

Abstract
We are accruing patients to a Phase I dose escalation cellular therapy trial (www.clinicaltrials.gov, NCT01144247) involving intratumoral placement of alloreactive cytotoxic T lymphocytes (alloCTL) for recurrent gliomas. The trial is being conducted to confirm the findings of a prior pilot study that indicated this adjuvant therapy may be beneficial in extending survival of recurrent WHO grade III gliomas. To reduce costs of the cellular therapy, we tested a number of synthetic tissue culture media and found the AIM-V growth medium superior for their growth. We also moved the production of the alloCTL from artificial capillary systems to less expensive tissue culture bags. To standardize alloCTL infusates used for therapy, release criteria of ≥60% CD3+ and ≥60% viability were established that consistently translated to a 4 hr cytotoxicity of ≥30% at a 30:1 effector to target ratio. To allow time for completion of quality control testing and transport to the infusion site, we determined that 30,000 IU of human recombinant Interleukin-2 in the cellular infusates sufficiently retained cell viability and cytotoxicity to allow a 10 hr expiration time to be placed on the infusates. We identified a cytotoxic T cell subset, CD3+/CD8+/CD69+, that demonstrated upregulated IFN-γ production upon exposure to relevant target cells. The phenotypic identification of this T cell subset was indicative of robust in vitro cytotoxic function and thus will be followed to determine if it correlates with patient immune response to treatment. Finally, other therapeutic agents routinely used for glioma treatment were integrated into an analysis of alloCTL cytotoxic functionality. Temozolomide and bevacizumab do not adversely affect cytotoxic function of the alloCTL in the short-term, thus providing rationale for further investigating combinatorial chemo-immunotherapy for gliomas.
Keywords: Astrocytoma, glioma, CTL, temozolomide, bevacizumab, adoptive immunotherapy, cellular therapy, immunotherapy
Introduction
Gliomas are the most common primary malignant brain tumor in adults [1]. Gliomas exhibit focal solid tumor growth, yet infiltrate extensively into surrounding normal brain tissue, making their complete removal by surgery impossible. While improvements to conventional radio-chemotherapy have occurred, these brain tumors still quickly progress, are debilitating and ultimately lethal. With these treatments the median survival of glioma patients increases modestly [2, 3]. This establishes a firm rationale for introducing novel experimental immune therapies early. Although patient enrollment numbers are small, documentation from small Phase I or II trials is available that indicates remarkable individual responses using approaches entailing passive cellular therapy or active immunotherapy with vaccines [4].
Alloreactive CTL (alloCTL), sensitized to the human leukocyte antigens (HLA) of the tumor-bearing host, demonstrated in vitro and in vivo promise in preclinical glioma model studies [5-9]. Also, safety and encouraging clinical results were documented in recurrent glioma patients receiving multiple intratumoral infusions of alloCTL made by one-way mixed lymphocyte reactions (MLR) that were derived from different donors at each treatment cycle [10, 11]. In the pilot study, expense of the cellular therapy was minimized by purchase of waste leucopaks from apheresis procedures used for platelet collection; their growth was optimized in artificial capillary systems [12]. The cellular therapy approach was based upon the observation that HLA expression is largely absent on normal brain neuroglia [13-15] but is present on brain tumor cells [15-17]. Thus, intralesionally-placed alloCTL should largely exert their effector function to tumor cells displaying HLA. While collateral damage to HLA-expressing microglia is possible from the instilled alloCTL, these cells should be capable of repopulating in the brain, and damage to HLA-expressing endothelial cells may engender beneficial effects by adversely affecting tumor vasculature [18].
The preclinical studies described herein supported reactivation of IND BB 5423 with the Food and Drug Administration and were performed to answer questions from the Institutional Review Board. The findings impacted the generation methods used for alloCTL production, our clinical implementation of growth factor amounts to the infusates, the latter of which may reduce patient toxicity to the infusates, and supported the expiration time placed on the infusates. Also, we identified a phenotypic cytotoxic subset that is upregulated and produces IFN-γ upon exposure to relevant target cells in vitro; it will be correlated with immune monitoring results and patient response to treatment. Finally, our data support the noninterference of other commonly used treatment agents, temozolomide and bevacizumab, that might later be given in parallel with adoptively transferred alloCTL. It is commonplace for newly diagnosed glioblastoma patients to continue receiving oral temozolomide after completion of radiotherapy [3]. Also, at relapse glioma patients are generally given bevacizumab, an antibody directed to vascular endothelial growth factor (VEGF) [19]. Temozolomide is an inhibitor of a DNA repair enzyme, methyl guanine, methyltransferase [20], and anti-VEGF is an anti-angiogenesis agent [19]. Thus, the anti-tumor mechanism of both these agents might be anticipated to be independent of that from CTL. If alloCTL retained anti-tumor functionality in the presence of these two therapeutic agents, combinatorial therapy might be possible.
Materials and methods
Glioma cell culture and characterization
Human glioma cell line U-87MG was derived from a glioblastoma patient; it was grown in Dulbecco's modified Eagles medium (DMEM) with 10% fetal bovine serum (FBS; Gemini-Bioproducts, Woodland, CA). The human 13-06-MG cell line that was derived from a glioblastoma surgical specimen obtained from a patient at initial diagnosis, was processed and the cells were established in culture according to methods developed in our laboratory [21, 22]. The glioma cell lines (13-06-MG and U-87MG) have been characterized [15, 17, 22-24]. Class I and II HLA display was determined by flow cytometric analysis using FITC-conjugated mouse anti-human HLA-ABC (eBioscience, San Diego, CA) and mouse anti human PerCP-conjugated HLA-DR (BD Bioscience, San Diego, CA). The percentage of positive cells and the relative antigen density indicated by median fluorescence intensity (MFI) were determined using FlowJo flow cytometric analysis software (Tree Star, Ashland, OR) [15]. The specific HLA-AB type determined by serological HLA typing for 13-06-MG cells was A1,2 and B44,57 and for U-87MG was A2 and B44.
