

Abstract
The molecular mechanisms regulating fate divergence of closely related, but distinct, layer 6 corticothalamic and layer 5 subcerebral projection neurons are largely unknown. We present evidence for central transcriptional mechanisms that regulate fate specification of corticothalamic (layer 6) and subcerebral (layer 5) projection neurons. We found that TBR1 promotes the identity of corticothalamic neurons and represses subcerebral fates through reducing expression of Fezf2 and CTIP2. These conclusions are based on the following: (1) In Tbr1−/− mice, the number of cells expressing layer 6 markers was reduced, and the number of cells expressing layer 5 markers was increased. Early-born (birthdated on E11.5) neurons ectopically expressed subcerebral neuronal markers, and extended their axons into subcerebral targets. (2) Ectopic Tbr1 expression in layer 5 neurons prevented them from extending axons into the brainstem and the spinal cord. (3) Chromatin immunoprecipitation analysis using TBR1 antibodies showed that TBR1 bound to a conserved region in the Fezf2 gene. (4) Analysis of Fezf2 mutants and Tbr1−/−; Fezf2−/− compound mutants provided evidence that Fezf2 blocks corticothalamic fate in layer 5 by reducing Tbr1 expression in subcerebral neurons. All neocortical regions appear to use this core transcriptional program to specify corticothalamic (layer 6) and subcerebral (layer 5) projection neurons.
Introduction
The cerebral cortex contains six layers of neurons. Neurons within each layer adopt specific identities and form appropriate local and long-distance connections (O'Leary and Koester, 1993; McConnell, 1995). Cortical projection neurons can be classified into two broad classes: corticocortical neurons and corticofugal neurons. The corticocortical neurons project axons to ipsilateral and contralateral cortices and are in layers 2 through 6. Corticofugal neurons are further divided into two groups: corticothalamic neurons, which reside in layer 6 and extend their axons into thalamus; and subcerebral neurons, which are confined to layer 5 and project axons into basal ganglia, diencephalon, midbrain, hindbrain, and spinal cord.
Despite intensive investigation, only recently have the molecular mechanisms regulating cortical neuron identity begun to be uncovered. FEZF2 and CTIP2, two transcription factors expressed in deep-layer neurons, regulate the identity and differentiation of subcerebral projection neurons (Arlotta et al., 2005; B. Chen et al., 2005, 2008; J. G. Chen et al., 2005; Molyneaux et al., 2005). The corticospinal tract (CST) is missing in Ctip2−/− and Fezf2−/− mice (Arlotta et al., 2005; B. Chen et al., 2005; Molyneaux et al., 2005). The absence of high CTIP2 expression in the Fezf2−/− mice suggests that Ctip2 is a downstream effector of Fezf2. Indeed, restoring Ctip2 expression in Fezf2−/− mice rescues CST development (Chen et al., 2008).
SATB2, an AT-rich DNA-binding protein, is specifically expressed in callosal projection neurons and regulates their identity (Alcamo et al., 2008; Britanova et al., 2008). In Satb2−/− mice, CTIP2 expression is upregulated, and the mutant neurons send their axons subcortically (Alcamo et al., 2008; Britanova et al., 2008). SATB2 protein binds to the Ctip2 locus and inhibits its expression (Alcamo et al., 2008; Britanova et al., 2008). Interestingly, SATB2 expression is increased in deep-layer neurons of Fezf2−/− mice (Chen et al., 2008), and some Fezf2−/− neurons switch their identity and adopt the electrophysiological and axonal targeting properties of callosal neurons (Chen et al., 2008).
Molecular mechanisms defining the identity of corticothalamic neurons are not defined. SOX5, an SRY box-containing transcription factor, is a likely candidate. SOX5 is highly expressed in early-born cortical neurons, including layer 6 neurons. Two groups reported that Sox5 regulates migration and identity of deep-layer neurons (Kwan et al., 2008; Lai et al., 2008). However, it is unclear whether Sox5 mutant neurons switch their axonal targets, as the Fezf2−/− or Satb2−/− neurons do.
TBR1, a T-box transcription factor, is highly expressed in preplate and layer 6 neurons, and regulates their development (Hevner et al., 2001). In Tbr1−/− mice, preplate and layer 6 neurons exhibit molecular and functional defects (Hevner et al., 2001). However, in that study, the authors did not explore whether the Tbr1−/− neurons adopted alternate neuronal identity, and did not provide insight into the mechanism of corticothalamic neuron fate specification.
In the present study, we aim to answer two questions: (1) Does TBR1 regulate corticothalamic neuron identity? (2) What are the central transcriptional mechanisms that regulate fate divergence of closely related but distinct corticothalamic neurons and subcerebral neurons?
Materials and Methods
Animals.
Generation of the Tbr1 (Bulfone et al., 1998) and Fezf2 (B. Chen et al., 2005) mutant mice was reported previously. Golli-τ-GFP mice were generously provided by Dr. Anthony T. Campagnoni at University of California, Los Angeles (Los Angeles, CA). The day of the vaginal plug detection was designated as embryonic day 0.5 (E0.5). The day of birth was designated as postnatal day 0 (P0). The genders of the embryonic mice and early postnatal mice were not determined. Experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at University of California at Santa Cruz and were performed in accordance with institutional and federal guidelines.
PLAP staining.
Human placental alkaline phosphatase (PLAP) staining was performed as described previously (B. Chen et al., 2005).
Immunohistochemistry.
Immunohistochemistry was performed using standard protocols. Primary antibodies used were as follows: rat anti-CTIP2 (Abcam); rabbit anti-DARPP32 (Abcam); rabbit anti-TBR1 (Millipore); rabbit anti-TBR1 (Abcam); rabbit anti-NFIB (Active Motif); goat anti-TLE4 (Santa Cruz Biotechnology); rabbit anti-NURR1 (Santa Cruz Biotechnology); goat anti-SOX5 (Santa Cruz Biotechnology); rabbit anti-FOXP2 (Abcam); mouse anti-βIII tubulin (TUJI) (Covance), rabbit anti-hPLAP (Accurate Chemical); goat anti-ChAT (Millipore); and sheep anti-BrdU (Abcam). Secondary antibodies were from Jackson ImmunoResearch and Invitrogen.
Image acquisition and analysis.
Images for quantitative analyses were acquired with a Zeiss LSM5 confocal microscope with detector gain set such that < 1% of pixels were saturated. Cell counting was performed on single z-slices. Bright-field and dark-field images were acquired with an Olympus BX51 microscope and Q-Imaging Retiga camera. The unpaired t test was used to determine statistical significance.
BrdU birthdating.
Timed pregnant mice were injected with a single dose of BrdU (50 mg/kg) at E11.5, E12.5, E13.5, E14.5, or E15.5. P0 brains were collected for BrdU staining. Strong BrdU-labeled cells were defined as the cells with ≥75% of their nuclei labeled with BrdU staining.
Quit fraction analysis.
Timed pregnant mice received a single injection of BrdU (50 mg/kg) at E10.5, E11.5, E12.5, or E13.5. Embryos were collected 24 h later and processed for BrdU staining and Ki67 staining. The number of cells that were stained with BrdU antibody but not with Ki67 (BrdU+Ki67−) and the number of all the cells that were stained with BrdU antibody (BrdU+) were counted. The quit fraction was calculated as the ratio of the number of BrdU+Ki67− cells to the number of BrdU+ cells.
Anterograde and retrograde tracing.
Anterograde tracing using biotinylated dextran amine (BDA) was performed as described previously (B. Chen et al., 2005). Littermate 2-month-old male mice were used in the tracing.
