithelial cells (17EM15, 21EM1, αTN4) and 293t cells were grown in DMEM supplemented with 10% fetal calf serum and penicillin/streptomycin. Cells were transfected at 70% confluence with FuGENE 6 (Roche) in 12 well plates. Sox2 (Ambrosetti et al., 1997) (100ng, 200ng, 400ng) and Oct-1 (Tanaka and Herr, 1990) (200ng, 400ng, 800ng) expressing plasmids were titrated in the presence of pGL2 (Promega) or pGL109, which contains the 109 base pair minimal element of the Pax6 EE upstream of the luciferase reporter gene in pGL2. pcDNA3 (Invitrogen) was added to standardize the concentration of DNA in each experiment. 50 ng of phRL-CMV (Promega) was included for standardization of the transfection efficiency. Cell lysates were assayed for firefly and renilla luciferase activity on a Veritas Microplate Luminometer using the Dual-Luciferase Reporter System (Promega). Each experiment was performed in triplicate and the data illustrated are the average of at least three experiments. Fold induction is the ratio of luciferase activity detected in experiments containing pGL109 or its derivatives versus comparable experiments with pGL2.
DNA Constructs
The plasmid pETSox2-HMG was constructed by PCR amplification of the HMG domain from Sox2, from pCMVSox2 (Ambrosetti et al., 1997). The PCR product was subcloned into pETBlue-1 (Novagen). Oct1-GST was generated by PCR amplification of the POU domain (POUS-linker-POUHD) from pCGOct-1 (Tanaka and Herr, 1990) and subcloning into pGEX-3X (Amersham Pharmacia) in frame with GST. Sox and POU mutant transgenic constructs were generated by PCR mutagenesis of pLNGLKS (Zhang et al., 2002) using the Quick Change™ Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer’s instructions.
Mouse genetics
All mouse work was performed in accordance with protocols approved by the Harvard Animal Care and Usage Committee. Oct-1+/− mice were maintained on a C57BL/6 background (Wang et al., 2004), while Sox2βgeo2 and Pax61-NeuSey mice were maintained on a C3H/HeN background (Ekonomou et al., 2005). Sox2βgeo/+ mice were interbred with Oct-1+/− mice to generate Oct-1/Sox2 compound mutants. Double heterozygous offspring from these matings were interbred. Appropriately staged embryos and their extra-embryonic tissues were collected, and genomic DNA from extra-embryonic tissue was screened by PCR (Nishiguchi et al., 1998; Wang et al., 2004; Ekonomou et al., 2005). The Pax61-NeuSey allele was identified by a PCR fragment polymorphism producing a HincII restriction site.
Transgenic mice were generated by standard methods (Nagy et al., 2003). Staged embryos were collected and stained for β-galactosidase activity (Nagy et al., 2003). Extra-embryonic tissue from transgenic embryos was processed for genomic DNA preparation (Nagy et al., 2003). DNA was genotyped by PCR with primers specific to the LacZ gene. β-galactosidase positive embryos were dehydrated, embedded in wax, and sectioned (10 μM, transverse plane) with a microtome. Sections were counterstained briefly with hematoxylin or eosin, mounted, and photographed on a Zeiss Axiophot microscope with a Leica DCF300 digital camera.
Immunofluorescence and In Situ Hybridization
For all paraffin processing, embryos were fixed overnight in 4% paraformaldehyde (PFA), washed in 1X PBS, dehydrated, and embedded in wax. 8–10 μM transverse sections were cut on a microtome. For all frozen sections, embryos were fixed for 1 hour in 4% PFA, equilibrated in 30% sucrose, and frozen in OCT (Tissue-Tek). 10 μm transverse sections were cut on a cryostat (Leica CM1900).
Pax6 immunofluorescence was performed on frozen sections as described (Purcell et al., 2005). Antibody staining for Sox2 (Chemicon International, 1:500), Oct-1 (Santa Cruz, 1:200), and E-cadherin (Zymed®, 1:1000) were performed on frozen sections. Detection of Pax6, Oct-1, and Sox2 was enhanced by boiling in VECTOR® Antigen Unmasking Solution. (For data on the specificity of the Oct-1 and E-cadherin antibodies, see Fig. S3). Indirect visualization for all immunolocalization was achieved using secondary antibodies with fluorescent conjugates. These antibodies included anti-rabbit, anti-mouse, anti-rat Cy3 (Jackson Immunologicals, 1:500) and anti-rabbit Alexa-Fluor 488 (Molecular Probes, 1:700). All fluorescent sections were mounted with Vectashield® plus DAPI (Vector Laboratories) and visualized on a Zeiss Axiophot microscope. A Leica DFC350F Digital Camera was used to record images.
In situ hybridization using DIG-labeled RNA probes was performed using standard procedures (Nagy et al., 2003). Alkaline phosphatase activity was detected with the substrate NBT/BCIP. Post-fixed embryos and sections were photographed using a Leica DFC300 Digital Camera.
Protein purification and EMSAs
The Sox2-HMG domain and Oct1-GST were purified from E. coli as described (Van Houte et al., 1995; Klemm et al., 1994). Each recombinant protein was greater than 90% pure on Coomassie blue stained gels. EMSAs were performed by pre-incubating pure proteins with cold, competitor oligonucleotides or antibody in 20 mM HEPES, pH 7.8; 10 mM EDTA, pH 8.0; 1 mM DTT; 2 mM MgCl2; 50 mM NaCl; 5% glycerol; and 50 ng/μL poly dG-dC. After addition of radiolabeled probes, reactions were loaded onto a running 8% native PAGE gel at 4°C. After resolution, gels were dried, exposed to film (Kodak XAR), and developed using an X-OMAT film processor. Titration of Sox2-HMG onto EE-3 began at 63 nM and increased in 1.5-fold concentration steps. For cooperative binding studies, the initial concentration of Oct1-GST was 15 nM and increased 1.2-fold in each subsequent lane. The concentration of Sox2-HMG was 328 nM. For IC50 ratio calculations, Sox2-HMG (328 nM) was incubated with cold competitor and radiolabeled EE-3 at ratios ranging from 0.1X to 100X. For IC50 ratio and cooperativity calculations, gels were analyzed using a Phosphor Imager (Molecular Dynamics). (For representative EMSAs see Fig. S6).
RESULTS
Sox2, Pax6, and Pou2f1 are co-expressed in developing lens and nasal placodes
To identify potential POU family co-factors for Sox2 in the lens, we performed RT-PCR on total RNA collected from E13.5 wild-type eyes and from several lens epithelial cell lines (Fig. S1). This screen identified Pou2f1, which encodes the Oct-1 protein, as a candidate POU cofactor for Sox2 in the lens. Interestingly, Pou5f1, which encodes Oct-3, a known Sox2 interacting factor (Ambrosetti et al., 1997) was not detected in lens epithelial cells (Fig. S1).
To determine the in vivo relationship amongst Sox2, Pax6 and Pou2f1, we examined their expression at pre-placodal stages. Pax6, Sox2, and Pou2f1 were broadly expressed in the E8.5 embryonic head (Fig. 1A–F). Upon cross section, Pou2f1 expression was detected in neural ectoderm (Fig. 1E′) and resembled that reported for both Pax6 and Sox2 at this time (Grindley et al., 1995; Wood and Episkopou, 1999).
FIG. 1.

Pax6, Sox2, and Pou2f1 are broadly co-expressed in anterior pre-placode stage E8.5 embryos. Whole mount in situ analysis of Pax6 (A–B), Sox2 (C–D) and Pou2f1 (E–F) in E8.5 embryos displayed in a frontal view (A, C, E), lateral view (B, D, F), or in cross section (E′). The white line in E indicates plane of section for E′. Abb: nf neural folds; ov optic vesicle; se surface ectoderm. Scale bars: 100 microns in all figures.
To definitely determine the relative onset of Pax6, Sox2 and Oct-1 expression in the presumptive lens ectoderm (PLE), we performed immunofluorescence on mouse embryos staged between 12- and 17-somites (E8.5). Pax6 protein was already observed in the PLE at the 12-somite stage and its expression was maintained in all subsequent stages (Fig. 2A–C). Sox2, on the other hand, was not detected at the 12-somite stage in the PLE (Fig. 2D). Sox2 protein was first detected in the PLE at the 15-somite stage and was maintained through subsequent pre-placodal stages (Fig. 2E–F). Similar to Sox2, Oct-1 was not detected in the PLE at the 12-somite stage, but was expressed by the 15-somite stage and maintained through subsequent stages (Fig. 2G–I). Thus, Pax6 expression precedes both Sox2 and Oct-1 in the PLE.
FIG. 2.

Pax6 precedes Sox2 and Oct-1 in the PLE. Immunofluorescence for Pax6 (red, A–C), Sox2 (green, D–F), and Oct-1 (red, G–I) are shown for embryos at the 12-somite (s) (A, D, G), 15s (B, E, H), or 17s stages (C, F, I). Arrows highlight nuclei positive for Sox2 or Oct-1 expression in the PLE. Abb: OV optic vesicle; PLE presumptive lens epithelium.
We next evaluated Pax6, Sox2 and Oct-1 protein expression in the lens placode and during early lens differentiation (Fig. 3A–I). At E9.5, Sox2 and Pax6 were strongly co-expressed in the lens placode (Fig. 3A–C), while Oct-1 protein was expressed in the ocular and peri-ocular region including the lens placode (Fig. 3G). Sox2 and Pax6 remained co-expressed throughout placode invagination and lens vesicle formation (Fig. 3D–F) until Sox2 was down-regulated at E12.5 (Nishiguchi et al., 1998; Kamachi et al., 1998). At E11.5, Oct-1 expression was excluded from the peri-ocular region but maintained in the lens vesicle (Fig. 3H). At E13.5, Oct-1 expression was down-regulated in lens fiber cell nuclei but maintained in the anterior epithelial layer (AEL) (Fig. 3I). Thus, Pax6, Sox2 and Oct-1 are expressed in the developing lens until epithelial and fiber cells differentiate. Upon differentiation, Sox2 expression is down-regulated throughout the lens, while Oct-1 and Pax6 expression are maintained in the AEL and down-regulated in the fiber cells of the posterior lens.