Isolation of peripheral blood mononuclear cells (PBMC) and generation of alloCTL enriched cultures by one-way MLR
Whole blood was collected in heparinized tubes from normal donors as a source of PBMC under a UCLA IRB-approved protocol. Heparinized whole blood was diluted 1:1 (v/v) in phosphate buffered saline (PBS) without calcium or magnesium (Life Technologies Inc., Gaithersburg, MD). The PBMC, a source of responder (R) or stimulator (S) lymphocytes, were isolated by density gradient (Lymphocyte Separation Media, Mediatech Inc., Manassas, VA) centrifugation and washed twice with PBS as described [12]. Stimulator lymphocytes were expanded with OKT3 and IL-2 in Lifecell tissue culture bags (Miltenyi Biotec, Inc., San Francisco, CA; former designation by Baxter Healthcare, Deerfield, IL was IL-PL732 tissue culture flasks) [12]. Harvested stimulator cells were cryopreserved in AIM-V medium mixed 1:1 (v/v) with human serum albumin containing 15% dimethyl sulfoxide. For inactivation we used either 127Cs or X-Ray sources. Unless detailed otherwise, the stimulator lymphocytes were resuspended at room temperature before their inactivation by an experimental X-ray irradiator (Gulmay Medical Inc. Atlanta, GA) at a dose rate of 2.789 Gy/min for the time required to apply a dose of 20 Gy (2000 rads). They were then washed twice more before combining them with responder PBMC (aka precursor alloCTL) at a R:S ratio of 10:1. The cell mixtures were placed into synthetic medium (AIM-V, unless specified) containing 5% serum and 60 IU interleukin-2 (IL-2)/ml (Proleukin, Novartis, San Carlos, CA). Again, the R:S lymphocyte mixtures were placed into tissue culture bags. Growth was monitored daily by taking measurements of lactic acid concentration (2300 STAT Plus, YSI Inc., Yellow Springs, Ohio). Plots of the rate of lactate production were extrapolated to determine medium feedings by keeping them optimally at a lactate concentration between 0.5-0.7 gm/liter [12]. At or about day 14 post-MLR, alloCTL cultures were exposed to relevant or partially-relevant glioma cells or lymphoblasts. The viability and cell numbers of the cultures were determined by trypan blue dye exclusion by counting on a hemocytometer. In one set of experiments alloCTL to U-87MG were made by one-way mixed lymphocyte tumor reaction (MLTR) where the HLA was upregulated on the stimulator tumor cells by IFN-γ incubation prior to inactivation as detailed [15].
Anti-tumor functionality assessments of alloCTL
Cytotoxicity Assay. Lytic activity of alloCTL preparations were assessed at various effector to target ratios (E:T) by 51Chromium release cytotoxicity assays at day 14 post-MLR [10]. Briefly, 5 × 106 lymphoblast or tumor target cells, suspended in 0.1 ml of their growth medium, were labeled with 100 μCi Na251CrO4 (Amersham, Park Ridge, IL) for 60 min at 37°C. Cells were washed twice with HBSS and suspended in AIM-V growth medium. In a final volume of 0.2 ml, 6 × 104 cells were placed into 96 -well round-bottom microtitration plate wells that contained different concentrations of al-loCTL. In blocking assays, neat conditioned medium from the W6/32 hybridoma making anti-HLA-ABC was also placed into wells containing the effectors and targets. In other experiments, temozolomide (15 μM, aka Temodar®, Schering -Plough, Berkeley Heights, NJ) was placed into the glioma cell target medium 12 hr prior to assay and as well was in the cytotoxicity assay medium. The plates were centrifuged at 200 × g for 5 min and then incubated for 4 hr at 37°C, in a humidified, 5% CO2 atmosphere. Following centrifugation at 200 × g for 10 min, 50% of the well volume was harvested and counted. Maximal release was produced by incubation of the targets with 0.1 M HCl. Spontaneous release was the radioactivity as cpm of targets in assay medium alone. The percentage specific release was calculated by the formula: [(51Crexperimental - 51Crspontaneous) / (51Crmaximal “ 51CRspontaneous)] × 100%. Data are given as the mean specific release of triplicate wells, provided that the standard error did not exceed 10% and the specific release was greater than 10%.
7-Amino Actinomycin D (7AAD) Cell Injury Assay. Tumor cell injury was detected by 7AAD incorporation. Glioma cells were seeded into 6-well plates and cultured for 48 hr before labeling for 1 hr with 0.25 mM carboxyfluorescein suc-cinimidyl ester (CFSE) in PBS following manufacturer instruction (Molecular Probes, Eugene OR). Bevacizumab (0.1 mg/ml, aka Avastin®, Genen-tech, So San Francisco, CA) or neat-conditioned medium from the hybridoma W6/32 producing anti-HLA, ABC was added to the assay medium right before the start of the 4 hr assay. AlloCTL effectors were added to the wells at various E:T ratios. After coincubation, adherent and nonad-herent cells were collected from each well. The collected cells were centrifuged at 200 × g for 5 min and incubated in 100 μL of 7AAD (20 μg/ mL, Sigma) in PBS for 20 min at 4°C. The percentages of CFSE-labeled glioma cells within the segregated live, apoptotic, and late apoptotic/ necrotic populations on scattergrams were determined as previously described [25] and plotted by bar diagram as the percentage of cells contained within the 3 segregated areas.
Phenotypic Analysis and Cytokine Production. The BD Fast Immune Kit contains antibodies with various fluorescent probes: surface markers for CD3-APC, CD8-PercP Cy5.5, CD69-PE and also provides for intracellular interferon-y (IFNy-FITC) determination. Labeling was performed according to manufacturer instruction and analyses were performed on a LSR II analytic flow cytometer.
Statistical analyses
Statistical analyses were obtained using Graph-Pad Prism software (version 4, GraphPad Software, San Diego, CA). p values of <0.05 were considered significant when determined by ANOVA.