Retrograde tracing was performed using Alexa Fluor 488-conjugated cholera toxin β subunit (CT-β) (Invitrogen) injections under ultrasound guidance (VisualSonics Vevo 770). Subcerebral projection neurons were labeled by CT-β injections at E16.5 or E17.5 into the cerebral peduncle at locations caudal from the hypothalamus, and rostral from the pyramidal decussation. Corticothalamic projection neurons were labeled by CT-β injections at P1 into the midthalamus. Injected mice were collected at P0 or P4 and processed for immunostaining.
In utero electroporation.
The full-length Tbr1 cDNA was amplified by PCR and inserted into pCAGGS-ires-Egfp vector using ClaI and XhoI restriction sites. In utero electroporation experiments were performed according to a published protocol (B. Chen et al., 2005). Plasmids encoding TBR1-ires-EGFP or EGFP were electroporated into E12.5 and E13.5 CD1 embryos. After electroporation, the embryos were allowed to survived to P0, P3, or P7, at which time CTIP2 expression was analyzed by immunostaining, and axonal projections were visualized with EGFP staining.
Nucleofection.
Cortices from E12.5 and E13.5 CD1 embryos were dissected and dissociated into single cells. The Tbr1-Myc expression plasmids or the Egfp plasmids were transfected into dissociated neurons and progenitor cells using nucleofection, which allowed the transfected plasmids to be delivered into cell nuclei (Lonza). The transfected cells were cultured for 2 d, fixed, and stained with antibodies against TBR1, MYC, GFP, and CTIP2.
Chromatin immunoprecipitation and quantitative PCR.
Chromatin immunoprecipitation (ChIP) was performed according to a published method (Dhillon et al., 2009). Briefly, cortices dissected from E13.5 embryos were fixed for 10 min with 1% formaldehyde and neutralized with glycine. The cells were lysed and the chromatin was sheared into ∼300 bp fragments. Immunoprecipitation reactions were performed in duplicate using antibodies against TBR1 (SCBT and Abcam) and a control antibody (Abcam). Immunoprecipitated and input DNA were quantified using the Picogreen dsDNA quantitation kit (Invitrogen) and a fluorescent spectrophotometer. Equal amounts of input and immunoprecipitated DNA (between 100 and 250 pg) were used for quantitative PCR (qPCR) using SYBR Green.
Only primer pairs with equivalent relative efficiencies of amplification were used, and their locations and sequences are provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Relative enrichment of DNA loci was calculated as a ratio of 2ΔCt (between immunoprecipitated and input DNA) of the experimental locus to 2ΔCt (between immunoprecipitated and input DNA) of a reference locus.
Luciferase assay.
The TBR1 binding region at the 3′ of Fezf2 gene was amplified by PCR using primer set GTTGAGCTGACCTCGTGG and TGCAGCGTTTGGGGGA, and cloned into the pGL4 luciferase vector (Promega), either upstream of the SV40 promoter or downstream of the firefly luciferase gene. Dissociated cells from dissected wild-type E13.5 cortices were nucleofected with either TBR1 expression plasmid or empty vector (control), firefly luciferase (plasmid) containing TBR1 binding region, and Renilla luciferase (pRL-SV40). Luciferase activities were assayed 48 h later. The ratio of firefly luciferase and Renilla luciferase for each batch of transfected cells was calculated.
Results
TBR1 is expressed in layer 6 corticothalamic projection neurons
To investigate whether TBR1 regulates the identity of deep-layer neurons, we first determined whether TBR1 is expressed in corticothalamic neurons, subcerebral projection neurons, or both, in neonatal and perinatal animals. The retrograde tracer CT-β was injected into either the thalamus (thal) of P1 or the cerebral peduncle (Cpd) of E17.5 wild-type animals, to label the cell bodies of corticothalamic or subcerebral projection neurons. We then stained brain sections with antibodies to either TBR1 or CTIP2, and analyzed their expression in the motor and somatosensory cortex. Staining with TBR1 antibodies revealed that layer 6 neurons expressed high levels of TBR1 protein, while few neurons in layer 5 expressed TBR1 strongly (P3 cortex in Fig. 1A,C, P0 cortex in G and I). Staining with CTIP2 antibodies revealed an opposite pattern: neurons in layer 5 expressed high levels of CTIP2 protein (CTIP2high), while CTIP2 expression level in layer 6 neurons was much lower (CTIP2low) (Fig. 1D,F,J,L).
Figure 1.
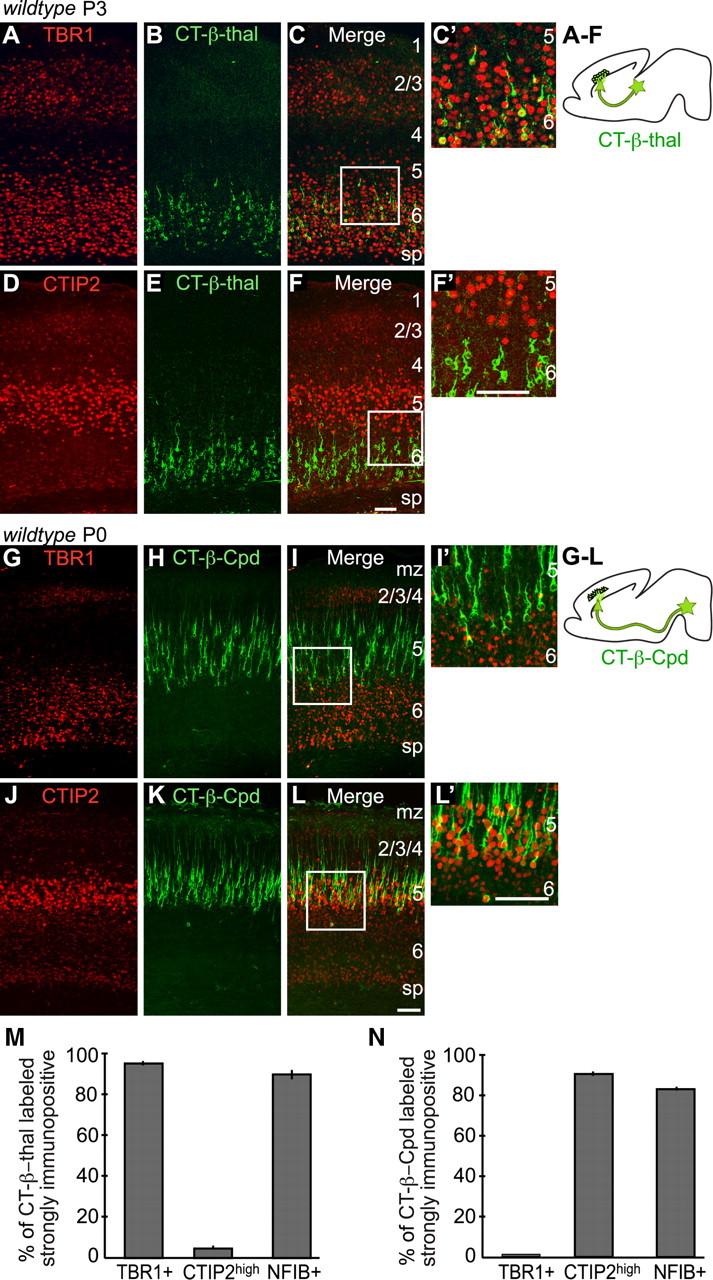
TBR1 and CTIP2 expression in corticothalamic and subcerebral projection neurons of the rostral neocortex at early postnatal stages in wild-type mice. A–F′, Corticothalamic projection neurons were labeled by injecting the retrograde tracer CT-β into thalamic nuclei (CT-β-thal) at P1. A, TBR1 staining. B, E, CT-β-labeled corticothalamic neurons. D, CTIP2 staining. G–L′, Subcerebral projection neurons labeled by injecting CT-β into cerebral peduncles (CT-β-Cpd) at E17.5. G, TBR1 staining. H, K, CT-β-labeled subcerebral neurons. J, CTIP2 staining. C′, F′, I′, and L′ showed the boxed areas in C, F, I, and L, respectively. M, N, Quantitative analysis of TBR1, CTIP2high, and NFIB expression in CT-β-thal- and in CT-β-Cpd-labeled neurons (CT-β-thal tracing: 634 TBR1+ among 662 labeled neurons, 96 ± 0.8%, n = 7 mice; 18 CTIPhigh among 446 labeled neurons, 4 ± 0.2%, n = 3 mice; 374 NFIB+ among 416 labeled neurons, 90 ± 0.2%, n = 3 mice. CT-β-Cpd tracing: 2 Tbr1+ among 233 labeled cells, 0.9%, n = 3 mice; 727 CTIP2high among 811 labeled neurons, 90 ± 0.3%, n = 8 mice; 174 NFIB+ among 212 labeled neurons, 82 ± 0.3%, n = 3 mice). The error bars represent SEM. mz, Marginal zone; sp, subplate. Scale bars, 50 μm.