FIG. 3.

Sox2, Pax6 and Oct-1 are co-expressed during lens placode induction and early eye morphogenesis. Immunofluorescence and nuclear co-localization (DAPI-blue) of Pax6 (red, A, C, D, F), Sox2 (green, B–C, E–F), and Oct-1 (red, G–I) at E9.5 (A–C, G), E11.5 (D–F, H) and E13.5 (I) in the ocular region. (For Oct-1 antibody controls, see Fig. S3). Abb: ael anterior epithelial layer; fc fiber cell compartment; lp lens placode; lv lens vesicle; nr neural retina; ov optic vesicle.
We also evaluated the expression of Sox2, Oct-1 and Pax6 proteins during nasal placode induction and early nasal morphogenesis (Fig. 4A–L). Pax6, Sox2, and Oct-1 were all expressed in the nasal placode at E9.0 (Fig. 4A–C). Thereafter, all three proteins continued to be expressed in most cells of the nasal pit (Fig. 4D–F) and in early derivatives of the placode, including the nasal epithelium and the developing vomero nasal organ (VNO) (Fig. 4G–L). Thus, similar to the lens, Pax6, Oct-1 and Sox2 are co-expressed in the nasal placode, during nasal pit formation and in early nasal placode derivatives.
FIG. 4.

Sox2, Pax6 and Oct-1 are co-expressed in the nasal placode, nasal epithelium and vomero nasal organ (VNO). Immunofluorescence and nuclear co-localization (DAPI, blue, D–F) of Pax6 (red, A, D) (green G, J), Sox2 (green, B, E, H, K), and Oct-1 (red, C, F, I, L) at E9.0 (AC), E10.5 (D–F) and E12.5 (G–L). Abb: ne nasal epithelium; np nasal placode; npt nasal pit; p-lp pre-lens placode; vno vomero nasal organ.
Pou2f1 and Sox2 loss-of-function alleles interact genetically in lens development
To determine if Sox2 affects lens placode induction and early lens morphogenesis in cooperation with either Pax6 or Oct-1, we genetically compounded Pax6Sey/+ and Sox2βgeo2/+ mice and Oct1+/− and Sox2βgeo2/+ mice (Wang et al., 2004; Ekonomou et al., 2005). Early embryonic lethality in Sox2 null mice precluded our assessment of lens placode induction in the Sox2 null state (Avilion et al., 2003). Sox2+/−, Pax6Sey/+ compound embryos, from E9.5 to E13.5, had patterns of Pax6 lens expression that were indistinguishable from those in Sox2+/− or Pax6Sey/+ lenses (Fig. S2). While lens dysmorphology due to reduced Pax6 gene dosage was evident in both Pax6Sey/+ and Sox2+/−, Pax6Sey/+ lenses from E11.5, Sox2+/−, Pax6Sey/+ lenses were no more severely affected than Pax6Sey/+ lenses (Fig. S2). Thus, superposition of Sox2βgeo2 heterozygosity does not exacerbate the Pax6Sey/+ lens phenotype. While Sox2 and Pax6 are co-expressed in the PLE from the 15-somite stage through early lens morphogenesis, no genetic interaction between Pax61-NeuSey and Sox2βgeo2 alleles was detected.
Since Oct-1 expression coincides with that of Sox2, we next tested whether Sox2 and Pou2f1 are important for lens placode induction and early lens morphogenesis by intercrossing Oct1+/− (Wang et al., 2004) and Sox2βgeo2/+ mice (Ekonomou et al., 2005). From E9.5 to E11.5, Oct-1+/−, Sox2+/− embryos exhibited grossly normal lens morphology and normal patterns of Pax6 expression (Fig. S2). However, unlike either Sox2+/− or Oct-1+/− mice, 100% (n=9) of Oct-1+/−, Sox2+/− double heterozygotes developed cataracts by 6 weeks of age (Fig. S2). Thus, similar to Pax6Sey/+ mice, Oct-1+/−, Sox2+/− compound mutant mice exhibit a discrete late defect in lens development, whereas mice heterozygous for either mutation alone do not.
Severely affected Sox2/Oct-1 compound mutants have anophthalmia
We next evaluated lens placode induction and early lens development in Oct-1−/− and Oct-1−/−, Sox2+/− embryos (Fig. 5A–I) up to E12.5, since Oct-1−/− embryos begin to die at this stage (Wang et al., 2004). At E10.5, Oct-1−/− lenses had normal morphology but were occasionally small (Fig. 5A–B), reflecting the reduced size of Oct-1−/− embryos (Wang et al., 2004). In contrast, Oct-1−/−, Sox2+/− lenses exhibited a variable phenotype that ranged from small lenses to a complete failure of lens induction (Fig. 5C, F). The variable phenotype in Oct-1−/−, Sox2+/− lens induction occurred in the same animal, and therefore did not reflect different genetic backgrounds. Mildly affected Oct-1−/−, Sox2+/− lenses resembled those of Pax6Sey/+ (Fig. 5C–D), whereas the entire peri-ocular region of severely affected Oct-1−/−, Sox2+/− embryos resembled that in Pax6Sey/Sey embryos (Fig. 5E–F). At E12.5, Oct-1+/−, Sox2+/− lenses were normal (Fig. 5G), while Oct-1−/−, Sox2+/− lenses were small and dysmorphic or absent (Fig. 5H–I).
FIG. 5.
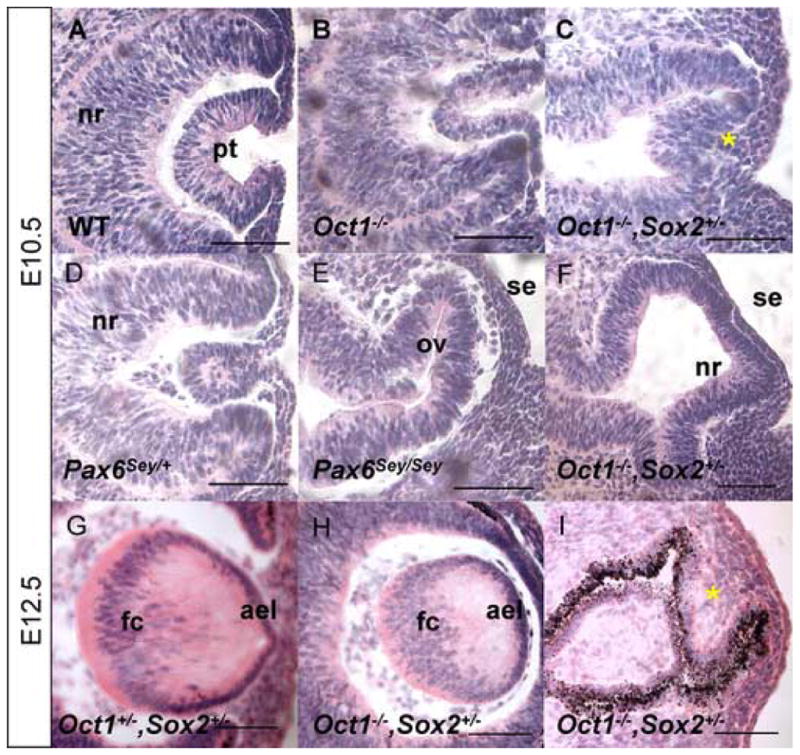
Compound Oct1−/−, Sox2+/− mutant embryos display severe ocular defects. Histological analyses of E10.5 wild type (A), Oct1−/− (B), Oct1−/−, Sox2+/− (C, F), Pax6Sey/+ (D) and Pax6Sey/Sey (E) embryos. Histological analyses of E12.5 Oct+/−, Sox2+/− (G) and Oct1−/−, Sox2+/− (H–I) embryos. * in panel C indicates lens primordium (see Fig. 6) and * in panel I indicates a lack of lens tissue (see Fig. S4 for marker analyses). Abb: ael anterior epithelial layer; fc fiber cell compartment; pt lens pit; nr neural retina; ov optic vesicle; se surface ectoderm.
Reciprocal signaling between the PLE and the optic vesicle (OV) is an integral part of early eye development and depends upon the close physical apposition of OV to PLE. The apposition of OV to PLE appears normal even in severely effected Oct-1−/−, Sox2+/− eyes (Fig. 5F), thus failed physical association between the OV with the PLE does not cause the observed anophthalmic phenotypes.
To better understand the defects in Oct-1−/−, Sox2+/− lenses, we evaluated the expression of the lens and surface ectoderm markers Pax6, E-cadherin and Sox2 (Fig. 6A–O). At E10.5, Oct-1−/− lenses had normal patterns of Pax6, E-cadherin, and Sox2 expression (Fig. 6A–C, see also Fig. S3). In contrast, Oct-1−/−, Sox2+/− embryos exhibited disrupted marker expression (Fig. 6G–L). In mildly affected eyes, a small domain positive for all three markers was observed separate from the surface ectoderm, indicating the presence of a small lens primordium (Fig. 6G–I, circles). In severely affected eyes, Pax6 and Sox2 expression were both absent from the surface ectoderm, while E-cadherin was maintained (Fig. 6J–L). These data indicate that, while the surface ectoderm was intact, lens placode induction failed.
FIG. 6.