Results
Testing growth of alloCTL in synthetic media and in tissue culture bags
We tested three synthetically-defined growth mediums commercially available for serum-free culture of lymphoid cells to determine if one provided for more optimal alloCTL growth. Re-sponder lymphocytes mixed with inactivated stimulator lymphocytes (R:S 2 × 107:0.2 × 107 cells) were placed into either AIM-V (Invitrogen, Carlsbad, CA), Invitrus (Cell Culture Technologies, LLC, Gravasano, Switzerland), or Cellgro Free (Mediatech, Manassas, VA) medium. The cultures were initially supplemented with 5% serum from the donor supplying the responder lymphocytes. Representative data are shown that were obtained from the growth mediums from the one-way MLRs that were sampled daily for their lactate concentrations (Figure 1a). A determination of the rates of lactate production was also plotted and 24 hr extrapolations of those values were used to determine feeding needs. Feedings were with medium without serum to wean the cells to a serum-free state. The lymphocyte growth was much better in AIM-V medium compared to that in the Invitrus and Cellgro mediums. The Cellgro culture lagged more so and was discontinued on day 11 post one-way MLR because of poor viability. The viability and cell numbers determined at days 11 and 13 post-MLR (Table 1) showed that the AIM -V medium provided much higher viabilities and cell numbers making it superior to the other two growth mediums for alloCTL production. Cytotoxic T lymphocyte assays performed at various effector to target (E:T) ratios on day 13 post one -way MLR show that alloCTL generated in AIM-V were functionally better than those grown in Invitrus (Figure 1b). While all 3 mediums may suffice for shorter-term LAK cell cultures, the AIM-V medium proved much more satisfactory than Invitrus and Cellgro for alloCTL cultures, which are typically expanded over 2-3 weeks before clinical administration.
Figure 1.

Growth and cytolytic effector function of alloCTL cultured in three synthetically-defined growth media. A) Lactate concentration (mg/L; closed symbols) was measured from medium aliquots taken from alloCTL cultures grown in Aim-V, Invitrus, or CellGro Free (diamonds, circles, and triangles, respectively) and the rate of lactate production (mg/day; open symbols) determined and plotted. B) Stimulator lymphoblast cytolysis by alloCTL grown in Aim-V or Invitrus media at various effector to target (E:T) ratios tested on day 13 post one-way MLR. Data are the mean of triplicate wells + SEM.
Table 1.
Choice of synthetic medium affects viability and total number of alloCTL generated following one-way MLR
| Day of culture | Media | % Viability | # of Cells |
|---|---|---|---|
| 11 | Aim V | 78.3 | 1.3 × 108 |
| Invitrus | 35.6 | 5.0 × 106 | |
| CellGro Free | 4.1 | 3.0 × 105 | |
| 13 | AimV | 79.5 | 1.2 × 108 |
| Invitrus | 36.7 | 6.6 × 106 |
The production of alloCTL for use in the pilot clinical study was refined in closed artificial capillary systems (Cellco Inc, Gaithersburg, MD) composed of cellulosic fibers [12]. The systems are no longer available and similar systems supplied by another company (Fibercell Systems, Gaithersburg, MD) constructed with polyvinyl acetate fibers were not found to be acceptable (data not shown). Fiber absorption of IL-2 from the medium likely prevented expansion of alloCTL following the one-way MLR (personal communication, John Cadwell, Fibercell Systems). Lifecell tissue culture bags, previously reported by other cellular therapy groups generating lym-phokine activated killer (LAK) cells or tumor infiltrating lymphocytes (TIL) for clinical application [26-28], were found to be an appropriate substitute for alloCTL generation. However, as the volume of medium increases in the bags, the culture conditions become more microaerophilic and generally result in lower cell viabilities. Thus, although the bags are less costly, more bags are needed for the longer term alloCTL cultures to maintain lower volumes; the alternative is to harvest the alloCTL a few days earlier to use for clinical administration.
Radiation Doses for Inactivation of Stimulator Lymphocytes That Do Not Affect Lysis by Responder alloCTL
We determined the viabilities of stimulator lymphocytes at various times after their exposure to various doses of irradiation (1700 to 2500 rads). At 24 hr the viabilities for all were sufficiently high at 48-70% to provide for good sensi-tization. At 48 hr the viabilities were between 12 -34%, and at 72 and 96 hr the viabilities of all ranged between 12-25% and 10-17%, respectively (Figure 2).
Figure 2.
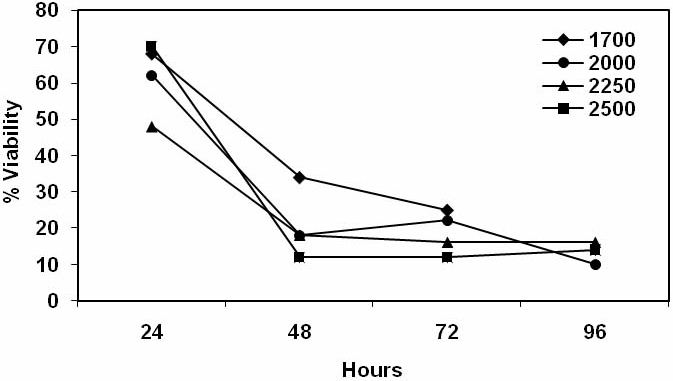
Viability of stimulator lymphocytes following irradiation. Stimulator lymphocytes were inactivated at various doses of irradiation ranging from 1700 to 2500 rads. Cell viabilities were determined each day for 4 days using trypan blue dye exclusion microscopic counts of cells. Representative data are shown that are the mean viabilities from quadruplicate counts.
To investigate whether incomplete inactivation of stimulators (effectively causing a short-lived two-way MLR) may affect the longer-term lytic potential of responder alloCTL, we started oneway MLRs where the stimulators varied in the dose of radiation received (1700 to 2250 rads). The cytotoxicities of the alloCTL responder cells to stimulator target lymphoblasts at two weeks following the MLRs were tested. Representative data are shown at 30:1 E:T ratios (Figure 3). Significantly lower lytic function was obtained when the stimulators received 2250 rads compared to those receiving 1700, or 1850 rads (p≤0.005 by ANOVA). Therefore, 2000 rads for inactivation of the stimulators had an acceptable viability profile over time to enact sensitization, yet not affect long term lytic function, i.e., cytotoxicity was not statistically distinct from the R:S mixtures where the stimulators received a 1700 and 1850 rad dose.
Figure 3.