Consistent with published studies, most corticothalamic neurons were located in layer 6 (Fig. 1B,C,E,F) (O'Leary and Koester, 1993). We found 96% of the CT-β-labeled corticothalamic neurons expressed high levels of TBR1 protein (Fig. 1A–C,C′,M). Whereas most CT-β-thal-labeled neurons appeared to express CTIP2low, 4% expressed CTIP2high, the majority of which were located in layer 5 (Fig. 1D–F,F′,M). Retrogradely labeled subcerebral projection neurons (including those projecting axons into the midbrain, hindbrain area and to the spinal cord) were found primarily in layer 5 (Fig. 1G–L,L′). Among all CT-β-Cpd-labeled cells, 90% expressed CTIP2high (Fig. 1J–L,L′,N), while <1% expressed high levels of TBR1 protein (Fig. 1G–I,I′,N).
These results demonstrate that, at an early postnatal stage, the majority of layer 6 neurons express high levels of TBR1 and low levels of CTIP2 protein, while only a subset of layer 5 neurons express TBR1, and show that TBR1 is strongly expressed by corticothalamic neurons, but not by subcerebral projection neurons.
Axonal defects in Tbr1−/− mice
It was reported that the corticothalamic and thalamocortical projections were both missing in the Tbr1−/− mice (Hevner et al., 2001). However, whether the mutant layer 6 neurons adopted alternate projection targets was not determined. To address this issue, we defined the trajectory of deep-layer axonal projections in Tbr1−/− mice by using a transgenic method to label these axons. To this end, we generated Tbr1+/−; Fezf2+/− and Tbr1−/−; Fezf2+/− mice, as well as Tbr1+/−; Golli-τ-GFP and Tbr1−/−; Golli-τ-GFP mice. FEZF2 is expressed in both corticothalamic and subcerebral projection neurons. By using a Fezf2 mutant allele in which an ires-PLAP cassette (ires, internal ribosomal entry site) replaced the Fezf2 open reading frame, we were able to visualize cell bodies and axons of corticothalamic and subcerebral projection neurons by PLAP staining (B. Chen et al., 2005, 2008). The Golli-τ-GFP allele directs expression and localization of GFP in the cell bodies and processes of deep-layer cortical neurons (Jacobs et al., 2007).
We examined PLAP-stained axonal projections in Tbr1−/−; Fezf2+/− mice at P1, and compared them with those in Tbr1+/−; Fezf2+/− littermates. PLAP-labeled corticothalamic axons were present in Tbr1+/−; Fezf2+/− mice, turning dorsally from the internal capsule and entering thalamic nuclei (supplemental Fig. S1A,B, arrows, available at www.jneurosci.org as supplemental material). Consistent with previous work (Hevner et al., 2001), PLAP-labeled axons failed to innervate the thalamus in Tbr1−/−; Fezf2+/− mice (supplemental Fig. S1E,F, arrows, available at www.jneurosci.org as supplemental material).
PLAP-labeled axons in P1 Tbr1−/−; Fezf2+/− were present in both the cerebral peduncle and the pyramidal tract, similar to those seen in Tbr1+/−; Fezf2+/− mice (Fig. 2A,B,D,G, arrows). In the region surrounding the cerebral peduncle (Fig. 2E,H, dashed boxes), however, the PLAP-stained area was increased to 2.6-fold in Tbr1−/−; Fezf2+/− mice (Fig. 2J) (p = 0.0036) compared to that in the Tbr1+/−; Fezf2+/− mice. In P1 Tbr1−/−; Fezf2+/− mice, ectopic PLAP+ axons extended dorsally and caudally from the cerebral peduncle, into the midbrain and toward the hindbrain [Fig. 2, compare arrowheads in A to B, E to H, F to I; supplemental Fig. S1 (available at www.jneurosci.org as supplemental material), compare B to F]. Similar tracts of PLAP+ axons projecting toward the midbrain and hindbrain were also present in Tbr1+/−; Fezf2+/− mice by P3, but were not present at P1 (Fig. 2A–C, compare arrowheads). Therefore, it seemed that some PLAP-labeled axons in the P1 mutant brains were prematurely projecting toward subcerebral targets in the midbrain and hindbrain.
Figure 2.

PLAP-labeled subcortical axons in early postnatal Tbr1+/−; Fezf2+/− and Tbr1−/−; Fezf2+/− mice (all the panels show P1 staining, except for C, which shows P3 staining). A–D, G, Sagittal sections. E, F, H, I, Coronal sections at the approximate anterior–posterior levels indicated in D and G. Arrowheads in A–I point to corticobulbar projection. Arrows point to cerebral peduncle unless labeled differently. J, Average dark PLAP staining area in the peripeduncular region (boxed regions in E and H). K–M, Coronal sections were immunostained with anti-ChAT (K) and anti-PLAP (L) antibodies. PLAP and ChAT expression was colocalized in a region of the hindbrain consistent with the position of the pedunculopontine tegmental nucleus in Tbr1−/−; Fezf2+/− mice at P1 (M). The area in K–M was taken from approximately the region boxed in I. hyp, Hypothalamus; pt, pyramidal tract; *p = 0.0036 t test, n = 7 matched sections from 2 litters; scale bars: A–C (in A), D, G (in D), 500 μm; E, H (in E), F, I (in F), 250 μm; K–M (in M), 50 μm. See also supplemental Figure S1 (available at www.jneurosci.org as supplemental material).
Similar results were observed when we compared GFP-labeled axons in the Tbr1+/−; Golli-τ-GFP and Tbr1−/−; Golli-τ-GFP mice. GFP-labeled axons were missing in the thalamus of the Tbr1−/−; Golli-τ-GFP mice (compare arrows in supplemental Fig. S1I,L, available at www.jneurosci.org as supplemental material). Interestingly, just as the PLAP-labeled axons, ectopic GFP-labeled axons were observed projecting into the midbrain and hindbrain of the P0 Tbr1−/−; Golli-τ-GFP mice (arrowheads in supplemental Fig. S1O, available at www.jneurosci.org as supplemental material).