Lens placode induction fails in severely affected Oct1−/−, Sox2+/− embryos. Immunofluorescence of E10.5 Oct1−/− (A–C), Pax6 Sey/+(D–F), Oct1−/−, Sox2+/− (G–L), and Pax6 Sey/Sey (M–O) embryos for Pax6 (green, A, D, G, J, M), E-cadherin (red, B, E, H, K, N), and Sox2 (green, C, F, I, L, O). DAPI nuclear stain is blue. Circles indicate small lenses. * in panels J and M indicate non-specific Pax6 antibody staining in embryos that have lost Pax6 expression. Arrowheads in panels L and O indicate the domain devoid of Sox2 expression in the distal OV, while the arrows in panels J–L indicate the surface ectoderm. Abb: pt lens pit; nr neural retina; ov optic vesicle; se surface ectoderm.
The ocular regions of severely affected Oct-1−/−, Sox2+/− embryos, including the lens and OV, closely resembled those of Pax6Sey/Sey embryos in both morphology and marker expression (Fig. 6J–O). Pax6 expression in the surface ectoderm was lost in both mouse models (Fig. 6J, M), and Sox2 expression was absent from the surface ectoderm and the distal OV (Fig. 6L, O, arrow heads). Thus, while lens placode induction and subsequent lens morphogenesis appear normal in Sox2+/− and Oct1−/− embryos, Oct1−/−, Sox2+/− embryos have severe defects in lens placode induction that closely resemble those in Pax6Sey/Sey mutants.
Since Oct-1 and Sox2 are both expressed in the OV and the ocular defects in Oct-1−/−, Sox2+/− embryos include altered expression of Pax6 and Sox2 in the distal OV (see above), it is possible that defects in lens placode induction can be exacerbated by defects in the OV. To evaluate the integrity of the OV we performed In Situ hybridization for Six3 (Fig. S5A–B) and secreted Frizzled-related protein 2 (sFrp2) (Fig. S5C–D), which are important for formation of the retinal anlage and influence the neural potential of the retina, respectively (Lagutin et al., 2001; Carl et al., 2002; Esteve et al., 2003; Van Raay et al., 2005). Expression of both Six3 and sFrp2 expression in the Oct-1−/−, Sox2+/− OV were maintained, while Six3 transcripts were absent from the surface ectoderm (Fig. S5B). Thus, while the Oct-1−/−, Sox2+/− mutant OV is abnormal, the expression of some genes important for retinal development is maintained.
Nasal placode induction fails in Sox2/Oct-1 compound mouse mutants
Additional evidence that Sox2 and Pou2f1 act synergistically to control placode induction was obtained by evaluating Oct-1−/−, Sox2+/− embryonic heads at E10.5. While the heads of Oct-1−/− and Sox2+/− embryos were indistinguishable from wild-type embryos (data not shown), the heads of Oct-1−/−, Sox2+/− embryos were abnormal (Fig. 7A–E). In E10.5 wild-type embryos, the ocular region and nasal pits were clearly visible (Fig. 7A, E). In contrast, Oct-1−/−, Sox2+/− embryos had no observable nasal pits and the ocular region was either not evident (Fig. 7C) or shifted towards the anterior (Figs. 7B, D–E).
FIG. 7.
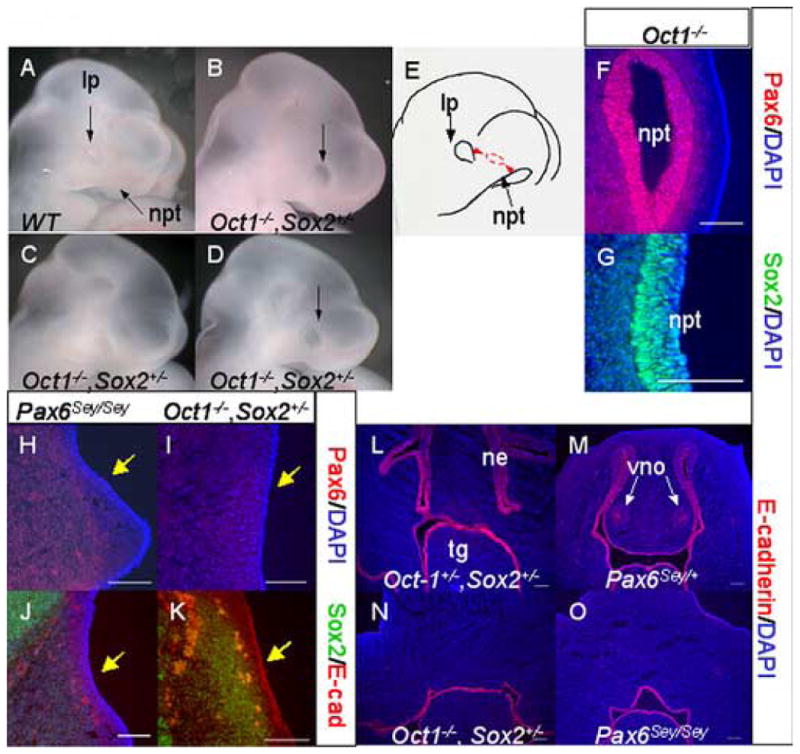
Nasal placode induction fails in Oct1−/−, Sox2+/− mutant embryos. (A–D) Photographs of the heads of E10.5 wild type (A), or Oct1−/−, Sox2+/− mutant embryos (B–D). (E) Diagram illustrating inappropriate location of a small lens placode in an Oct1−/−, Sox2+/− embryo between the normal sites of lens and nasal placode formation. Immunofluorescence for Pax6 (red, F) and Sox2 (green, G) overlaid with DAPI (blue, F–G) in Oct1−/− E10.5 nasal pits. Immuno-fluorescence for E10.5 Pax6 (red, H–I), Sox2 (green, J–K), and E-cadherin (red, J–K) in Pax6Sey/Sey (H, J) and Oct1−/−, Sox2+/− (I, K) embryos. DAPI nuclear stain is blue (H–J). (L–O) Epithelial derivatives of the nasal placode are absent at E12.5. E-cadherin (red, L–O) immunofluorescence is shown with DAPI nuclear stain for Oct1+/−, Sox2+/− (L), Pax6Sey/+ (M), Oct1−/−, Sox2+/− (N), and Pax6Sey/Sey (O) embryos. Abb: lp lens placode; ne nasal epithelium; npt nasal pit; tg tongue; vno vomero nasal organ.
Given the abnormal head and placode appearance, we evaluated nasal placode induction and early nasal morphogenesis in Oct-1−/− and Oct-1−/−, Sox2+/− embryos (Fig. 7F–O). In Oct-1−/− mice, nasal placodes were induced and Pax6 and Sox2 expression were normal (Fig. 7F–G). Unlike Oct-1−/− mice, however, Oct-1−/−, Sox2+/− embryos had neither detectable nasal placodes nor detectable Pax6 expression (Fig. 7I). Sox2 expression was also lost in Oct-1−/−, Sox2+/− embryos (Fig. 7K). The lack of nasal placodes combined with the loss of Pax6 and Sox2 expression were similarly observed in Pax6Sey/Sey embryos (Fig. 7H, J). Thus, the nasal ectoderm of Oct-1−/−, Sox2+/− embryos resembles that of Pax6Sey/Sey embryos. Unlike the variable phenotype observed in the lens placodes of Oct-1−/−, Sox2+/− embryos, however, nasal placodes were never detected.
To confirm that nasal placode induction completely failed, we assessed Oct-1−/−, Sox2+/− embryos at E12.5 for nasal placode derivatives. Although E-cadherin was uniformly expressed throughout the nasal epithelium and developing VNO in Oct-1+/−, Sox2+/− and Pax6Sey/+ embryos (Fig. 7L–M), no E-cadherin expression was detected in Oct-1−/−, Sox2+/− or Pax6Sey/Sey embryos (Fig. 7N–O). Thus, similar to Pax6Sey/Sey embryos, the induction of the nasal placodes fail and all nasal placode derivatives are absent in Oct-1−/−, Sox2+/− embryos.
The Pax6 EE contains Sox2 and Oct-1 binding sites
Given the severity of the Oct-1−/−, Sox2+/− eye and nasal phenotypes, we hypothesized that Sox2 and Oct1 act cooperatively to control the expression of genes required for lens and nasal placode induction. Given the similarity between the phenotype of Oct-1−/−, Sox2+/− and Pax6Sey/Sey embryos, an obvious candidate for such regulation is Pax6 (Hogan et al., 1988; Hill et al., 1991; Glaser et al., 1994; Hanson et al., 1994; Grindley et al., 1995; Sisodiya et al., 2001). Two Pax6 lens enhancers, the ectoderm enhancer (EE) and the SIMO enhancer, have been identified (Williams et al., 1998; Kammandel et al. 1999, Kleinjan et al., 2001) (Fig. 9A). Moreover, while Aota et al. (2003) previously identified three Sox binding sites in the EE (Table 1A–B, Sox sites A–C), our analysis of the sequence in and around these Sox sites also revealed the presence of a nearly perfect octamer element (5′-ATTCAAAT-3′) which overlaps with Sox site C (Table 1A–B, Octamer).
FIG. 9.