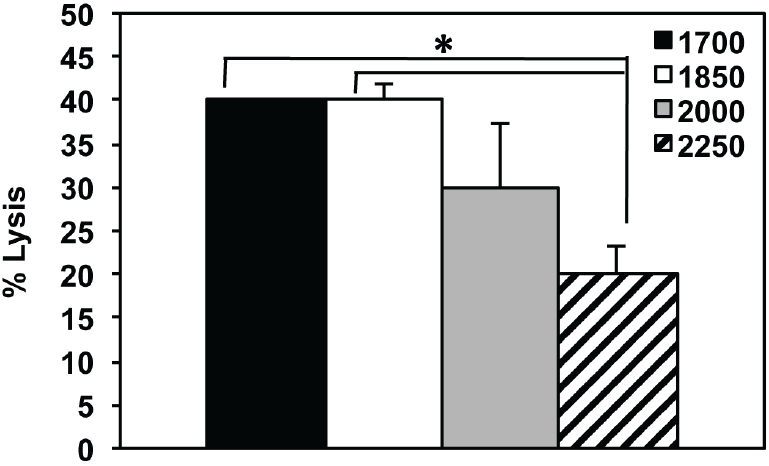
The effects of irradiation dose delivered to stimulator cells at initiation of one-way MLR to cytotoxicity of stimulator target lymphoblasts by alloCTL. Stimulator cells were inactivated by exposure to a doses of irradiation ranging between 1700 to 2250 rads and then co-incubated with responder cells to generate alloCTL. Two weeks later, the cytolytic ability of the alloCTL preparations were tested using a 51Cr release assay at an E:T ratio of 30:1. Data are average of duplicate wells + SEM. *p < 0.005 by ANOVA pairwise comparisons.
Establishing release criteria and expiration time of alloCTL infusates
We examined various parameters that impacted the release of the cultured alloCTL for therapy and the time required to process, qualify, and deliver the alloCTL for patient injection. The precursor alloCTL cultures were phenotyped by flow cytometry with a fluorescently-labeled CD3 monoclonal antibody to determine if the IL-2 (aka T cell growth factor) in the culture medium effectively converted the alloCTL culture to primarily a T-cell phenotype over time [10-12]. The precursor alloCTL preparations generally had an initial CD3 positivity that ranged from 48 to 74%. At two weeks post-MLR the cultures were generally 67 to 90% CD3+. We also validated that alloCTL cultures that exhibit high viability and CD3+ content also exhibit acceptable cyto-toxicity. If the viability of the alloCTL infusates is ≥ 60% and the CD3+ content is also ≥ 60% they can be released for treatment, since we have established that alloCTL with these characteristics routinely exhibit injury ≥ 30% to stimulator lymphoblasts after a 4 hr coincubation with them at a 30:1 E:T ratio. Establishing release criteria of ≥ 60% viability and ≥ 60% CD3+ phenotypic content was reasonable and helped to standardize the cellular therapeutic.
Infusates of alloCTL with rIL-2
When alloCTL are harvested, washed and readied for infusion they are placed into Hanks balanced salt solution (HBSS, 2.5 ml) without phenol red. To allow time for testing of release criteria, transport, and completion of quality control testing, we also add human recombinant IL-2 to the infusates. In the pilot clinical study we added 60,000 IU of IL-2 in the glucose-containing buffer to keep their IL-2 receptors loaded and to maintain their cytolytic function [10, 11]. To reduce the potential for toxicity developing from inflammation and vascular leak syndrome, in addition to reducing IL-2 induced transient changes to neuroimages that may be erroneously interpreted as tumor flare, we investigated whether the amount of IL-2 could be reduced without affecting viability and cytotoxicity. When the alloCTL were freshly harvested and prepared as infusates, we tested the percentage viabilities ± SEM by trypan blue dye exclusion at 4, 8, 10 and 20 hr after their placement onto cold packs. Representative data are given for one alloCTL preparation at 4 time points when they are placed at 60,000, 30,000, or 10,000 International Units (IU) of IL-2 (Figure 4a). The viabilities of the alloCTL at all IL-2 concentrations tested did not fall below 15% of the freshly prepared alloCTL value, however decreases in the alloCTL viabilities for all IL-2 concentrations at the 20 hr timepoint were significant (p < 0.01). In addition, the cytotoxicities to target lymphoblasts determined by 4 hr chromium release assay at a 30:1 E:T are shown at various time points for all IL-2 concentrations (Figure 4b). The cytotoxicities also did not fall below 15% of the value obtained with freshly-prepared alloCTL. The percentage lysis obtained at 10,000 IU IL-2 at 20 hr was the only one that was significantly less (p < 0.01) than that of the freshly isolated alloCTL. Therefore, it was generously safe and reasonable for us to place a 10 hr expiration time on the infusates when 30,000 IU were placed into the alloCTL infusate buffer for this Phase I study. With more preclinical work, it is likely possible to further extend the expiration time placed on the infusates, especially if multi-institutional studies were to be undertaken, as opposed to the current manufacture and clinical trial conduct at a single site.
Figure 4.

Viabilities and cytotoxicities of alloCTL at various times after their placement into infusate buffer containing various amounts of IL-2. alloCTL viabilities (a) and their cytolysis of stimulator target lymphoblasts (b) when they were prepared for infusion by placement into HBSS infusate buffer containing different amounts of IL-2 (10,000 IU, gray bar; 30,000 IU, dotted bar; 60,000 IU, striped bar). Values from freshly harvested alloCTL (black bar) are compared to those determined at times between 4 and 20 hr. Representative data from quadruplicate counts (a) or cpm from triplicate wells (b) show average percentages + SEM. *p < 0.01 by two-way ANOVA with Bonferroni post-test.
alloCTL cytotoxic subset producing IFN-γ
By flow cytometry, a cytotoxic subset within alloCTL preparations was identified by phenotypic markers to produce high levels of IFN-γ within alloCTL preparations generated by one-way MLR. Representative data gathered at 14 days post-MLR (Table 2) show the mean fluorescence intensities (MFI) for IFN-y in the CD3+ T cell population, and for the CD3+/CD8+, CD3+/ CD8+/CD69+, CD3+/CD4+, and CD3+/CD4+/ CD69+ subsets within that population. While one might have anticipated that the T helper CD4+ population would produce higher levels of proinflammatory cytokine, a distinct CD69+ activated subset of CD8+ cells displayed the highest MFI levels for IFN-y. The CD3+/CD8+/ CD69+/IFN-Y+ subset had an MFI of 8950 compared to the MFI for the CD3+/CD4+/CD69+/ IFN-Y+subset at 3792.
Table 2.