To identify targets of the ectopic PLAP or GFP-labeled axons in the midbrain and hindbrain region of Tbr1−/−; Fezf2+/− or Tbr1−/−; Golli-τ-GFP mice, we compared the PLAP staining pattern with the expression pattern of choline acetyltransferase (ChAT), a marker of midbrain and hindbrain motor nuclei that are targets of layer 5 corticobulbar projections (Valverde, 1962; Satoh et al., 1983; O'Leary and Koester, 1993; Desbois et al., 1999). We found that ectopic, mutant PLAP projections overlapped with ChAT immunostaining in what appeared to be the pedunculopontine tegmental nucleus (Fig. 2K–M). Compared to the Tbr1+/−; Fezf2+/− mice, many more PLAP-labeled axons approached ChAT-positive nuclei in the midbrain and hindbrain in Tbr1−/−; Fezf2+/− mice (Fig. 2, compare arrowheads in F and I; Fig. 2K–M; supplemental Fig. S1D,H, available at www.jneurosci.org as supplemental material).
These data, together with the loss of corticothalamic projections, suggested that some axons from layer 6 neurons in Tbr1 mutant mice joined subcerebral projections from layer 5 in reaching midbrain and hindbrain targets.
Fewer neurons express layer 6 genes and more neurons express layer 5 genes in Tbr1−/− mice
The absence of corticothalamic projections and increased axonal projection in the midbrain and hindbrain areas suggested that neuronal identity was misspecified in Tbr1−/− mice. Thus we assayed the expression of CTIP2 and a panel of molecular markers of deep-layer neurons. The images presented here were from somatosensory and motor cortex, but the conclusion applied to all neocortical regions.
There are two populations of CTIP2-expressing neurons in normal mouse brains at birth: those in layer 6 express low levels of CTIP2 protein (CTIP2low), and those in layer 5 express high levels of CTIP2 protein (CTIP2high). Staining with CTIP2 antibodies demonstrated a broader distribution of CTIP2high cells throughout the cortex in Tbr1−/− mice compared to controls (Fig. 3A,D,G,J,M,P,S,V), consistent with reported defects in neuronal migration (Hevner et al., 2001). We were struck, however, by an apparent increase in the number of CTIP2high neurons in Tbr1−/− brains (Fig. 3A,D,G,J,M,P,S,V). We found a 90% increase in the number of CTIP2high cortical neurons in Tbr1−/− versus control mice (Fig. 3Y).
Figure 3.
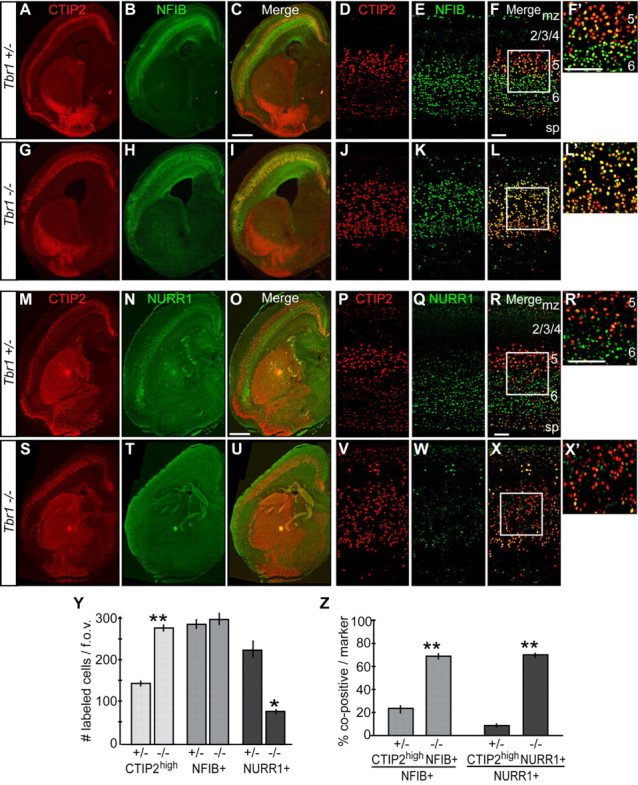
Expression of layer 5 and layer 6 markers. A–F′, CTIP2 and NFIB staining in Tbr1+/− mice. G–L′, CTIP2 and NFIB staining in Tbr1−/− mice. A–C, G–I, Low magnification. D–F, J–L, High magnification. F′ and L′ show the boxed areas in F and L, respectively. M–R′, CTIP2 and NURR1 staining in Tbr1+/− mice. S–X′, CTIP2 and NURR1 staining in Tbr1−/− mice. M–O, S–U, Low magnification. P–R, V–X, High magnification. R′ and X′ show the boxed areas in R and X, respectively. Y, Numbers of CTIP2high, NFIB+, and NURR1+ neurons in Tbr1+/− and Tbr1−/− mice. Z, Percentages of CTIP2highNFIB+ or CTIP2highNURR1+ cells among total NFIB+ or NURR1+ cells in Tbr1+/− and Tbr1−/− mice. sp, Subplate; mz, marginal zone. *p < 0.001 and **p < 0.0001 by t test; n = 54 matched fields of view (f.o.v.) from 12 litters for CTIP2high counts in Y; n ≥ 13 from 4 of more litters for other quantification in Y and Z; error bars indicate the SEM; scale bars, 50 μm. See also supplemental Figure S2 (available at www.jneurosci.org as supplemental material).
We assayed Fezf2 expression by in situ hybridization. Both layer 5 and layer 6 neurons expressed Fezf2, with expression in layer 5 higher than in layer 6 (see Fig. 5A). Tbr1−/− mutants had increased Fezf2 mRNA expression at P0 (see Fig. 5A–C); thus both CTIP2 and Fezf2 expression showed a similar change in molecular properties of Tbr1−/− layer 6 neurons.
Figure 5.

Fezf2 expression was increased in Tbr1−/− mice and TBR1 bound to the Fezf2 gene in vivo. A, B, In situ hybridization with 35S-labeled RNA probes showing Fezf2 mRNA expression in wild-type (A) and Tbr1−/− (B) P0 brains. C, Northern hybridization showing increased Fezf2 mRNA levels in Tbr1−/− brains. Gapdh was used as a loading control. Samples in each lane represent total RNA from cortices of an individual mouse. D, E, ChIP analysis shows TBR1 binding to Fezf2 gene in vivo. E, To determine whether TBR1 directly bound the Fezf2 locus, we designed primer sets to cover 15 regions containing all highly conserved sequences >100 bp. Where possible, two unique primer pairs were used per conserved region. The conserved regions 1–15 are aligned with Fezf2, the 434 enhancer element, and the mammalian conservation track from the UCSC's genome browser. Each histogram bar in D represents ChIP data from one primer pair. The fold enrichment was determined by normalizing ΔCt values for each amplicon against the ΔCt value from 3′ amplicon at region 13. Error bars represent the SEM obtained from 4 biological replicates. F, The region enriched in TBR1 ChIP-qPCR assays (black box) was cloned either upstream (reporter 1) or downstream (reporter 2) of the firefly luciferase reporter gene. G, Luciferase assay showing that overexpressing TBR1 significantly inhibits the firefly luciferase activity. Compared to no TBR1 control (value = 1), the relative luciferase activity was 0.57 ± 0.04 (SEM) when the TBR1 binding region in Fezf2 was cloned downstream of the firefly luciferase gene, and the relative luciferase activity was 0.65 ± 0.02 (SEM) when the TBR1 binding region in Fezf2 was cloned upstream of the firefly luciferase gene. p < 0.001, t test (3 biological replicates, with >5 technical replicates each). See also supplemental Figure S4 and supplemental Table S1 (available at www.jneurosci.org as supplemental material).
