

Statistical imputation of genotype data is an important statistical technique that uses patterns of linkage disequilibrium observed in a reference set of haplotypes to computationally predict genetic variants in silico1. Currently, the most popular reference sets are the publicly available International HapMap2 and 1000 Genomes datasets3. While these resources are valuable for imputing a sizeable fraction of common SNPs, they may not be optimal for imputing data for the next generation of genome-wide association studies (GWAS) and SNP arrays, which explore a fraction of uncommon variants.
We have built a new resource for imputation of SNPs for existing and future GWAS, known as the Division of Cancer Epidemiology and Genetics (DCEG) Reference Set. The dataset includes 728 cancer-free individuals of European ancestry from three large prospectively sampled studies4-6, 98 African-American individuals from the Prostate, Lung, Colon, and Ovary Cancer Screening Trial (PLCO), 74 Chinese individuals from a clinical trial in Shanxi, China (SHNX)7, and 349 individuals from the HapMap Project (see Table 1). The final harmonized dataset includes 2.8 million autosomal polymorphic SNPs on 1,249 subjects after rigorous quality control metrics were applied (see Supplementary Methods).
Table 1. Samples included in the DCEG Reference Set.
Subjects passing quality control metrics for the SNP arrays indicated in the right hand columns. This table reports the content of Build 1.
| Populations | Illumina Array | |||||||
|---|---|---|---|---|---|---|---|---|
| Group | European AmericanAfrican | American | African | Asian | Hap660 | Hap1 | Omni1 | Omni2.5 |
| ATBC | 246 | ✓ | ✓ | ✓ | ||||
| CPSII | 227 | ✓ | ✓ | ✓ | ||||
| PLCO | 255 | ✓ | ✓ | ✓ | ||||
| PLCO | 98 | ✓ | ✓ | |||||
| SHNX | 74 | ✓ | ✓ | |||||
| HapMap | ||||||||
| CEU | 116 | ✓ | ||||||
| CHB | 44 | ✓ | ||||||
| JPT | 44 | ✓ | ||||||
| TSI | 86 | ✓ | ||||||
| YRI | 59 | ✓ | ||||||
|
| ||||||||
| Total | 930 | 98 | 59 | 162 | ||||
We compared the imputation performance of the DCEG Reference Set to the International HapMap and 1000 Genomes reference sets, which were available from the IMPUTE2 web site (URL below). We assessed imputation accuracy using directly genotyped SNP data from the DCEG Reference Set and masking subsets to simulate data from two common low-cost commercial genotyping arrays used in GWAS studies (Illumina Human Hap660 and OmniExpress). Probabilistic genotypes were imputed using both IMPUTE2 (ref. 8) and BEAGLE (ref. 9) software and compared with the unmasked genotyped SNPs. Accuracy was measured using the squared-Pearson correlation coefficient (R2) under a trend/dosage model (see Supplementary Methods). We observed higher imputation accuracy compared to the combination of 1000 Genomes and HapMap data across a spectrum of minor allele frequencies (MAF) (see Figure 1). Accuracy in individuals of European ancestry imputed from Hap660 or OmniExpress arrays, measured by the proportion of variants imputed with R2>0.8, improved by 34%, 23% and 12% for variants with MAF of 3%, 5% and 10%, respectively. We estimated the difference in power to detect associations in GWAS design between an imputed dataset and one composed of directly genotyped SNPs in the DCEG Reference Set based on a model of Park et al.10. When using Hap660 data to impute, we observed detection rates of 92.9% when imputing with the DCEG Reference Set and 84.7% with the 1000 Genomes and HapMap reference relative to the detection rate attained with directly genotyped SNPs; for OmniExpress data, we observed detection rates of 93.9% and 86.2%, respectively.
Figure 1. Imputation accuracy for individuals of European ancestry with DCEG and public reference set.
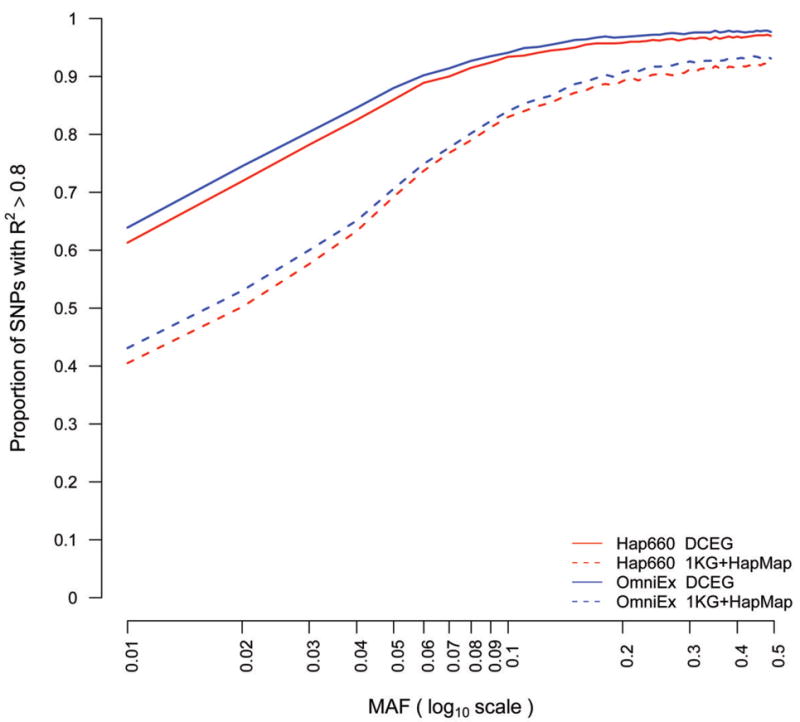
The proportion of SNPs with allelic dosage R2 > 0.8 by MAF, is shown on the log scale to emphasize differences at smaller values. Solid red depicts imputation of Hap660 data using the DCEG Reference Set. Dashed red depicts imputation of Hap660 using the 1000 Genome plus HapMap3 reference. Solid blue depicts imputation of OmniExpress data using the DCEG Reference Set. Dashed blue depicts imputation of OmniExpress using the 1000 Genome plus HapMap3 reference.