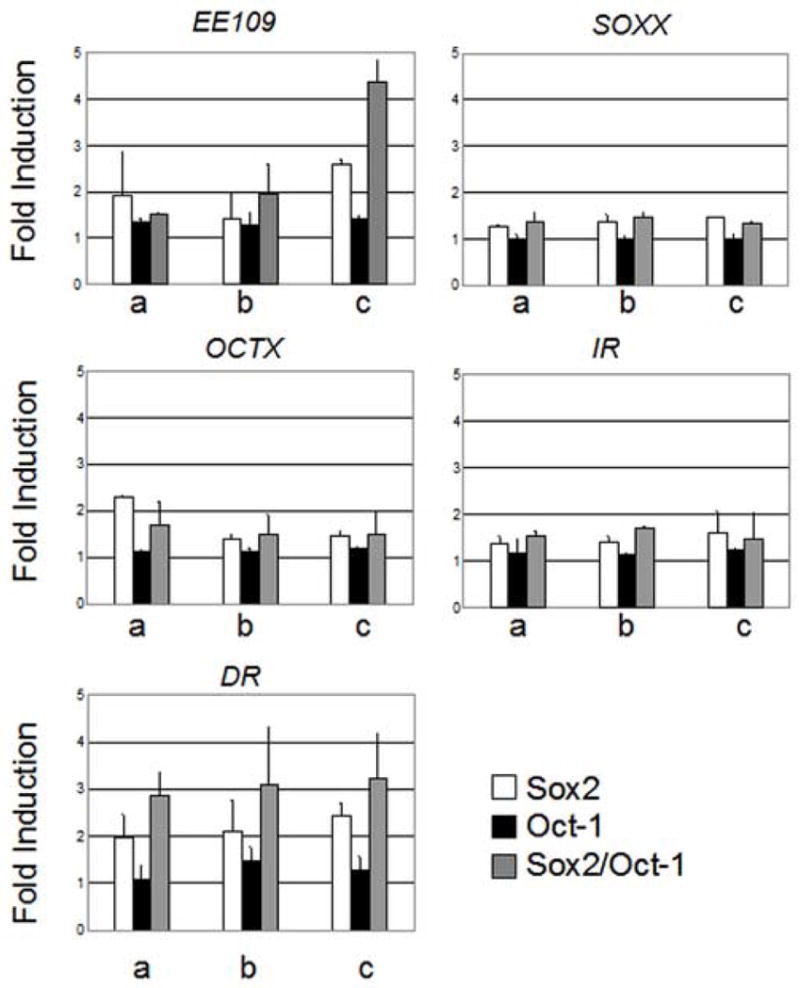
Oct-1 and Sox2 synergistically transactivate the Pax6 EE in epithelial cells. Fold induction of luciferase activity in the presence of increasing concentrations of plasmids encoding Sox2 (a 100ng, b 200ng, c 400 ng) and Oct-1 (a 200 ng, b 400 ng, c 800 ng), in isolation or together on EE109, SOXX, OCTX, IR, and DR luciferase reporter constructs in 293t epithelial cells.
Table 1.
Sequences of EE subfragments, Consensus binding sites, EMSA probes, luciferase mutants and Transgenic constructs

|
Wild-type sequence is lower case, while mutated nucleotides are upper case and bold.
identified herein
To confirm that Sox2 protein binds the EE, we purified the HMG domain of Sox2, Sox2-HMG, and titrated it onto an EE subfragment, EE-3, in EMSAs (Table 1B). These experiments demonstrated that at least two Sox HMG domains could simultaneously occupy the Sox sites in the EE, with occupation of the second binding site producing a slower migrating dimeric complex (Fig. 8B, lanes 1–16). Thus, Sox2 can directly bind multiple sites in the EE.
FIG 8.

Sox2 and Oct-1 proteins interact cooperatively with the Pax6 EE. (A) Schematic representation of Pax6 showing the two major promoters (P0, P1), the 5′ ectoderm enhancer (EE) and the 3′ SIMO lens enhancer. (B) Purified Sox2-HMG protein forms monomeric and dimeric protein-DNA complexes when titrated onto radiolabeled EE-3 (for sequence, see Table 1B) in EMSA (lanes 1–16). Purified Oct1-GST forms a protein-DNA complex with EE-3 (Oct1). This complex is diminished and super-shifted (SS) by an Oct-1 specific antibody (lanes 18–19) but unaffected by a Brn3a antibody (lane 20). (C) Titration of Oct1-GST onto EE-3 in the absence (lanes 1–8) or presence of constant amounts of Sox2-HMG protein (lanes 9–15). Shifts due to Oct1-GST binding (Oct1), Sox2-HMG (Sox2) and the co-complex (Oct1/Sox2) are indicated.
To test the ability of the Octamer element to bind Oct-1, we performed EMSAs with the POU domain of Oct-1 fused to GST, Oct1-GST (Fig. 8B, lanes 17–20). The combination of Oct1-GST and probe EE-3 resulted in the formation of a specific protein-DNA complex. The specificity of this protein-DNA interaction was demonstrated with an antibody specific to Oct-1 that produced a weak super-shift complex (SS) and largely eliminated the Oct1-GST-EE3 protein-DNA complex (Fig. 8B, lanes 18–19). In contrast, an antibody against the related POU factor, Brn3a, had no affect on protein-DNA complex formation (Fig. 8B, lane 20).
We also tested whether Sox2 and Oct-1 bound the EE cooperatively. Previous reports have demonstrated both cooperative DNA binding between Sox2-HMG and the POU domain of Oct-3 and an interaction between Sox2-HMG and the Oct-1 POU domain in the absence of DNA (Ambrosetti et al., 1997). EMSAs in which Oct1-GST was titrated onto EE-3 alone or in the presence of constant amounts Sox2-HMG were performed (Fig. 8C). The concentration of Sox2-HMG utilized was sufficient to bind the EE-3 as a monomer in the absence of Oct1-GST (Fig. 8C, lane 9). As the concentration of Oct1-GST increased in the presence of Sox2, an Oct-1/Sox2 complex of extremely slow mobility became increasingly abundant (Fig. 8C, lanes 10–15). The amount of complex formation in the presence of both Oct-1 and Sox2 exceeded the sum of the complexes formed when Oct-1 and Sox2 were incubated with EE-3 separately. Thus, in vitro, Oct-1 and Sox2 bind the EE in a positively cooperative manner.
Mutations in Sox and Octamer binding sites disrupt complex formation on the Pax6 EE
To test the importance of the Sox and Octamer DNA binding sites for the activity of the EE in vivo, we first analyzed the ability of specific EE mutants to eliminate or weaken Sox2 and Oct-1 binding in vitro. Since the Sox2 binding site in the EE is minimally a duplex site, DNA mutations that affect Sox2 binding had to be identified empirically. This was particularly critical given the overlap between Sox Site C and the Octamer element (Table 1A–B). Based on the single site in vitro selection data of Mertin et al. (1999), a series of oligonucleotides containing mutations in and around the Sox sites in EE-3 were generated (Table 1C). The simplest mutations were single nucleotide substitutions at position 4 of Sox binding sites A and B (Table 1C, SOXAM4 and SOXBM4). DM4 combined these two position 4 mutations (Table 1C), while SOXX contained two mutations in each of the same two Sox binding sites, (Table 1C). In EE-3C, the central region where the Sox sites A and B overlap was altered, while the mutant EE-3E obliterated the center of Sox Site C.
Each mutant Sox site was tested in the form of an unlabeled competitor DNA for its ability to disrupt monomeric Sox2 complex formation on wild type EE-3 probe in EMSAs. The molar ratio of cold mutant competitor to radiolabeled EE-3 at which 50% of the shifted complex was lost was defined as the IC50 ratio (Table 2: for the representative EMSAs, see Fig. S6). The number of Sox site nucleotide substitutions correlated directly with decreased competitor function in EMSA (Table 2). Mutation of Sox site C, in EE-3E, had no effect on the affinity of Sox2-HMG for the EE-3 subfragment (Table 2). This is consistent with the observation of Aota et al. (2003) that Sox site C did not contribute to Sox-dependent transactivation of the EE in cell culture. Thus, we discounted the Sox site C as a candidate Sox2 binding site.
Table 2.
IC50 ratios for Sox2-HMG and EE-3 variants
| Competitor | IC50 ratio |
|---|---|
| EE-3 | 4 |
| EE-3E | 5 |
| SOXAM4 | 48 |
| SOXBM4 | 21 |
| DM4 | 52 |
| SOXX | 41 |
| EE-3C | 56 |
We also tested the importance of sequences in the Octamer element for Oct1-GST-EE protein-DNA complex formation by mutating nucleotides necessary for Oct-1 protein-DNA interaction (Di Rocco et al., 2001) (Table 1C). As expected, mutation of either the POU-specific (POUS, OCTPM) or POU-homeodomain (POUHD, OCTHM) DNA recognition sites dramatically reduced complex formation in competitive EMSAs (Fig. S6).
Sox2 and Oct-1 synergistically transactivate the Pax6 EE
Consistent with the observations of Aota et al. (2003) and Zhang et al. (2002), the introduction of the minimal 109 base pair Pax6 EE upstream of the luciferase gene in cell culture experiments results in transcriptional repression compared to controls. We observed transcriptional repression in each of the lens epithelial cell lines tested 17EM15, 21EM1, αTN4, B3, N/N1003A, and LEP2 (data not shown). The strong repression mediated by the Pax6 EE forced the evaluation of the Pax6 and Sox2 binding sites out of context of the EE (Aota et al., 2003) and the analysis of the Meis binding site with a fusion protein containing the exogenous transcriptional activation domain from VP16 (Zhang et al., 2002). While both of these approaches were useful in providing information about the regulation of the EE, we believe they limit the value of a cell culture system in representing in vivo interactions. Since there is no cell culture model that accurately recapitulates lens placode induction, we utilized a non-lens epithelial cell line, 293t, in which EE109 is weakly repressed compared to controls (0.8-fold). This allowed us to assess transcriptional activation in the context of the endogenous enhancer and eliminated the need for a non-native transcriptional activation domain.