Deep-layer subcortical projection neurons also express the transcription factors NFIB and SOX5 (Kwan et al., 2008; Lai et al., 2008; Plachez et al., 2008). We assayed their expression, in combination with CTIP2, in Tbr1 mutant and control mice. Within the deep layers of control sections, strong NFIB expression and SOX5 expression were observed in the subplate, layer 6 neurons, and layer 5 (Fig. 3B,E; supplemental Fig. S2A,C, available at www.jneurosci.org as supplemental material). Within layer 5 of control animals, most NFIB+ or SOX5+ cells coexpressed CTIP2high (Fig. 3A–F,F′; supplemental Fig. S2B–D,2D′, available at www.jneurosci.org as supplemental material). Within layer 6 of control animals, very few NFIB+ or SOX5+ cells coexpressed CTIP2high, and many NFIB+ or SOX5+ cells coexpressed CTIP2low (Fig. 3A–F,F′; supplemental Fig. S2B–D,D′, available at www.jneurosci.org as supplemental material). Thus, the NFIB+CTIP2high or SOX5+CTIP2high neurons of layer 5 could be distinguished from the NFIB+CTIP2low or SOX5+ CTIP2low neurons of layer 6. When we examined NFIB and SOX5 staining in the cerebral cortex of Tbr1−/− mice, we found no significant changes in the numbers of either NFIB+ or SOX5+ neurons (Fig. 3B,E,H,K,Y; supplemental Fig. S2A,C,E,G, available at www.jneurosci.org as supplemental material). However, among the NFIB+ or SOX5+ cells, there was a more than threefold increase in the percentage that strongly coexpressed CTIP2high [Fig. 3, compare F and F′ to L and L′; supplemental Fig. S2 (available at www.jneurosci.org as supplemental material), compare D and D′ to H and H′]: 22% of NFIB+ cells coexpressed CTIP2high in controls, whereas 69% of NFIB+ cells coexpressed CTIP2high in the Tbr1−/− cortex (Fig. 3Z).
The reduced numbers of NFIB+CTIP2low and SOX5+ CTIP2low neurons, together with increased numbers of NFIB+ CTIP2high and SOX5+CTIP2high cells, suggested that layer 6 neuronal identities were affected in Tbr1−/− mice. To further assess the identities of layer 6 cells, we examined the expression of the layer 6-specific markers NURR1 (Zetterström et al., 1996; Lai et al., 2008), FOG2 (Kwan et al., 2008), FOXP2 (Ferland et al., 2003), DARPP32 (Molyneaux et al., 2005), and TLE4 (Koop et al., 1996; B. Chen et al., 2005), in combination with CTIP2.
In control animals, CTIP2high marked layer 5 neurons, while NURR1, FOG2, FOXP2, DARPP32, and TLE4 expression was largely restricted to the subplate and layer 6, and overlapped with a few CTIP2high cells in layer 5 (Fig. 3M–R,R′; supplemental Fig. S2I–N,U–W′,AA–CC′,GG–II′, available at www.jneurosci.org as supplemental material). The expression of each layer 6 marker was substantially reduced in the Tbr1−/− neocortex (Fig. 3S–X′; supplemental Fig. S2O–T,X–Z′,DD–FF′,JJ–LL′). Strongly labeled NURR1+ cells were decreased to 34% (Fig. 3Y). Of note, most cells that retained NURR1, FOG2, FOXP2, DARPP32, and TLE4 expression coexpressed CTIP2high (Fig. 3S–X,X′; supplemental Fig. S2O–T,X–Z′,DD–FF′,JJ–LL′). For instance, in Tbr1−/− brains, 70% of the NURR1+ cells coexpressed CTIP2high, whereas in controls, only 8.7% did so (Fig. 3Z). The remaining expression of layer 6 markers (Fig. 3; supplemental Fig. S2, available at www.jneurosci.org as supplemental material) demonstrated that some layer 6 properties were maintained in Tbr1 mutants.
Next we estimated the percentage of layer 6 neurons that became CTIP2high in the Tbr1−/− mice. NFIB is expressed by both corticothalamic neurons in layer 6 and subcerebral neurons in layer 5 (Fig. 1M,N). Our retrograde tracing experiment showed that in wild-type mice, ∼90% of the CT-β-thal-traced neurons in layer 6 expressed NFIB (Fig. 1M) and >80% of the CT-β-Cpd-traced subcerebral neurons in layer 5 expressed NFIB (Fig. 1N). Since the number of NFIB-expressing neurons in layers 5 and 6 was not significantly affected by Tbr1 mutation (Fig. 3Y), we determined the percentage of CTIP2lowNFIB+ layer 6 neurons that became CTIP2highNFIB+ subcerebral neurons. Analysis of NFIB and CTIP2 expression demonstrated that >60% of the CTIP2lowNFIB+ neurons (correlating to layer 6 neurons in control mice) switched their identity to CTIP2highNFIB+-expressing subcerebral neurons in Tbr1−/− mice. This result, combined with the reduced expression of layer 6 markers in Tbr1−/− mice, demonstrated that in Tbr1−/− mice, even though some layer 6 neuronal character remained, overall, across all neocortical areas, most of the layer 6 neurons, including the corticothalamic neurons, switched to subcerebral identity.
More neurons born on E11.5 and E12.5 in Tbr1−/− mice express CTIP2high
During murine cortical neurogenesis, layer 6 corticothalamic neurons start to be generated at E11.5 and reach their neurogenic peak at E12.5, while layer 5 neurogenesis peaks 24 h later (Polleux et al., 1997, 1998). To test whether the increased numbers of CTIP2high neurons in the Tbr1−/− mice originated from misspecified corticothalamic neurons, we performed BrdU birthdating to identify the embryonic ages during which the CTIP2high neurons were generated. Timed-pregnant mice were injected once with BrdU at E11.5, E12.5, E13.5, E14.5, or E15.5, brains were collected at P0 and stained with BrdU and CTIP2 antibodies.
We counted strongly BrdU-labeled (BrdU+) neurons (defined as nuclei appearing >75% saturated by BrdU immunostaining), CTIP2high neurons, and BrdU+CTIP2high neurons in matched littermate sections, and determined the percentage of neurons born on each embryonic day that expressed CTIP2high at birth. In control animals the highest percentage of BrdU+ neurons that expressed CTIP2high at birth were from injections performed on E13.5, consistent with the peak time for layer 5 neurogenesis (Fig. 4A). In addition, some neurons born on E11.5 and E12.5 in control mice expressed CTIP2high at birth as well (Fig. 4A). However, in Tbr1−/− mice, more than twofold more neurons born at E11.5 and E12.5 became CTIP2high (Fig. 4A).
Figure 4.