IFN-γ expression by CD3+ T cells and subsets of it within an alloCTL preparation
| alloCTL Phenotypic Subset | IFN-γ MFI |
|---|---|
| CD3+ | 2503 |
| CD3+/CD8+ | 1720 |
| CD3+/CD8+/CD69+ | 8950 |
| CD3+/CD4+ | 2600 |
| CD3+/CD4+/CD69+ | 3792 |
The IFN-y producing cytotoxic subset within alloCTL preparations is upregulated upon exposure to relevant target cells
A different preparation of alloCTL was restimulated at day 12 with relevant target glioma cells at an E:T of 10:1 and was analyzed 18 hr later for the percentages of CD3+/CD8+/CD69+ and CD3+/CD4+/CD69+ subsets that did or did not produce IFN-y (Table 3). The coincubation of the alloCTL with relevant glioma cell targets induced a greatly upregulated IFN-y MFI by the CD8+/ CD69+ subset, whereas the induction of IFN-y by the CD4+/CD69+ was not robust. Interestingly, the percentage of the CD3+ cells within the alloCTL preparation that also expressed CD8+ and CD69+ totaled 10.5% and half of those cells (52.1%) also expressed IFN-y at an MFI of 500. After 18 hr of incubation with relevant glioma target cells the CD3+/CD8+/CD69+ subset increased six-fold and over a third of those cells (34.7%) produced IFN-y at a five-fold higher level. In contrast, the CD3+/CD4+/ CD69+ subset within the same alloCTL preparation that also produced IFN-y was less than 1% of the total CD3+ population and after restimulation remained less than 1%. Therefore, the activated memory T cell subset (CD8+/CD69+) reacted in response to seeing relevant target antigen by upregulating IFN-y, more so than the activated helper (CD4+/CD69+) T cell subset.
Table 3.
T cell phenotypic subsets producing IFN-γ within an alloCTL preparation before and after restimulation with relevant target glioma cells.
| alloCTL ± relevant target glioma | T-cell subset phenotype | % of the total CD3+ cells | % of CD3+ subset also IFN-γ+ | IFN-γ MFI |
|---|---|---|---|---|
| alloCTL | CD3+/CD8+/CD69+ | 10.5% | 52.1% | 500 |
| alloCTL + target glioma | CD3+/CD8+/CD69+ | 62.7% | 34.7% | 2543 |
| alloCTL | CD3+/CD4+/CD69+ | 35.3% | 0.70% | 254 |
| alloCTL + target glioma | CD3+/CD4+/CD69+ | 80.0% | 0.90% | 1605 |
Commonly used treatment agents, temo-zolomide (Temodar®) or bevacizumab (Avastin®), do not inhibit the cytotoxic function of alloCTL
Standard of care treatment for patients diagnosed with high grade gliomas consists of de-bulking surgery followed by radiation therapy with concurrent temozolomide administered at 75 mg/m2 for 6 weeks followed by 6 months of pulse temozolomide administered daily for 5 days a month at a dose of 150-200 mg/m2 [3]. Patients who then recur with high grade glioma are generally treated with bevacizumab administered at 10-15 mg/kg every 2-3 weeks [19, 29-32]. Due to the modest increase in efficacy obtained with the chemo-radiation and anti-angiogenic therapies, modification of those regimens with other synergistic agents should be explored. To consider addition of adjuvant immunotherapy to standardized therapy, we conducted experiments to determine if temozolomide or bevacizumab interfere with the cytotoxic function of the alloCTL. Target 13-06-MG glioma cells were preincubated with temozolomide in the medium for 12 hr prior to assay and it was also contained within the cytotoxicity assay buffer when the alloCTL were added. Representative results from a 4 hr chromium release cytotoxicity assay are shown with day 14 alloCTL made by MLR that were directed towards the HLA of 13-06-MG cells (Figure 5). No statistically significant differences were obtained in the lysis of the glioma target cells at the 3 different E:T ratios tested, indicating temozolomide does not adversely affect lysis by the alloCTL.
Figure 5.

Effects of temozolomide on alloCTL cytotoxicity to glioma cells. Day 14 alloCTL were harvested and mixed at various E:T ratios with target 13-06-MG glioma cells that were preincubated with (white bars) or without (black bars) temozolomide 12 hr prior to assay and during the 4 hr cytotoxicity assay. Cytoly-sis was quantified by 51Cr-release and displayed as the average percentage lysis ± SEM at the 3 E:T ratios shown, calculated from wells in triplicate.
In another set of cytotoxicity experiments using 7AAD uptake as a measure of cell injury, CFSE-labeled target U-87MG glioma cells were coincubated at various E:T ratios for 4 hr with day 14 alloCTL made by MLTR that were directed towards the HLA of the U-87MG cells. The cell injury induced by the alloCTL alone, when bevacizumab was placed into the assay medium, or when anti-HLA-ABC was placed into the assay medium is shown (Figure 6). As a baseline, when no alloCTL are added (0:1 E:T), the viable tumor cell percentage is 90%. When alloCTL are added significant tumor cell injury (early apoptotic, late apoptotic/necrotic cells = 72-93%) was engendered at all the E:T ratios tested. The addition of bevacizumab largely does not affect the degree of injury to the target cells by the alloCTL (early apoptotic, late apoptotic/necrotic = 66-86%). For comparison purposes, at the 10:1 E:T, the average necrotic/ dead cells totaled 29% with alloCTL alone, 33% when bevacizumab was added, and 17% when anti-HLA-ABC was added. The protective effect provided by addition of the anti-class I to the E:T coincubates indicated alloreactive T cells were present in the effector cell preparation.
Figure 6.

Effects of bevacizumab on alloCTL induced cell injury to glioma target cells. Day 14 alloCTL directed toward the HLA of U-87MG glioma cells were made by MLTR, harvested and mixed with CFSE-labeled glioma cells at various E:T ratios. al-loCTL:glioma cells were either coincubated alone, or had bevacizumab, or class I HLA, ABC blocking antibody added to them. The uptake of 7AAD was quantified after 4 hr by flow cytometric scattergram analysis. The average percentages of CFSE-labeled glioma cells that excluded 7AAD were considered viable (black). The cell percentages that took up less 7AAD are early apopototic (gray), and those that took up more are late apopototic/necrotic (white). The viable and cell injury stages are displayed in the bar diagram as the average percentages from duplicate wells + SEM. Uptake of 7AAD by target cells without the addition of effector cells (E:T of 0:1) is also shown.