We transfected 293t cells with increasing concentrations of Sox2 or Oct-1 encoding plasmids, alone or in combination in the presence of pGL109 or pGL2. Sox2 and Oct-1, in isolation, elicited 2.6-fold and 1.4-fold activation of the pGL109, respectively (Fig. 9, EE109). The combination of Sox2 and Oct-1 resulted in 4.4-fold increase in luciferase activity. The increase in activation observed is consistent with Sox2 and Oct-1 having a slightly synergistic effect on Pax6 EE activity. To ensure that the observed transcriptional activation was dependent upon the Sox and Octamer elements identified herein, we utilized two mutant luciferase constructs, SOXX and OCTX (Fig 9, see also Table 1C). SOXX, derived from pGL109, has the same sequence substitutions as the SOXX mutation described for IC50 ratio calculations. OCTX is similarly derived from pGL109, but contains four base pair substitutions (two in each half-site of the Octamer element, Table 1C). Transcriptional activation in the presence of Sox2 and Oct-1, individually or in combination was compromised on both of these altered enhancers. Indeed, Sox2-dependent activation of the EE was largely abolished by mutation of either the Sox sequence (maximum activation 1.5-fold) or the Octamer element (maximum activation 1.5-fold) (Fig. 9, OCTX), indicating that Sox2-dependent activation, in the absence of any exogenous Oct-1, is dependent upon the presence of an intact Octamer element and endogenous Oct-1 or a comparable POU factor. The sum of Sox2-dependent activation and Oct-1-dependent activation was statistically significantly lower than the Sox2/Oct-1-dependent activation of the EE (p < 0.005, ttest). Thus, Sox2/Oct-1 activation of the Pax6EE through the Sox and Octamer sites is synergistic.
To determine the relative contribution of the two Sox sites (A and B) to the Oct-1/Sox2-dependent activation of the EE, we utilized two constructs, IR and DR (Fig. 9, IR and DR, see also Table 1C). IR contains mutations in Sox site A: the Octamer element and the Sox site B are intact. DR contains mutations in Sox site B such that the Octamer element and Sox site A are intact. Transcriptional activation was not observed on the IR construct (Fig. 9, IR). Sox2-dependent and Sox2/Oct-1 co-activation, however, were observed on the DR template (Fig 9, DR). These results indicate that Sox2 and Oct-1 synergistically activate the Pax6 EE by interaction with the Octamer element and Sox site A.
Mutations in Sox and Octamer DNA sites reduce the activity of the Pax6 EE in vivo
Since cell lines do not accurately recapitulate the environment of the developing lens placode or early differentiating lenses, we assessed the consequences of mutating the Sox binding sites on Pax6 EE activity in transgenic mouse embryos. The wild-type transgene used in these studies contained 526 base pairs of the Pax6 P0 upstream region, including the EE, and yielded highly efficient transgene expression that recapitulated the endogenous Pax6 expression pattern in 5 out of 5 and 10 of 14, E10.0 and E12.5 embryos, respectively (Fig. 10A–B; Table 3). In contrast, the singly mutant transgenes, SOXAM4 and SOXBM4, each exhibited a reduced efficiency of transgene expression at E12.5 (1 of 12 and 4 of 10, respectively), while SOXBM4 also exhibited an altered pattern of expression (Table 3). Specifically, activity of the EE in the presumptive cornea and glandular ectoderm was abolished in SOXBM4 transgenic embryos (Fig. 10B). Mutation of both Sox sites in the DM4 transgene essentially eliminated the activity of the EE at both E10.0 and E12.5 (Table 3); the two transgenic embryos that expressed β-galactosidase at E12.5 exhibited patchy, weak expression that was not detectable in serial cross-sections (data not shown). The transgene SOXX, with the most severe mutations in the duplex Sox sites by in vitro criteria, had no detectable in vivo activity (Table 3). Thus, the effects of the various mutations on Sox binding in vitro correlate directly with their effects on EE expression in vivo. Furthermore, the duplex Sox sites A and B are necessary for EE activity in the lens pit, the differentiating lens, the presumptive cornea and ocular glandular ectoderm.
FIG. 10.

Sox and Octamer DNA binding elements are required for the in vivo transcriptional activity of the Pax6 EE. (A) Schematic representation of transgenic constructs utilized. The Pax6 EE directs expression of LacZ through the TATA element of the human β-globin (βg) gene. (B) Whole mount photo of EE526 transgenic embryo at E10.0 and representative sections of E12.5 embryos transgenic for EE526, SOXAM4, SOXBM4, OCTPM, and OCTHM. The arrows indicate the lens at E10.0, the non-lens epithelium of EE526, SOXAM4 AND SOXBM4 sections and the AEL of OCTPM and OCTHM images.
Table 3.
β-galactosidase expression in Transgenic embryos
| Transgene | No. expressing in:
|
||||
|---|---|---|---|---|---|
| Stage | Lens | Cornea | Ectoderm | No. Tg | |
| EE526 | E10.0 | 5 | NA | NA | 5 |
| E12.5 | 10 | 10 | 10 | 14 | |
| SOXAM4 | E12.5 | 1 | 1 | 1 | 12 |
| SOXBM4 | E12.5 | 4 | 0 | 0 | 10 |
| DM4 | E10.0 | 0 | NA | NA | 2 |
| E12.5 | 2* | 1 | 0 | 8 | |
| SOXX | E10.0 | 0 | NA | NA | 4 |
| E12.5 | 0 | 0 | 0 | 23 | |
| OCTHM | E10.0 | 0 | NA | NA | 9 |
| E12.5 | 9 | 0 | 0 | 12 | |
| OCTPM | E12.5 | 1 | 0 | 0 | 3 |
| MPAX6 | E12.5 | 0 | 0 | 0 | 7 |
patchy, weak expression
NA, not applicable
We also assayed the in vivo activity of the EE with a mutated Octamer element. At E10.0, mutation of the POUHD half-site abolished enhancer activity in the lens (Table 3, OCTHM). At E12.5, transgenic embryos with mutations in either the POUS or POUHD half-site behaved similarly, with the absence of all ectoderm expression outside the lens (Fig. 10B; Table 3). Interestingly, the effect of these mutations in the differentiating lens was not uniform. Expression in the AEL was maintained in OCTPM and OCTHM transgenic embryos, while expression in the fiber cell compartment was lost (Fig. 10B). The Octamer element is therefore necessary for Pax6 EE activity in the lens pit, the lens primary fiber cells and in the ectoderm surrounding the lens at E12.5. It is not, however, necessary for the activity of the Pax6 EE in the AEL. In total, our in vivo transgenic data demonstrate that the Octamer and Sox DNA binding sites are each required for activity of the Pax6 EE in the early lens, the differentiating lens and the peri-ocular ectoderm and that the utilization of these sites is distinct in the differentiating lens epithelium and primary fiber cells.
DISCUSSION
Sox2 and Oct-1 are co-factors required for lens and nasal placode induction
While Sox transcription factors can bind similar DNA sequences (Kamachi et al., 1999), individual Sox proteins control distinct downstream targets (Kamachi et al., 1999, 2000). Thus, it is believed that Sox factors affect transcription by forming complexes with other proteins, and that accessory co-factors provide transcriptional specificity. Pax6 and Oct-3 are two co-factors that have been characterized for Sox2 (Ambrosetti et al., 1997; Kamachi et al., 2001; Aota et al., 2003; Rodda et al., 2005; Okumura-Nakanishi et al., 2005). Here, we identify Oct-1 as an important co-factor for Sox2 during lens and nasal placode induction.
In contrast to previous reports that Oct-1 is ubiquitously expressed (reviewed in Ryan and Rosenfeld, 1997), we find that during embryonic development Oct-1 exhibits a developmentally dynamic and tissue-specific pattern of expression. The dynamic profile of Oct-1 expression overlaps extensively with that of Sox2 in the early neurepithelium, in the lens and nasal placodes and their derivatives. While heterozygous mutation of either Pou2f1 or Sox2 alone has no phenotypic consequences for lens or nasal development, compound heterozygous mutation of Pou2f1 and Sox2 produces ocular cataracts. Cataracts are a sensitive readout of subtle disturbances in lens development, and are a cardinal feature observed in Pax6Sey/+ mice.
Homozygous mutation of Pou2f1 results in small embryos ~4% of the time (Wang et al., 2004), and consistent with this, we observe a morphologically normal but small lens pits in a small fraction of Oct-1−/− embryos. However, in Oct-1−/−, Sox2+/− embryos, while lens phenotypes within a single embryo were variable (potentially due to the hypomorphic nature of the Oct-1 allele and see below) (Wang et al., 2004), severe defects occurred in both lens and nasal placode induction. These embryos generally exhibit microphthalmia in one eye and anophthalmia in the other, and a complete failure of nasal placode induction. Since compound mutations of Sox2 and Pou2f1 in Oct-1+/−, Sox2+/− or in Oct-1−/−, Sox2+/− embryos produce phenotypes not observed when the genes are mutated independently, Pou2f1 and Sox2 act cooperatively during the induction of the lens and nasal placodes.
Although we provide evidence for Oct-1/Sox2-dependent activation of the Pax6 EE, it is likely that another POU factor in the lens also interacts with Sox2 to activate Pax6 expression through the EE. The activity of the Pax6 EE is strictly dependent upon an intact Octamer element, but is not strictly dependent upon Oct-1, since Pax6 expression and lens morphogenesis are normal in Oct-1−/− embryos. Alternatively, Oct-1 expression from the hypomorphic Pou2f1 allele, even in the homozygous condition, may be sufficient for activation of the EE and maintenance of Pax6 expression. Homozygous mutation of Pou2f1 in conjunction with heterozygosity for Sox2, however, prevents Oct/Sox2 complex formation on the EE and disrupts expression of Pax6.