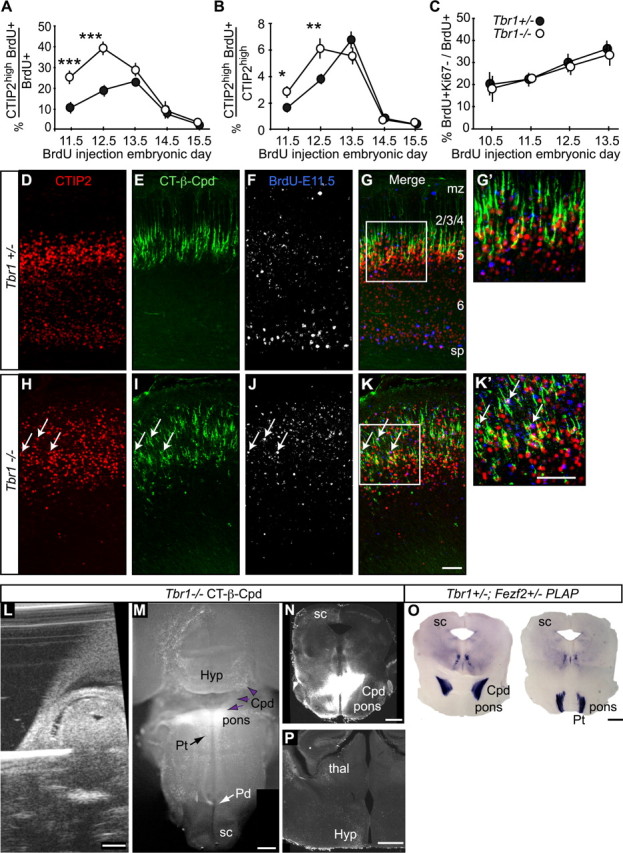
Early-born cortical neurons expressed CTIP2high and projected to subcerebral targets in Tbr1−/− mice. A, Among the cells born at E11.5, E12.5, E13.5, E14.5, or E15.5, the percentages that expressed CTIP2high at P0. B, The percentage of the total CTIP2high population present at P0, that was born on E11.5, E12.5, E13.5, E14.5, or E15.5. C, Quit fractions in Tbr1+/− and Tbr1−/− brains were not significantly different during early and middle stages of cortical neurogenesis. D–K′, Retrograde tracing from the cerebral peduncles (CT-β-Cpd) was combined with E11.5 BrdU birthdating and CTIP2 immunostaining. Arrows in H–K′ mark BrdU-labeled, CT-β-Cpd-traced, and CTIP2high neurons. L, An ultrasound image of the CT-β-Cpd injection. M, A ventral view of an injected Tbr1−/− brain showing that the injection was at the pontine level, caudal to the hypothalamus, and rostral to the pyramidal decussation. N, Image of a Tbr1−/− brain section showing the injection site in the cerebral peduncle. O, PLAP staining of Tbr1+/−; Fezf2+/− brain sections at similar rostral–caudal levels as in N showed the positions of PLAP-labeled cerebral peduncle and pyramidal tract. P, An image of a Tbr1−/− brain section containing the thalamus. CT-β did not spread into the thalamus; the labeling in the thalamus was due to retrogradely labeled cell bodies in the thalamus. M, N, and P show the same Tbr1−/− brain. sp, Subplate; mz, marginal zone; Hyp, hypothalamus; sc, superior colliculus; Pd, pyramidal decussation; Pt, pyramidal tract; *p = 0.032 in B, **p = 0.0072 in B, ***p < 0.0001 in A, by t test; n = 12 matched sections/age from >3 litters; error bars indicate the SEM; scale bars: D–K′, 50 μm; L–P, 500 μm. See also supplemental Figure S3 (available at www.jneurosci.org as supplemental material).
At P0, Tbr1−/− mice had many more CTIP2high cells than Tbr1+/− controls. To better understand the cause of this phenotype, we determined the fraction of P0 CTIP2high neurons that were born at each embryonic age (Fig. 4B). In control mice, the percentage of CTIP2high cells born on E11.5 and E12.5 increased sharply to a peak on E13.5 (Fig. 4B). In Tbr1 mutants, however, significantly greater fractions of the CTIP2high cells were generated on E11.5 and E12.5, and peak CTIP2high neurogenesis was sustained through E13.5 (Fig. 4B).
These data demonstrated that significantly more CTIP2high neurons were born at E11.5 and E12.5 in the Tbr1−/− cortex (Fig. 4A,B). To assess when increased CTIP2 expression first appeared in the developing cerebral cortex, we examined E12.5 brains. As early as E12.5 more CTIP2high cells were present in the Tbr1−/− mutants than in control mice (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material).
CTIP2high neurons born on E11.5 and E12.5 in Tbr1−/− mice project axons into subcerebral targets
CTIP2 acts downstream of Fezf2 (Chen et al., 2008), and its expression is necessary for the development of the CST (Arlotta et al., 2005). When ectopically expressed, CTIP2 is sufficient to redirect the axons from layer 2/3 neurons to project subcortically (Chen et al., 2008). We, therefore, wanted to determine whether the early-born CTIP2high neurons in Tbr1−/− brains projected to layer 5 targets. To do this, we combined birthdating, retrograde axonal tracing, and immunostaining. We injected timed-pregnant Tbr1+/− mice with BrdU on E11.5, to minimize labeling later-born subcerebral neurons. On E16.5, we injected CT-β into the cerebral peduncle (CT-β-Cpd) to label subcerebral projection neurons, under the guidance of an ultrasound imaging system (Fig. 4L–O). P0.5 brains were collected and processed for immunohistochemistry.
As expected, in control mice, neurons strongly labeled with BrdU were mostly located in the subplate and marginal zone (Fig. 4F,G), and neurons labeled by CT-β-Cpd injections were located in layer 5 and expressed CTIP2high (Fig. 4D,E,G,G′). Only 7% of BrdU+ neurons were retrogradely labeled by CT-β-Cpd injection and expressed CTIP2high (Fig. 4D–G,G′). In the Tbr1−/− mice, consistent with published migration defects (Hevner et al., 2001), BrdU-labeled cells were scattered in the cortex (Fig. 4J,K). Not surprisingly, we found that the CT-β-Cpd traced neurons in Tbr1 mutants expressed CTIP2high (Fig. 4H,I,K,K′). Significantly, however, in the mutant brains, ∼22% early-born (E11.5) BrdU-labeled neurons both expressed CTIP2high and were labeled by CT-β-Cpd injections (arrows in Fig. 4H–K′). This result, in combination with the increased PLAP-labeled axons in the midbrain and hindbrain of Tbr1−/−; Fezf2+/− mice (Fig. 2A,B,E,F,H,I), demonstrated that in the absence of TBR1 many early-born CTIP2high neurons projected their axons into subcerebral targets.
Cell cycle exit and neurogenesis is not accelerated in the Tbr1−/− mice
Our data demonstrated that, in Tbr1−/− mice, neurons exhibiting layer 5 molecular and projection characteristics were born at the time when layer 6 neurons were normally generated. Two possible mechanisms may explain this phenotype: (1) a switch in neuronal identity; (2) precocious cell cycle exit leading to the premature generation of layer 5-like neurons. To investigate the second mechanism, we analyzed the percentage of cells exiting the cell cycle in the neocortex at E10.5, E11.5, E12.5 and E13.5, which represented the early- and mid-stages of cortical neurogenesis. BrdU was injected into timed-pregnant Tbr1+/− mice carrying embryos at each of these stages. Twenty-four hours later, embryos were collected and brain sections were stained with antibodies against BrdU and Ki67. BrdU staining labeled cells that were in S-phase 24 h previously (supplemental Fig. S3C–F, green, available at www.jneurosci.org as supplemental material), and Ki67 staining labeled cells that continued cycling (supplemental Fig. S3C–F, red, available at www.jneurosci.org as supplemental material). Cells that became postmitotic during the 24 h were BrdU+ Ki67−. The percentage of cells that exited the cell cycle (quit fraction) was calculated by dividing the number of BrdU+Ki67− cells by the total number of BrdU+ cells. We compared the quit fraction for control and Tbr1−/− embryos, and found no significant difference at any of the above ages (Fig. 4C).
To ascertain whether cortical neurogenesis was affected by Tbr1 mutation in the early stages, we also examined the expression of a pan-neuronal marker, βIII tubulin, in the Tbr1−/− and control mice at E11.5 and E12.5 (supplemental Fig. S3G,H, available at www.jneurosci.org as supplemental material), and found no significant change. Thus, neurogenesis and cell cycle exit was not accelerated in Tbr1−/− mice. Therefore, the generation of subcerebral neuron-like cells on E11.5 and E12.5 in Tbr1−/− embryos was due to a fate switch.