Discussion
While surgery effectively eliminates bulk tumor, adjuvant therapies must eliminate the tumor arising from infiltrative cells migrating from the main tumor mass. Refinement of experimental immunotherapies has led to increased numbers of passive cellular and active immunotherapy approaches available for patient enrollment [4]. One reason a higher response to immune therapies is being obtained might be due to additional steps taken to decrease immunosuppression and because adjuvant immunotherapies are being offered upfront to newly diagnosed, recently resected glioma patients [33].
Our localized cellular therapy with alloresponsive T lymphocytes holds advantage because T cells are able to move through tissue as part of their immunosurveillance function. Ex-vivo activated T cells have the capability of trafficking to and making direct contact with tumor cells to effect their lysis, or if coming within close proximity to them can produce cytokines that can induce tumor cell apoptosis [6, 15]. Thus, adoptive transfer of CTL may initially cause tumor cell injury, then endogenous immune cells may take up and present tumor antigens further inducing beneficial long-term immune effects.
A small pilot study with intratumoral alloCTL was conducted before to test clinical feasibility and safety [10, 11]. We have recently reactivated the IND (BB IND 5423) with the FDA and designed a dose escalation trial (www.clinicaltrials.com, NCT01144247) to confirm our earlier findings of possible patient response to the immunotherapy by recurrent WHO grade III glioma patients, who all are either still alive or survived much longer than expected.
The study is predicated on the fact that freshly isolated normal brain cells display little or no human leukocyte antigens, whereas primary glioma cell explants exhibit high expression of class I HLA [15]. As such, alloCTL derived from healthy individuals that are sensitized to the HLA of the brain tumor patient display specificity to patient glioma cells. At UCLA we are enrolling recurrent glioma patients into a Phase I dose-escalation trial. Modifications to the initial pilot study and the present Phase I trial are detailed (Table 4). Our preclinical studies tested various other alternative, less-costly synthetic mediums, but the more costly AIM-V medium was optimal for alloCTL growth. We did adapt generation of the alloCTL from artificial capillary systems to tissue culture bags, which effectively reduced the cost of cellular therapy. In the pilot study, we contained costs by purchasing the waste leucopak product from platelet collection as a source of precusor alloCTL. In the present dose escalation study, where therapeutic alloCTL cell numbers must be guaranteed and where alloCTL are required for other correlative laboratory studies, we are performing more costly leukapheresis of donor subjects. When we determine the maxmimum tolerated dose and/or beneficial infusion schedule, we may find we can revert to less costly precursor alloCTL sources or fewer leukapheresis procedures. In addition, in the pilot study we used a dose of 60,000 IU of IL-2 in the infusates to maintain cytolytic function of the alloCTL. Because of the UCLA Data Safety Monitoring Board's perception of potential for toxicity and vascular leak caused by the proposed 60,000 IU of IL-2 placed in the alloCTL infusates, we were asked to investigate whether the amount could be lowered. The study indicated the infusate dose could be reduced by half (30,000 IU) for the present clinical study. We may find that lower amounts of IL-2 in the alloCTL infusates will result in less vascular leak or other proinflammatory immune effects that transiently cause changes to follow-up neuroimages. We have experienced problems using conventional radiologic criteria when assessing clinical response to immunotherapy. Response may be associated with immune cell infiltration and inflammation that appears as “tumor flare"; this has been noted in the clinical setting with other experimental immunotherapy studies [4, 34, 35]. The enhancement gives the impression of an enlarging lesion by radiological imaging and can be erroneously interpreted as progressive disease. This has resulted in removal of patients from potentially beneficial experimental immunotherapy protocols. In the present clinical study we are also trying to provide more flexibility in how the alloCTL are first administered and also better standardize the alloCTL therapeutic used for treatment. We have established release criteria, and now better control the maximum number of infusate doses and time between doses. In the present study we will typify patient immune response to treatment by monitoring for pre vs post-treatment increases in patient PBMC producing T helper 1:T helper 2 cytokines. Also, the alloCTL will be monitored in vitro for increases in the IFN-γ producing activated cytotoxic subset upon exposure to relevant target cells to determine if the increases correspond to acute patient toxicity and/or long-term response. Finally, we are studying the molecular HLA disparities between donor and patient to assess what mismatches are permissive for production of functionally-robust alloCTL.
Table 4.
Differences between Pilot Clinical Trial and Current Phase I Clinical Trial
| Pilot Clinical Trial [10-11] | Phase I Clinical Trial (NCT01144247) |
|---|---|
| alloCTL generated in artificial capillary systems | alloCTL are generated in tissue culture bags |
| Source of donor responder cells derived from apheresis waste leucopaks from platelet collection | Source of donor responder cells are derived by leukapheresis |
| alloCTL infusates contained 60,000 IU recombinant IL-2 | alloCTL infusates contain 30,000 IU recombinant IL-2 |
| 1-3 alloCTL infusates/cycle, infusates given over 7-10 days, all cells generated used for therapy, doses varied | 2 alloCTL infusates/cycle, infusates given 7 days apart, doses uniform with 2-fold dose escalation |
| 1st alloCTL injection at surgery by blunt brain cannula; subsequent infusions given through subgaleally-placed reservoir/ catheter | 1st alloCTL injection at surgery or up to a few weeks later through subgaleally-placed reservoir/ catheter |
| Patient immune responses were not characterized | Patient immune responses are characterized; phenotypic subset response to relevant antigen monitored in vitro and correlated with response |
| Patients and donors were serologically HLA typed and disparate at2HLAA,Bloci | Patients and donors are molecularly HLA typed and disparities at class I and II loci typified, i.e., HLA-ABC, DR,DQ |
The neuro-oncology field has evolved in the last ten years. Temozolomide is an orally available drug that is converted spontaneously to 5-(3-methyltriazen-1-l)imidazole-4-carboximide, the active metabolite of dacarbazine. The FDA approved it for the treatment of high grade gliomas early after diagnosis and also at recurrence [3]. As well, the FDA has sanctioned bevacizu-mab, a monoclonal antibody to VEGF that induces VEGF-blockade, for glioblastoma treatment [19]. Nondetrimental and possibly even synergistic effects of these two agents to immunotherapy regimens may be possible because their mechanisms of action are distinct from immune mediated effects, or the agents may act to enhance immune function. For instance, there is evidence that T regulatory cells were markedly depressed in patients receiving temozolomide, indicating it may have specific effects to T cells with an inhibitory immunosuppressive phenotype [36, 37]. Furthermore, other reports suggest that immune responses may be dramatically increased in the context of VEGF inhibition. For example, decreased levels of VEGF are associated with an increase in T-cell infiltration into tumors [38]. Bevacizumab may also abrogate immunosuppressive effects [39]. Strategies that inhibit this classically angiogenic axis have shown synergistic effects with anti-tumor immunotherapy [40, 41]. Our preliminary data support the feasibility of combining adoptive immunotherapy with alloCTL with these two therapeutic agents that have either become standard of care or used off-label to treat glioma patients. Indeed, our examination shows alloCTL to retain anti-tumor functionality when placed in the presence of these therapeutic agents. Temozolomide, at a concentration found to be present in CSF [42], did not appear to negatively influence glioma cell lysis by alloCTL in 4 hr Cr-51 release assays, and bevacizumab also did not appear to inhibit alloCTL cell injury to glioma cells. Successful treatment of gliomas may require more aggressive stances to be taken in the clinic. It is necessary to test combinatorial therapies to find those that display pre-clinical efficacy and advance them to the clinic [4,43,44].