In addition to lens and nasal placode defects in Oct1−/−, Sox2+/− mice, we have observed dysmorphology of Rathke’s pouch and the adenohypophysis (ALD and RLM, unpublished data). Thus, all Pax6-dependent PPR derivatives are disrupted in these embryos. These observations are consistent with the utilization of a common molecular pathway, involving Pou2f1, Sox2 and Pax6 in mammalian lens, nasal and Rathke’s pouch placode induction and differentiation.
Sox2 and Oct-1 maintain Pax6 expression through the EE
Sox2 and Oct-1 are important for lens and nasal placode induction because they directly control the expression of Pax6. Sox2 and Oct-1 proteins are first detectable in the mouse PLE at the 15-somite stage, after the onset of Pax6 expression. Thus, Sox2 and Oct-1 cannot be required for the initial activation of Pax6. However, Sox2 and Oct-1 are both expressed prior to mouse lens specification, which occurs at the 23-somite stage (Furuta and Hogan, 1998). Furthermore, Pax6 expression is dynamically restricted from a broad domain of the head surface ectoderm to the PLE during pre-specification stages (Grindley et al., 1995). Thus, Sox2 and Oct-1 may stabilize Pax6 expression in the PLE during lens specification.
Sox2 and Oct-1 bind cooperatively to the Pax6 EE and the disruption of Sox or Octamer DNA binding sites abolishes Pax6 EE-dependent transcriptional activation in cell culture. Octamer and Sox site mutations also eliminate Pax6 EE-dependent expression in the lens of transgenic mouse embryos. Thus, the Octamer and Sox sites identified herein are required for the in vivo activity of the Pax6 EE.
The Pax6 EE, however, is not the only enhancer required for appropriate Pax6 expression in the early lens (Dimanlig et al., 2001). Indeed Pax6 expression in the head surface ectoderm, which initiates at E8.0 (Grindley et al., 1995), precedes the earliest detectable activity of the Pax6 EE at E8.75 (Dimanlig et al., 2001). Thus, the Octamer and Sox elements described herein may be necessary for the activity of the Pax6 EE without controlling the pre-15-somite stage head surface ectoderm expression of Pax6.
Homozygous deletion of the Pax6 EE in Pax6ΔEE/ΔEE mice reduces Pax6 expression, delays lens placode induction and results in microphthalmia (Dimanlig et al., 2001). Mildly effected Oct-1−/−, Sox2+/− lenses resemble Pax6ΔEE/ΔEE lenses, while severely effected Oct-1−/−, Sox2+/− lenses are more severely affected than Pax6ΔEE/ΔEE lenses. There are several possible explanations for this result. As mentioned previously, there are at least two enhancers that control Pax6 expression in the lens. The SIMO enhancer has been shown to direct expression to the lens placode and its derivatives in transgenic mice (Kleinjan et al., 2001). In the most highly conserved domain of the SIMO element there are at least eight sites that bind Sox proteins in vitro and one Octamer element (ALD and RLM, unpublished data). Thus, it is possible that Oct1 and Sox2 control Pax6 lens expression through other Pax6 enhancers such as the SIMO enhancer. Secondly, loss of Oct-1 and Sox2 in the lens may reduce or eliminate the expression of genes other than Pax6 that are important for lens placode induction and subsequent differentiation. Finally, the loss of Oct-1 and Sox2 in the OV may result in non-tissue autonomous defects in lens development. While we have assessed expression of several OV markers, tissue-specific deletion of Pou2f1 and Sox2 will be required to fully address this possibility since many of the molecules required for OV-PLE signaling are not known. However, given the existence of multiple placode defects in the Oct-1−/−, Sox2+/− embryos and the normal physical apposition between the OV with PLE, it is unlikely that a defect in the OV is the exclusive cause of the observed lens placode defect.
Sox2 and Pax6 are interdependent factors required for eye development
Sox2 and Pax6 proteins act together to regulate the transcription of the chicken δ-crystallin gene and the Pax6 EE (Kamachi et al., 2001; Aota et al., 2003). In Sox2βgeo2/+, Pax6Sey/+ double heterozygous mice, however, we did not detect synergistic effects of Sox2 and Pax6 on lens placode induction or lens differentiation. This may indicate that Sox2/Pax6 complex formation is not essential for murine lens placode induction or the early activity of the Pax6 EE. Alternatively, the lack of phenotypic evidence for this interaction may derive from the alleles utilized in this study. Embryos homozygous for the Sox2βgeo2 allele alone cannot be evaluated for lens defects due to the severity of the early phenotype (Avilion et al., 2003; Ekonomou et al., 2005). Embryos homozygous for the Pax61-NeuSey allele already fail to induce a lens placode; thus the phenotypic consequence of Pax6Sey/Sey compounded with Sox2 mutation on lens development cannot be meaningfully evaluated. Furthermore, mice heterozygous for the Pax61-NeuSey allele have cataracts. Thus, the Pax6 heterozygous mutation in isolation has the same phenotype as Oct1+/−, Sox2+/− mice, and compounding heterozygosity for the alleles Pax61-NeuSey and Sox2βgeo2 has no additional phenotypic consequences.
It is notable that the Oct-1/Sox2 complex that we identify herein and the Pax6/Sox2 complex characterized in Aota et al. (2003) both predominately utilize Sox Site A. Thus, alternative Sox2 complexes may control different aspects of Pax6 regulation. For example, during lens differentiation the Sox sites are required for Pax6 EE activity in the AEL and the fiber cell compartment, while the Octamer site is only necessary in the fiber cell compartment. Thus, the Pax6/Sox complex might function in the AEL, while the Oct-1/Sox2 complex is utilized during primary fiber cell differentiation.
Sox2 is activated downstream of Pax6, since Sox2 expression is lost in homozygous Pax6 null embryos (Furuta and Hogan, 1998; Ashery-Padan and Gruss, 2001). Indeed, we observe the loss of Sox2 expression in our compound Oct-1−/−, Sox2+/− mice in both the surface ectoderm and the distal OV. Since Sox2 expression is detectable in both Oct-1−/− and Sox2+/− mice, we presume that this is a consequence of disrupted Pax6 expression in the compound mutants.
Sox2 also acts upstream of Pax6 during lens induction. Our current analyses provide evidence that Sox2, acting through the EE, is required for the maintenance of Pax6 expression in the mouse PLE for lens placode induction and subsequent lens morphogenesis. Kamachi et al. (2001) demonstrated that ectopic expression of Sox2 up-regulated Pax6 in chick head ectoderm. Based on these data, Kondoh and colleagues proposed that Pax6 maintenance in the chick lens is Sox2-dependent (Kondoh et al., 2004). Our data are consistent with the model of Kondoh (2004) and expand it to include the co-factor, Oct-1, which acts in concert with Sox2 to maintain Pax6 expression.
Supplementary Material
Fig. S1. RT-PCR identification of Pou2f1 in E13.5 mouse eyes and lens epithelial cell cultures (A) RT-PCR primers specific for the linker-encoding region of Pou2 genes amplify a specific product from E13.5 eyes. (B) RT-PCR primers specific for Sox2, Pax6, and Pou2f1 amplify a product from lens epithelial cell lines, while primers for Pou2f2 and Pou5f1 do not.
Fig. S2. Compound heterozygous mutations of Oct1 and Sox2 in mice disrupt lens morphogenesis. (A–I) Immunofluorescence for Pax6 (red) is shown with DAPI (blue) in mice bearing mutations in Sox2 (A, D, G), Pax6 (B, E, H), or both (C, F, I) at E9.5 (A–C), E11.5 (D–F), and E13.5 (G–I). (J–M) Immunofluorescence for Pax6 (red) is shown with DAPI in mice mutant for Oct-1 (J, L) or Oct-1 and Sox2 (K, M) at E9.5 (J–K) and E11.5 (L–M). (N–O) Eyes of 6 week old Oct1+/− (N) or Oct1+/−, Sox2+/− (O) mice are shown with a lens cataract in the latter. Abbreviations: ael, anterior epithelial layer; lp, lens placode; lv, lens vesicle; nr, neural retina; ov, optic vesicle
Fig. S3. Specificity of the Oct-1 and E-cadherin antibodies (A–B)
The Oct-1 antibody is specific for Oct-1. Oct-1 (red) and DAPI (blue) are shown for wild-type (A) and Oct-1−/− (B) lenses at E10.5. The staining observed in Oct-1−/− embryos (*) is due to non-specific interaction of the secondary antibody.
(C–D) E-cadherin is a marker for epithelial cells. E-cadherin (red) and DAPI are shown for wild-type embryos at E10.5 (C) and E12.0 (D). E-cadherin is expressed in all cells of the lens vesicle and surface ectoderm (arrow in C) at E10.5. Prospective cornea (arrow in D) and AEL express E-cadherin at E12.0, while fiber cells do not (D).
Abbreviations: ael, anterior epithelial layer; lv, lens vesicle; pc, prospective cornea; se, surface ectoderm
Fig. S4. Oct-1−/−, Sox2+/− eyes are dysmorphic through E12.5. (A–H) Pax6 (red) is shown with DAPI (blue) for Oct1−/− (A), Oct-1−/−, Sox2+/− (C, E), and Pax6Sey/Sey (G) E11.5 lenses. E-cadherin (red) is shown with DAPI for Oct1−/− (B), Oct-1−/−, Sox2+/− (D, F), and Pax6Sey/Sey (H) lenses. Both Oct-1−/−, Sox2+/− lenses from the same embryo are shown to illustrate phenotypic variability. Circles highlight small lenses (C, D). (I–J) Whole mount photos of both eyes from an Oct-1−/−, Sox2+/− embryo at E12.5. (K–L) Immunoflourescence for Pax6 (red) and DAPI are shown for each eye, respectively. (M–N). E-cadherin (red) and DAPI staining are shown for the same. The + indicates lack of a lens (K, M).