TBR1 protein binds the Fezf2 gene in vivo
To investigate the molecular mechanisms underlying the cell fate switch in the Tbr1−/− mice, we examined the expression of Fezf2 and Ctip2, two genes essential for subcerebral neuronal identity and their axonal projections, using a combination of in situ hybridization, Northern hybridization and Western analysis. The cortical expression of Fezf2 and Ctip2 were increased in Tbr1−/− mice compared to controls at P0 (Fig. 5A–C; supplemental Fig. S4A–D, available at www.jneurosci.org as supplemental material). When normalized to Gapdh gene, Fezf2 mRNA level was increased to twofold in the Tbr1−/− cortices (p < 0.001, t test, n = 7 wild-type and Tbr1−/− P0 cortices), and CTIP2 protein level was increased to 1.7-fold (supplemental Fig. S4D, available at www.jneurosci.org as supplemental material). To test whether TBR1 protein directly regulates Fezf2 and Ctip2 in vivo, we performed ChIP.
Mouse Fezf2 mRNA is encoded by a 3.7 kb genomic region (Fig. 5E). As important regulatory sequences are conserved among mammalian species, we used the University of California at Santa Cruz (UCSC) genome browser to identify 15 highly conserved regions (excluding protein coding sequences) within a 40 kb region surrounding the Fezf2 gene (21 kb upstream of Fezf2, 18 kb downstream of Fezf2, the intronic regions, and the 5′ and 3′ untranslated regions, UTRs) (Fig. 5D,E). We tested the binding of TBR1 protein to each of these conserved regions by ChIP. ChIP with TBR1 antibodies led to specific enrichment of two neighboring conserved regions (regions 7 and 8) near the 3′ end of Fezf2 gene (Fig. 5D). These regions were not enriched in the ChIP performed with control antibodies. TBR1 binding to highly conserved noncoding regions of Fezf2 suggests that TBR1 may directly repress Fezf2 transcription in vivo.
To determine whether the TBR1-binding region of Fezf2 gene confers transcription regulation by TBR1 protein, we performed the luciferase assay. The TBR1 binding region of Fezf2 was cloned either upstream or downstream of the firefly luciferase reporter gene (Fig. 5F). We found that in either position, overexpressing TBR1 inhibits the luciferase activity (Fig. 5G). This result further suggests that TBR1 may directly inhibit Fezf2 transcription.
Mouse Ctip2 mRNA is encoded by a 93 kb genomic region. We identified 26 conserved non-protein-coding regions in a 180 kb region (including 64 kb upstream, 23 kb downstream regions, and the intronic regions) (supplemental Fig. S4E, available at www.jneurosci.org as supplemental material). We tested the binding of TBR1 protein to each of these regions by ChIP. We did not find any enrichment of any of these 26 conserved regions in the ChIP assay with either of the TBR1 antibodies (data not shown).
Overexpressing TBR1 inhibits subcerebral axonal projection but not the expression of CTIP2 protein
The phenotype of the Tbr1−/− mice demonstrated that Tbr1 is necessary to specify the corticothalamic neuron identity and to repress subcerebral projection neuron fate. To ascertain whether TBR1 protein is sufficient to inhibit subcerebral neuronal identity in the early-born cortical neurons, we ectopically expressed Tbr1 in both cultured cortical cells and in developing mouse embryos, and assessed the effect on gene expression and axonal projections.
We generated two different TBR1-expression constructs: one tagged with a MYC epitope (Tbr1-Myc), and one bicistron construct expressing both TBR1 and EGFP (Tbr1-ires-EGFP) under the control of the chicken β-actin promoter. We first transfected Tbr1-Myc or control GFP plasmids into acutely dissociated E12.5 or E13.5 cortical cells using nucleofection (Lonza), which allowed transfecting genes into both neural progenitor cells and postmitotic neurons. Two days after transfection, we stained the cultured cortical cells with MYC, GFP, and TBR1 antibodies (Fig. 6A,C,D,E,G,H). Since we did not have FEZF2 antibodies that worked in immunostaining, we stained the cells with CTIP2 antibodies to assess the expression of subcerebral neuronal genes (Fig. 6B,F). We counted the numbers of cells expressing each protein, and calculated the percentages of MYC+ or GFP+ cells that expressed CTIP2. For both E12.5 and E13.5 cortical cells, we found no significant difference in CTIP2 expression when Tbr1-Myc or GFP plasmids were transfected (Fig. 6I).
Figure 6.
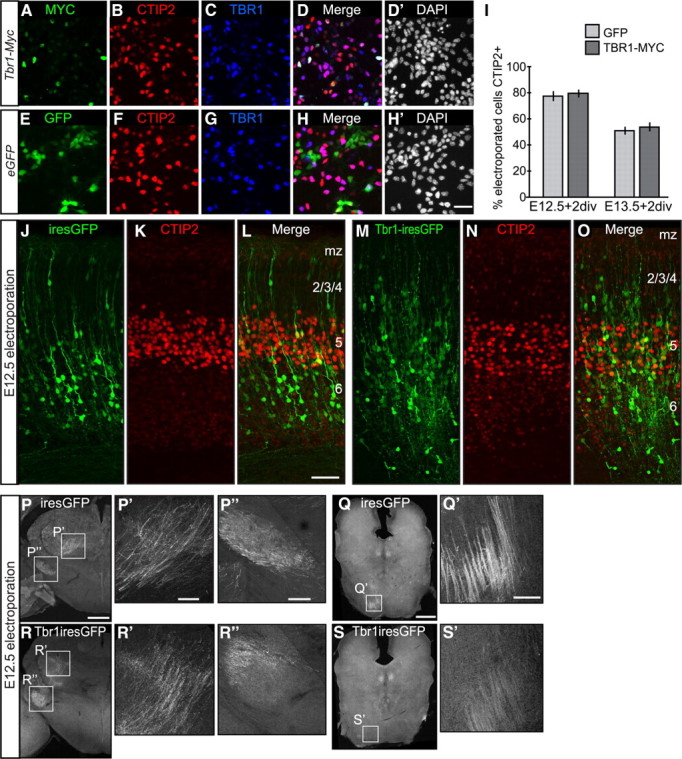
Overexpressing TBR1 in subcerebral neurons prevented their axons from reaching subcerebral targets, but did not affect CTIP2 expression. A–H′, Expression of MYC (A), GFP (E), CTIP2 (B, F), TBR1 (C, G), and DAPI-labeled nuclei (D′, H′) in dissociated E13.5 cortical cells nucleofected with Tbr1-Myc or eGFP plasmids and cultured 2 d in vitro (div). I, The percentages of CTIP2+ neurons among Tbr1-Myc or eGFP nucleofected cells were not significantly different. For cells nucleofected at E12.5, 324/417 GFP+ cells were CTIP2+ (78 ± 4.0%) compared to 359/449 MYC+ cells that coexpressed CTIP2 (80 ± 2.9%). For cells nucleofected at E13.5, 415/807 GFP+ cells were CTIP2+ (51 ± 3.3%) and 673/1254 MYC+ cells coexpressed CTIP2 (53 ± 3.5%). The error bars represent SEM. J–L, GFP and CTIP2 expression in P0 wild-type brains in which CAG-ires-GFP plasmid was electroporated into cortex on E12.5. M–O, GFP and CTIP2 expression in P0 wild-type brains in which CAG-Tbr1-ires-GFP plasmid was electroporated into cortex on E12.5. P–S′, GFP-labeled axons in P3 brains. P, P′, P″, Q, Q′, CAG-ires-GFP plasmid was electroporated into cortex on E12.5. R, R′, R″, S, S′, CAG-Tbr1-ires-GFP plasmid was electroporated into cortex on E12.5. P′, P″, Q′, R′, R″, and S′ showed the boxed areas in P–S. P′, R′, Posterior thalamus. P″, R″, Cerebral peduncle. Q′, S′, Hindbrain area. mz, Marginal zone. Scale bars: A–H′ (in H′), 25 μm; J–O (in L), P′, P″, Q′, R′, R″, S′ (in P′, P″, Q′), 50 μm; P–S (in P, Q), 500 μm. See also supplemental Figure S5 (available at www.jneurosci.org as supplemental material).