Acknowledgments
Supported in part by R01CA125244 (CAK/LML/ RMP), R01CA154256 (CAK/RMP), R01 CA121258 (CAK), USAMRMC W81XWH-08-1-0734 (CAK), the Joan S. Holmes Memorial Research Fund (CAK/LML), and the Joan S. Holmes Memorial Postdoctoral Fellowship (MJH).
Glossary
Abbreviations
- (alloCTL)
alloreactive cytotoxic T lymphocytes
- (aka)
also known as
- (CTL)
cytotoxic T lymphocyte
- (E:T)
effector to target
- (HLA)
human leukocyte antigens
- (IL-2)
interleukin-2
- (IU)
international units
- (IFN-γ)
interferon-gamma
- (MLR)
mixed lymphocyte reaction
- (MLTR)
mixed lymphocyte tumor reaction
- (PBMC)
peripheral blood mononuclear cells
- (R)
responder
- (R:S)
responder to stimulator ratio
- (TMZ)
temozolomide
- (S)
stimulator
- (SEM)
standard error of the mean
Conflict of Interest
None.
References
- 1.CBTRUS Statistical report: Primary brain and central nervous system tumors diagnosed in the United States, 2004-2006, Central Brain Tumor Registry of the United States. CBTRUS. 2010:p1–61. [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, All-geier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Hickey MJ, Malone CC, Erickson KL, Jadus MR, Prins RM, Liau LM, Kruse CA. Cellular and vaccine therapeutic approaches for gliomas. J Transl Med. 2010;8:100–110. doi: 10.1186/1479-5876-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleshner M, Watkins LR, Redd JM, Kruse CA, Bellgrau D. A 9L gliosarcoma transplantation model for studying adoptive immunotherapy into the brains of conscious rats. Cell Transplant. 1992;1:307–312. doi: 10.1177/096368979200100408. [DOI] [PubMed] [Google Scholar]
- 6.Kruse CA, Kong Q, Schiltz PM, Kleinschmidt-DeMasters BK. Migration of activated lymphocytes when adoptively transferred into cannulated rat brain. J Neuroimmunol. 1994;55:11–21. doi: 10.1016/0165-5728(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 7.Kruse CA, Lillehei KO, Mitchell DH, Kleinschmidt-DeMasters B, Bellgrau D. Analysis of interleukin 2 and various effector cell populations in adoptive immunotherapy of 9L rat gliosarcoma: allogeneic cytotoxic T lymphocytes prevent tumor take. Proc Natl Acad Sci USA. 1990;87:9577–9581. doi: 10.1073/pnas.87.24.9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruse CA, Schiltz PM, Bellgrau D, Kong Q, Kleinschmidt-DeMasters BK. Intracranial administrations of single or multiple source allogeneic cytotoxic T lymphocytes: chronic therapy for primary brain tumors. J Neurooncol. 1994;19:161–168. doi: 10.1007/BF01306458. [DOI] [PubMed] [Google Scholar]
- 9.Redd JM, Lagarde AC, Kruse CA, Bellgrau D. Allogeneic tumor-specific cytotoxic T lymphocytes. Cancer Immunol Immunother. 1992;34:349–354. doi: 10.1007/BF01741557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kruse CA, Cepeda L, Owens B, Johnson SD, Stears J, Lillehei KO. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunol Immunother. 1997;45:77–87. doi: 10.1007/s002620050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruse CA, Rubinstein D. Cytotoxic T Lymphocytes Reactive to Patient Major Histocompatibility Proteins for Therapy of Recurrent Primary Brain Tumors. In: Liau LM, Cloughesy TF, Becker DP, Bigner DD, editors. Brain Tumor Immunotherapy. Totowa: Humana Press; 2001. pp. 149–170. [Google Scholar]
- 12.Kruse CA, Beck LT. Artificial-capillary-system development of human alloreactive cytotoxic T-lymphocytes that lyse brain tumours. Biotechnol Appl Biochem. 1997;25(Pt 3):197–205. [PubMed] [Google Scholar]
- 13.Lampson LA. Interpreting MHC class I expression and class I/class II reciprocity in the CNS: reconciling divergent findings. Microsc Res Tech. 1995;32:267–285. doi: 10.1002/jemt.1070320402. [DOI] [PubMed] [Google Scholar]
- 14.Lampson LA, Hickey WF. Monoclonal anti body analysis of MHC expression in human brain biopsies: tissue ranging from "histologically normal” to that showing different levels of glial tumor involvement. J Immunol. 1986;136:4054–4062. [PubMed] [Google Scholar]
- 15.Read SB, Kulprathipanja NV, Gomez GG, Paul DB, Winston KR, Robbins JM, Kruse CA. Human alloreactive CTL interactions with gliomas and with those having upregulated HLA expression from exogenous IFN-γ or IFN-γ gene modification. J Interferon Cytokine Res. 2003;23:379–393. doi: 10.1089/107999003322226032. [DOI] [PubMed] [Google Scholar]
- 16.Kuppner MC, Hamou MF, de Tribolet N. Activation and adhesion molecule expression on lymphoid infiltrates in human glioblastomas. J Neuroimmunol. 1990;29:229–238. doi: 10.1016/0165-5728(90)90166-k. [DOI] [PubMed] [Google Scholar]
- 17.Bigner DD, Bigner SH, Ponten J, Westermark B, Mahaley MS, Ruoslahti E, Herschman H, Eng LF, Wikstrand CJ. Heterogeneity of genotypic and phenotypic characteristics of fifteen permanent cell lines derived from human gliomas. J Neuropathol Exp Neurol. 1981;40:201–229. doi: 10.1097/00005072-198105000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Kulprathipanja NV, Kruse CA. Microglia phagocytose alloreactive CTL-damaged 9L gliosarcoma cells. J Neuroimmunol. 2004;153:76–82. doi: 10.1016/j.jneuroim.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblas-toma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 20.Marzolini C, Decosterd LA, Shen F, Gander M, Leyvraz S, Bauer J, Buclin T, Biollaz J, Lejeune F. Pharmacokinetics of temozolomide in association with fotemustine in malignant melanoma and malignant glioma patients: comparison of oral, intravenous, and hepatic intra-arterial administration. Cancer Chemother Pharmacol. 1998;42:433–440. doi: 10.1007/s002800050842. [DOI] [PubMed] [Google Scholar]