Since imputation accuracy depends on similarity of haplotypes between reference and study populations, we examined an extreme scenario in which we used a reference population from Finland (ATBC) to impute genotypes using OmniExpress data from a US population of European ancestry (PLCO) (Supplementary Figure 1). For common SNPs, there was minimal loss of imputation accuracy when using the reference population from Finland, relative to another US based study (CPSII) or to HapMap CEPH and Northern Italian reference (CEU+TSI populations). This suggests that for common variants a reference set of sufficient size can adequately predict common SNPs when there is a discrepancy in population genetics history, provided that comparable haplotypes are sufficiently represented. This observation could enable investigators to proceed more confidently with imputation without additional genotyping in related but not identical populations.
Although the current build of the DCEG Reference Set is primarily intended for use in European populations, we tested the accuracy of imputing OmniExpress data on an African-American sample set11 (Supplementary Figures 2, 3) finding the accuracy lower than with the European samples, but still superior to the publicly available reference data for African-American populations. We attribute the lower accuracy to the relatively small number of individuals of African descent in the reference panels, though we observed that adding a sufficiently large sample set of individuals of European background can improve imputation in African-Americans. Although frequencies of variants may differ among population groups and subpopulations, our findings suggest that genotype imputation is relatively robust to these differences provided that a sufficient number of matching haplotypes appear in the reference data. These results could have an impact on the design of future studies in other populations with distinct substructures.
We compared the imputation performance using OmniExpress data from individuals of European background with a reference dataset that combined the DCEG Reference Set and 1,000 Genomes and HapMap data and observed no increase in accuracy over that achieved using the DCEG Reference Set alone. One explanation for the lack of improvement is that SNP array and sequencing data have distinct patterns of non-random errors. While suboptimal for imputation, it may also be necessary to preserve these errors when combining directly genotyped and imputed data in order to recapitulate the patterns of differential misclassification and perhaps retain statistical validity for association testing. Thus, SNP array data should be superior for imputation of genotypes obtained from SNP arrays, though not necessarily superior at imputing the true genotypes. While low-pass sequencing data may capture more variants, the cumulative effects of both higher false-positive and false-negative rates may also decrease imputation accuracy.
The DCEG Reference data set should be a valuable resource for the next-generation of GWAS with denser assays that include uncommon variants and it should enable investigators to conduct meta-analyses across SNPs arrays more efficiently. The DCEG Reference Set is available from the Database of Genotypes and Phenotypes (dbGaP accession number phs000396.v1.p1) in several formats along with documentation on how to use the data with the IMPUTE2 and BEAGLE programs. We anticipate generating subsequent data builds that will both expand the number of subjects from diverse populations and add new assay content from the Affymetrix 6.0/Axiom, Illumina Omni5 arrays, and other future commercial genotyping products.
Supplementary Material
Acknowledgments
The genotyping in the MEC was supported by a Department of Defense Breast Cancer Research Program Era of Hope Scholar Award to CA Haiman [W81XWH-08-1-0383], and National Institutes of Health grant CA132839.
Footnotes
References
- 1.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 2.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prorok PC, et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials. 2000;21:273S–309S. doi: 10.1016/s0197-2456(00)00098-2. [DOI] [PubMed] [Google Scholar]
- 5.The alpha-tocopherol, beta-carotene lung cancer prevention study: design, methods, participant characteristics, and compliance. The ATBC Cancer Prevention Study Group. Ann Epidemiol. 1994;4:1–10. doi: 10.1016/1047-2797(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 6.Calle EE, et al. The American Cancer Society Cancer Prevention Study II Nutrition Cohort: rationale, study design, and baseline characteristics. Cancer. 2002;94:2490–501. doi: 10.1002/cncr.101970. [DOI] [PubMed] [Google Scholar]
- 7.Ke L. Mortality and incidence trends from esophagus cancer in selected geographic areas of China circa 1970-90. Int J Cancer. 2002;102:271–4. doi: 10.1002/ijc.10706. [DOI] [PubMed] [Google Scholar]
- 8.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–23. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park JH, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–5. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolonel LN, et al. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. Am J Epidemiol. 2000;151:346–57. doi: 10.1093/oxfordjournals.aje.a010213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.