Fig S5. Six3 and sFRP2 are maintained in the Oct-1−/−, Sox2+/− retina. In Situ hybridzation is shown for Six3 (A–B) and sFRP2 (C–D) in wild-type (A, C) or Oct-1−/−, Sox2+/− eyes (B, D). Both genes are expressed in the OV or retina, while Six3 expression is lost from the surface ectoderm (arrows in A–B). Abbreviations: lpt lens pit, nr neural retina, se surface ectoderm
Fig. S6. Mutational analyses of Sox2 binding specificity to the EE. (A) Representative EMSA experiments are shown for Sox2-HMG interaction with radiolabeled EE-3 in the presence of unlabeled competitors EE-3, EE-3C, SOXAM4, SOXBM4, or DM4. (B) Representative EMSA experiments are shown for OCT1-GST interaction with radiolabeled EE-3 in the presence of competitors EE-3, OCTHM, or OCTPM.
Acknowledgments
The authors are grateful to Lisa Dailey, Winship Herr, Hisato Kondoh, Robin Lovell-Badge, John Reddan, Phil Sharp, and Dean Tantin for reagents. Lena Du from the BWH transgenic facility performed the transgenic injections. The Pax6 antibody developed by A. Kawakami was obtained from the Developmental Studies Hybridoma Bank (NICHD and University of Iowa). This work was supported by grant R01 EY010123-10 to RLM. ALD was supported by fellowships F32-DE05735 from NIDCR and T32-EY07145 from NEI administered by the Postdoctoral Training Program in the Molecular Bases of Eye Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahrens K, Schlosser G. Tissues and signals involved in the induction of placodal Six1 expression in Xenopus laevis. Dev Biol. 2005;288:40–59. doi: 10.1016/j.ydbio.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Ambrosetti DC, Basilico C, Dailey L. Synergistic activation of the Fibroblast Growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol Cell Biol. 1997;17:6321–6329. doi: 10.1128/mcb.17.11.6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aota S, Nakajima N, Sakamoto R, Wantabe S, Ibarake N, Okazaki K. Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev Biol. 2003;257:1–13. doi: 10.1016/s0012-1606(03)00058-7. [DOI] [PubMed] [Google Scholar]
- Ashery-Padan R, Gruss P. Pax6 lights-up the way for eye development. Curr Opin Cell Biol. 2001;13:706–714. doi: 10.1016/s0955-0674(00)00274-x. [DOI] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AP, Bhattacharyya S, Bronner-Fraser M, Streit A. Lens specification is the ground state of all sensory placodes, from which FGF promotes olfactory identity. Dev Cell. 2006;11:505–517. doi: 10.1016/j.devcel.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Bronner-Fraser M. Hierarchy of regulatory events in sensory placode development. Curr Opin Genet Dev. 2004;14:520–526. doi: 10.1016/j.gde.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Botquin V, Hess H, Fuhrmann G, Anastassiadis C, Gross MK, Vriend G, Scholer HR. New POU dimer configuration mediates antagonistic control of an osteopontin preimplantation enhancer by Oct-4 and Sox-2. Genes Dev. 1998;12:2073–90. doi: 10.1101/gad.12.13.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugmann SA, Moody SA. Induction and specification of the vertebrate ectodermal placodes: precursors of the cranial sensory organs. Biol Cell. 2005;97:303–319. doi: 10.1042/BC20040515. [DOI] [PubMed] [Google Scholar]
- Carl M, Loosli F, Whittbrodt J. Six3 inactivation reveals its essential role for the formation and patterning of the vertebrate eye. Development. 2002;129:4057–4063. doi: 10.1242/dev.129.17.4057. [DOI] [PubMed] [Google Scholar]
- Dimanlig PV, Faber SC, Auerbach W, Makarenkova HP, Lang RA. The upstream ectoderm enhancer in Pax6 has an important role in lens induction. Development. 2001;128:4415–4424. doi: 10.1242/dev.128.22.4415. [DOI] [PubMed] [Google Scholar]
- Di Rocco G, Gavalas A, Pöpperl H, Krumlauf R, Mavilio F, Zappavigna V. The recruitment of SOX/OCT complexes and the differential activity of HOXA1 and HOXB1 modulate the hoxb1 auto-regulatory enhancer function. J Biol Chem. 2001;276:20506–515. doi: 10.1074/jbc.M011175200. [DOI] [PubMed] [Google Scholar]
- Dong S, Leung KKH, Pelling AL, Lee PYT, Tang ASP, Heng HHQ, Tsui LC, Tease C, Fisher G, Steel KP, Cheah KSE. Circling, deafness, and yellow coat displayed by yellow submarine (Ysb) and light coat and circling (Lcc) mice with mutations on chromosome 3. Genomics. 2002;79:777–784. doi: 10.1006/geno.2002.6783. [DOI] [PubMed] [Google Scholar]
- Ekonomou A, Kazanis I, Malas S, Wood H, Alifragis P, Denaxa M, Karagogeos D, Constanti A, Lovell-Badge R, Episkopou V. Neuronal migration and ventral subtype identity in the telencephalon depend on SOX1. PloS Biology. 2005;3:1111–1122. doi: 10.1371/journal.pbio.0030186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve P, Trousse F, Rodríguez J, Bovolenta P. SFRP1 modulates retina cell differentiation through a β-catenin-independent mechanism. J Cell Sci. 2003;116:2471–2481. doi: 10.1242/jcs.00452. [DOI] [PubMed] [Google Scholar]
- Faber SC, Dimanlig P, Markarnkova HP, Shirke S, Ko K, Lang RA. Fgf receptor signaling plays a role in lens induction. Development. 2001;128:4425–38. doi: 10.1242/dev.128.22.4425. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Hogan BLM. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998;12:3764–75. doi: 10.1101/gad.12.23.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia, and central nervous system defects. Nat Genet. 1994;7:463–471. doi: 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- Grindley JC, Davidson DR, Hill RE. The role of Pax-6 in eye and nasal development. Development. 1995;121:1433–1442. doi: 10.1242/dev.121.5.1433. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, Pauer GJ, Reid J, Simpson E, Crowe S, Maumenee IH, Traboulsi EI. SOX2 mutation causes anophthalmia, hearing loss, and brain anolamlies. Am J Med Genet. 2005;138:95–8. doi: 10.1002/ajmg.a.30803. [DOI] [PubMed] [Google Scholar]
- Hanson IM, Fletcher JM, Jordan T, Brown A, Taylor D, Adams RJ, Punnett HH, van Heyningen V. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat Genet. 1994;6:168–73. doi: 10.1038/ng0294-168. [DOI] [PubMed] [Google Scholar]
- Hill RE, Favor J, Hogan BLM, Ton CT, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse Small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Hirst EM, Horsburgh G, Hetherington CM. Small eye (Sey): a mouse model for the genetic analysis of craniofacial abnormalities. Development. 1988;103(Suppl):115–119. doi: 10.1242/dev.103.Supplement.115. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Uchikawa M, Collignon J, Lovell-Badge R, Kondoh H. Involvement of Sox1, 2 and 3 in the early and subsequent molecular events of lens induction. Development. 1998;125:2521–2532. doi: 10.1242/dev.125.13.2521. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Cheah KS, Kondon H. Mechanism of regulatory target selection by SOX high-mobility-group domain proteins as revealed by comparison of SOX1/2/3 and SOX9. Mol Cell Biol. 1999;19:107–20. doi: 10.1128/mcb.19.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamachi Y, Uchikawa M, Kondoh H. Pairing Sox off: with partners in the regulation of embryonic development. Trends Genet. 2000;16:182–7. doi: 10.1016/s0168-9525(99)01955-1. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Uchikawa M, Tanouchi A, Sekido R, Kondoh H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001;15:1272–1286. doi: 10.1101/gad.887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammandel B, Chowdhury K, Stoykova A, Aparicio S, Brenner S, Gruss P. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev Biol. 1999;205:79–97. doi: 10.1006/dbio.1998.9128. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KKH, Tang ASP, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KSE. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison, and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet. 2001;10:2049–2059. doi: 10.1093/hmg/10.19.2049. [DOI] [PubMed] [Google Scholar]
- Klemm JD, Rould MA, Aurora R, Herr W, Pabo CO. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell. 1994;77:21–32. doi: 10.1016/0092-8674(94)90231-3. [DOI] [PubMed] [Google Scholar]
- Kondoh H, Uchikawa M, Kamachi Y. Interplay of Pax6 and Sox2 in lens development as a paradigm of genetic switch mechanisms for cell differentiation. Int J Dev Biol. 2004;48:819–827. doi: 10.1387/ijdb.041868hk. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Wright TJ, Moon AM, Mansour SL, Schoenwolf GC. FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 2005;19:603–13. doi: 10.1101/gad.1273605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagutin O, Zhu CC, Furuta Y, Rowitch DH, McMahon AP, Oliver G. Six3 promotes the formation of ectopic optic vesicle-like structures in mouse embryos. Dev Dyn. 2001;221:342–349. doi: 10.1002/dvdy.1148. [DOI] [PubMed] [Google Scholar]
- Litsiou A, Hanson S, Streit A. A balance of FGF, BMP and WNT signaling positions the future placode territory in the head. Development. 2005;132:4051–62. doi: 10.1242/dev.01964. [DOI] [PubMed] [Google Scholar]