- 21.Gomez GG, Kruse CA. Isolation and culture of human brain tumor cells. Methods Mol Med. 2004;88:101–109. doi: 10.1385/1-59259-406-9:101. [DOI] [PubMed] [Google Scholar]
- 22.Gomez GG, Varella-Garcia M, Kruse CA. Isolation of immunoresistant human glioma cell clones after selection with alloreactive cytotoxic T lymphocytes: cytogenetic and molecular cytogenetic characterization. Cancer Genet Cytogenet. 2006;165:121–134. doi: 10.1016/j.cancergencyto.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez GG, Hickey MJ, Tritz R, Kruse CA. Immunoresistant human glioma cell clones selected with alloreactive cytotoxic T lymphocytes: Downregulation of multiple proapoptotic factors. Gene Ther Mol Biol. 2008;12:101–110. [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S, Ma W, Hoa N, Minev B, Delgado C, Wepsic HT, Okada H, Jadus MR. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res. 2007;13:566–575. doi: 10.1158/1078-0432.CCR-06-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez GG, Read SB, Gerschenson LE, Santoli D, Zweifach A, Kruse CA. Interactions of the allogeneic effector leukemic T cell line, TALL-104, with human malignant brain tumors. Neuro Oncol. 2004;6:83–95. doi: 10.1215/S1152851703000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillman RO, Duma CM, Schiltz PM, DePriest C, Ellis RA, Okamoto K, Beutel LD, De Leon C, Chico S. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. 2004;27:398–404. doi: 10.1097/00002371-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Yannelli JR, Thurman GB, Dickerson SG, Mrowca A, Sharp E, Oldham RK. An improved method for the generation of human lymphokine activated killer cells. J Immunol Methods. 1987;100:137–145. doi: 10.1016/0022-1759(87)90182-7. [DOI] [PubMed] [Google Scholar]
- 28.Topalian SL, Muul LM, Solomon D, Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods. 1987;102:127–141. doi: 10.1016/s0022-1759(87)80018-2. [DOI] [PubMed] [Google Scholar]
- 29.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Wagner M, Bailey L, Bigner DD, Friedman AH, Friedman HS. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 30.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 31.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, Park J, Albert PS, Fine HA. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raizer JJ, Grimm S, Chamberlain MC, Nicholas MK, Chandler JP, Muro K, Dubner S, Rade-maker AW, Renfrow J, Bredel M. A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer. 2010;116:5297–5305. doi: 10.1002/cncr.25462. [DOI] [PubMed] [Google Scholar]
- 33.Gomez GG, Kruse CA. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene Ther Mol Biol. 2006;10:133–146. [PMC free article] [PubMed] [Google Scholar]
- 34.Yang I HN, Smith ZA, Han SJ, Parsa AT. Distinguishing glioma recurrence from treatment effect after radiochemotherapy and immunotherapy. Neurosurg Clin N Am. 2010;21:181–186. doi: 10.1016/j.nec.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Floeth FW, Wittsack HJ, Engelbrecht V, Weber F. Comparative follow-up of enhancement phenomena with MRI and proton MR spectroscopic imaging after intralesional immunotherapy in glioblastoma–Report of two exceptional cases. Zentralbl Neurochir. 2002;63:23–28. doi: 10.1055/s-2002-31579. [DOI] [PubMed] [Google Scholar]
- 36.Sakaguchi S, Takahashi T, Yamazaki S, Kuniyasu Y, Itoh M, Sakaguchi N, Shimizu J. Immunologic self tolerance maintained by T-cell-mediated control of self-reactive T cells: implications for autoimmunity and tumor immunity. Microbes Infect. 2001;3:911–918. doi: 10.1016/s1286-4579(01)01452-6. [DOI] [PubMed] [Google Scholar]
- 37.Su YB, Sohn S, Krown SE, Livingston PO, Wolchok JD, Quinn C, Williams L, Foster T, Sepkowitz KA, Chapman PB. Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: a toxicity with therapeutic implications. J Clin Oncol. 2004;22:610–616. doi: 10.1200/JCO.2004.07.060. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 39.Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, Hurwitz HI, Dev I, Nixon AB, Lyerly HK, Clay T, Morse MA. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother. 2008;57:1115–1124. doi: 10.1007/s00262-007-0441-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, Prell R, VanRoey MJ, Simmons AD, Jooss K. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res. 2006;12:6808–6816. doi: 10.1158/1078-0432.CCR-06-1558. [DOI] [PubMed] [Google Scholar]
- 41.Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, Hicklin DJ, Jaffee EM, Emens LA. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–3959. doi: 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 42.Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, Stupp R. Plasma and cerebrospinal fluid population pharma-cokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10:3728–3736. doi: 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- 43.Desjardins A, Reardon DA, Herndon JE, 2nd, Marcello J, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Bailey L, Bigner DD, Friedman AH, Friedman HS, Vreden-burgh JJ. Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res. 2008;14:7068–7073. doi: 10.1158/1078-0432.CCR-08-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling R, Shi W, Vredenburgh JJ, Bigner DD, Heimberger AB. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–333. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]