- Ma Y, Certel K, Gao Y, Niemitz E, Mosher J, Mukherjee A, Mutsuddi M, Huseinovic N, Crews ST, Johnson WA, Nambu JR. Functional interactions between Drosophila bHLH/PAS, Sox, and the POU transcription factors regulate CNS midline expression of the slit gene. J Neurosci. 2000;20:4596–4605. doi: 10.1523/JNEUROSCI.20-12-04596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Groves AK. Competence of cranial ectoderm to respond to Fgf signaling suggests a two-step model of otic placode induction. Development. 2005;133:877–87. doi: 10.1242/dev.02267. [DOI] [PubMed] [Google Scholar]
- Mertin S, McDowall SG, Harley VR. The DNA-binding specificity of SOX9 and other SOX proteins. Nuc Acids Res. 1999;27:1359–1364. doi: 10.1093/nar/27.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2003. [Google Scholar]
- Nishiguchi S, Wood H, Kondoh H, Lovell-Badge R, Episkopou V. Sox1 directly regulates the gamma-crystallin genes and is essential for lens development in mice. Genes Dev. 1998;12:776–81. doi: 10.1101/gad.12.6.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto M, Fukushima A, Okuda A, Muramatsu M. The gene for the embryonic stem cell coactivator UTF1 carries a regulatory element which selectively interacts with a complex composed of Oct-3/4 and Sox-2. Mol Cell Biol. 1999;19:5453–65. doi: 10.1128/mcb.19.8.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama T, Mohamed OA, Taketo MM, Dufort D, Groves AK. Wnt signals mediate a fate decision between otic placode and epidermis. Development. 2006;133:865–75. doi: 10.1242/dev.02271. [DOI] [PubMed] [Google Scholar]
- Okumura-Nakanishi S, Saito M, Niwa H, Ishikawa F. Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J Biol Chem. 2005;280:5307–17. doi: 10.1074/jbc.M410015200. [DOI] [PubMed] [Google Scholar]
- Phillips BT, Storch EM, Lekven AC, Riley BB. A direct role for Fgf but not Wnt in otic placode induction. Development. 2004;131:923–31. doi: 10.1242/dev.00978. [DOI] [PubMed] [Google Scholar]
- Purcell P, Oliver G, Mardon G, Donner AL, Maas RL. Pax6-dependence of Six3, Eya1, and Dach1 expression during lens and nasal placode induction. Gene Expr Patterns. 2005;6:110–118. doi: 10.1016/j.modgep.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin RO, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, FitzPatrick DR. SOX2 anophthalmia syndrome. Am J Med Genet. 2005;135A:1–7. doi: 10.1002/ajmg.a.30642. [DOI] [PubMed] [Google Scholar]
- Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, Robson P. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280:24731–7. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Rosenfeld MG. POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev. 1997;11:1207–25. doi: 10.1101/gad.11.10.1207. [DOI] [PubMed] [Google Scholar]
- Schlosser G. Induction and specification of the cranial placodes. Dev Biol. 2006;294:303–351. doi: 10.1016/j.ydbio.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Sisodiya SM, Free SL, Williamson KA, Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson IM, van Heyningen V. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet. 2001;28:214–216. doi: 10.1038/90042. [DOI] [PubMed] [Google Scholar]
- Smith AN, Miller LA, Song N, Taketo MM, Lang RA. The duality of beta-catenin function: a requirement in lens morphogenesis and signaling suppression of lens fate in periocular ectoderm. Dev Biol. 2005;285:477–89. doi: 10.1016/j.ydbio.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Streit A. Early development of the cranial sensory nervous system: from a common field to individual placodes. Dev Biol. 2004;276:1–15. doi: 10.1016/j.ydbio.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Herr W. Differential transcriptional activation by Oct-1 and Oct-2: interdependent activation domains induce Oct-2 phosphorylation. Cell. 1990;60:375–386. doi: 10.1016/0092-8674(90)90589-7. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kamachi Y, Tanouchi A, Hamada H, Jing N, Kondoh H. Interplay of SOX and POU factors in regulation of the Nestin gene in neural primordial cells. Mol Cell Biol. 2004;24:8834–46. doi: 10.1128/MCB.24.20.8834-8846.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006;20:1187–1202. doi: 10.1101/gad.1407906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houte LPA, Chuprina VP, van der Wetering M, Boelens R, Kaptein R, Clevers H. Solution structure of the sequence-specific HMG box of the lymphocyte transcriptional activator Sox-4. J Biol Chem. 1995;270:30516–524. doi: 10.1074/jbc.270.51.30516. [DOI] [PubMed] [Google Scholar]
- Van Raay TJ, Moore KB, Iordanova I, Steele M, Jamrich M, Harris WA, Vetter ML. Frizzled 5 signaling governs the neural potential of progenitors in the developing Xenopus retina. Neuron. 2005;46:23–36. doi: 10.1016/j.neuron.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Wang VEH, Schmidt T, Chen J, Sharp PA, Tantin D. Embryonic lethality, decreased erythropoiesis, and defective octamer-dependent promoter activation in Oct-1-deficient mice. Mol Cell Biol. 2004;24:1022–1032. doi: 10.1128/MCB.24.3.1022-1032.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawersik S, Purcell P, Rauchman M, Dudley AT, Robertson EJ, Maas R. BMP7 acts in murine lens placode development. Dev Biol. 1999;207:176–188. doi: 10.1006/dbio.1998.9153. [DOI] [PubMed] [Google Scholar]
- Williams SC, Atlmann CR, Chow RL, Hemmati-Brivanlou A, Lang RA. A highly conserved lens transcriptional control element from the Pax-6 gene. Mech Dev. 1998;73:225–229. doi: 10.1016/s0925-4773(98)00057-4. [DOI] [PubMed] [Google Scholar]
- Wood HB, Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Dev. 1999;86:197–201. doi: 10.1016/s0925-4773(99)00116-1. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Gascon-Guzman G, Tovilla-Canales JL. Bilateral anophthalmia and brain malformations caused by a 20-bp deletion in the SOX2 gene. Clin Genet. 2005;68:564–6. doi: 10.1111/j.1399-0004.2005.00518.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Friedman A, Heaney S, Purcell P, Maas RL. Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev. 2002;16:2097–2107. doi: 10.1101/gad.1007602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. RT-PCR identification of Pou2f1 in E13.5 mouse eyes and lens epithelial cell cultures (A) RT-PCR primers specific for the linker-encoding region of Pou2 genes amplify a specific product from E13.5 eyes. (B) RT-PCR primers specific for Sox2, Pax6, and Pou2f1 amplify a product from lens epithelial cell lines, while primers for Pou2f2 and Pou5f1 do not.
Fig. S2. Compound heterozygous mutations of Oct1 and Sox2 in mice disrupt lens morphogenesis. (A–I) Immunofluorescence for Pax6 (red) is shown with DAPI (blue) in mice bearing mutations in Sox2 (A, D, G), Pax6 (B, E, H), or both (C, F, I) at E9.5 (A–C), E11.5 (D–F), and E13.5 (G–I). (J–M) Immunofluorescence for Pax6 (red) is shown with DAPI in mice mutant for Oct-1 (J, L) or Oct-1 and Sox2 (K, M) at E9.5 (J–K) and E11.5 (L–M). (N–O) Eyes of 6 week old Oct1+/− (N) or Oct1+/−, Sox2+/− (O) mice are shown with a lens cataract in the latter. Abbreviations: ael, anterior epithelial layer; lp, lens placode; lv, lens vesicle; nr, neural retina; ov, optic vesicle
Fig. S3. Specificity of the Oct-1 and E-cadherin antibodies (A–B)
The Oct-1 antibody is specific for Oct-1. Oct-1 (red) and DAPI (blue) are shown for wild-type (A) and Oct-1−/− (B) lenses at E10.5. The staining observed in Oct-1−/− embryos (*) is due to non-specific interaction of the secondary antibody.
(C–D) E-cadherin is a marker for epithelial cells. E-cadherin (red) and DAPI are shown for wild-type embryos at E10.5 (C) and E12.0 (D). E-cadherin is expressed in all cells of the lens vesicle and surface ectoderm (arrow in C) at E10.5. Prospective cornea (arrow in D) and AEL express E-cadherin at E12.0, while fiber cells do not (D).
Abbreviations: ael, anterior epithelial layer; lv, lens vesicle; pc, prospective cornea; se, surface ectoderm
Fig. S4. Oct-1−/−, Sox2+/− eyes are dysmorphic through E12.5. (A–H) Pax6 (red) is shown with DAPI (blue) for Oct1−/− (A), Oct-1−/−, Sox2+/− (C, E), and Pax6Sey/Sey (G) E11.5 lenses. E-cadherin (red) is shown with DAPI for Oct1−/− (B), Oct-1−/−, Sox2+/− (D, F), and Pax6Sey/Sey (H) lenses. Both Oct-1−/−, Sox2+/− lenses from the same embryo are shown to illustrate phenotypic variability. Circles highlight small lenses (C, D). (I–J) Whole mount photos of both eyes from an Oct-1−/−, Sox2+/− embryo at E12.5. (K–L) Immunoflourescence for Pax6 (red) and DAPI are shown for each eye, respectively. (M–N). E-cadherin (red) and DAPI staining are shown for the same. The + indicates lack of a lens (K, M).
Fig S5. Six3 and sFRP2 are maintained in the Oct-1−/−, Sox2+/− retina. In Situ hybridzation is shown for Six3 (A–B) and sFRP2 (C–D) in wild-type (A, C) or Oct-1−/−, Sox2+/− eyes (B, D). Both genes are expressed in the OV or retina, while Six3 expression is lost from the surface ectoderm (arrows in A–B). Abbreviations: lpt lens pit, nr neural retina, se surface ectoderm
Fig. S6. Mutational analyses of Sox2 binding specificity to the EE. (A) Representative EMSA experiments are shown for Sox2-HMG interaction with radiolabeled EE-3 in the presence of unlabeled competitors EE-3, EE-3C, SOXAM4, SOXBM4, or DM4. (B) Representative EMSA experiments are shown for OCT1-GST interaction with radiolabeled EE-3 in the presence of competitors EE-3, OCTHM, or OCTPM.