The effect of TBR1 overexpression in vivo was investigated using in utero electroporation. To specifically label the axons of the TBR1 overexpressing cells, we generated TBR1-ires-EGFP expression plasmids and confirmed protein expression by electroporating the plasmids into E14 Tbr1−/− embryos. TBR1 expression was detected in GFP+ cells, but not in the nonelectroporated cells, confirming that TBR1 protein indeed was expressed (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
To assay the effect of overexpressing TBR1 on the development of subcerebral projection neurons, plasmids containing TBR1-ires-EGFP or EGFP were electroporated into wild-type embryos at E12.5 and E13.5. The brains from the electroporated mice were collected at P0 and P3, and stained with GFP and CTIP2 antibodies. Electroporated GFP+ cells were present in the deep layers of the cerebral cortex (Fig. 6J–O); there was no apparent difference in the location of neurons electroporated with TBR1-ires-EGFP or EGFP plasmids. Consistent with our in vitro overexpression results (Fig. 6A–I), we observed no effect of overexpressing TBR1 on the expression of CTIP2high in layer 5 neurons or CTIP2low in layer 6 neurons (Fig. 6L,O).
Staining with GFP antibodies revealed the axonal projections from the electroporated cells. In the brains electroporated with either EGFP or Tbr1-ires-EGFP plasmids, GFP-labeled axon bundles were present in the internal capsule (data not shown), thalamus (Fig. 6P,P′,R,R′), and cerebral peduncle (Fig. 6P,P″,R,R″). Thick bundles of GFP-labeled axons were present in the hindbrain when EGFP plasmids were electroporated (Fig. 6Q,Q′). However, in the brains electroporated with Tbr1-ires-EGFP plasmids, barely any GFP-labeled axons were detected in the hindbrain (Fig. 6S,S′) (n = 6 mice for each construct). Thus, overexpression of TBR1 in deep-layer cortical neurons prevented them from projecting their axons into subcerebral targets.
To investigate whether overexpressing TBR1 in layer 2/3 neurons would lead to development of deep-layer neuronal characteristics, we electroporated both Tbr1-Myc and Tbr1-ires-EGFP expression plasmids into E15.5 wild-type embryos, and examined the electroporated brains on P0, P3, and P8. No effect on cell migration, upper-layer neuron gene expression, or axonal projection was observed for either plasmid (data not shown).
Ectopic TBR1-expressing neurons in layer 5 of Fezf2−/− mice express other layer 6 neuronal markers and project their axons into the thalamus
Fezf2 regulates fate specification of layer 5 subcerebral projection neurons. Molyneaux et al. (2005) reported that TBR1 expression expanded into layer 5 neurons in Fezf2−/− mice, and we found that more axons entered the thalamus in Fezf2 mutants [supplemental Fig. S6A–D, available at www.jneurosci.org as supplemental material; and B. Chen et al. (2005)]. Given our evidence that TBR1 regulates the identity of layer 6 neurons, we asked whether the ectopic TBR1-expressing layer 5 neurons in Fezf2−/− mice switched their fate to become corticothalamic neurons. Thus we examined whether these ectopic TBR1-expressing layer 5 neurons projected their axons into the thalamus by combining BrdU birthdating, retrograde axonal tracing, and immunostaining. BrdU was injected into pregnant Fezf2+/− mice on E13.5 to label the layer 5 neurons, and CT-β was injected into the thalamus (CT-β-thal) on P1 (Fig. 7I–M). Three days later, the traced brains were collected and stained with BrdU and TBR1 antibodies.
Figure 7.
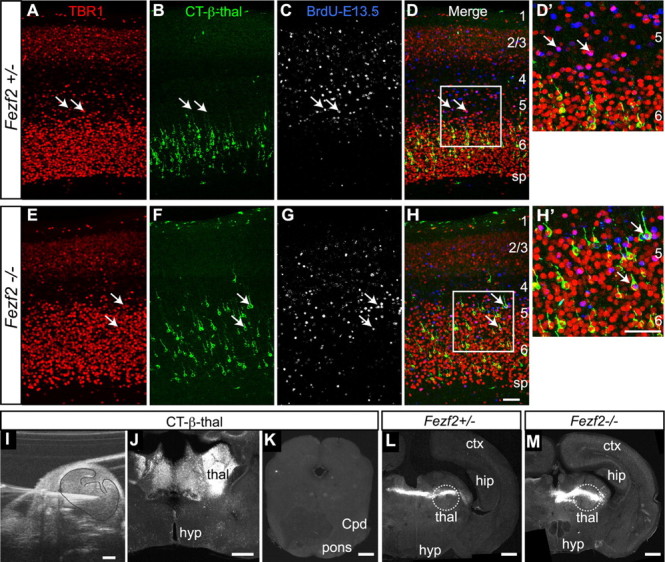
Supernumerary TBR1+ cells in layer 5 of Fezf2−/− mice were born on E13.5 and projected axons to thalamus. Results of combined birthdating, CT-β-thal tracing and immunostaining experiments. Analysis in P4 brains. A–D′, Fezf2+/− mice. Arrows pointed to cells that were born on E13.5 and expressed TBR1, but they were not traced by CT-β-thal injections. E–H′, Fezf2−/− mice. CT-β-thal injections labeled many TBR1+ cells in layer 5, some of which were BrdU-birthdated (arrows in E–H and H′). I, An ultrasound image of a CT-β-thal injection. J, Image of a typical CT-β-thal injection site. K, CT-β injection into the thalamus did not cause the tracer to spread into midbrain and hindbrain area. L, M, The injection sites for CT-β-thal injection in a Fezf2+/− (L) and a Fezf2−/− (M) brain. ctx, Cortex; hip, hippocampus; hyp, hypothalamus; sp, subplate. Scale bars: A–H′, 50 μm; I–M, 500 μm. See also supplemental Figure S6 (available at www.jneurosci.org as supplemental material).
Consistent with published data (Molyneaux et al., 2005), many more layer 5 neurons expressed TBR1 in the Fezf2−/− mice, while TBR1 expression in layer 6 was similar to control (Fig. 7A,E). In both control and Fezf2−/− mice, E13.5 BrdU-birthdated neurons were concentrated in layer 5 (Fig. 7C,D,G,H). In the control mice, neurons labeled by CT-β-thal injections were located in layer 6; they expressed high levels of TBR1 and were rarely labeled with BrdU (Fig. 7A–D,D′). Only 4.7% of the E13.5 BrdU-birthdated neurons were labeled by CT-β-thal injection. However, in the Fezf2−/− brains, TBR1-expressing layer 5 neurons were strongly BrdU+ (Fig. 7E,G,H,H′), indicating that these ectopic TBR1+ cells were born when subcerebral projection neurons were normally born. Furthermore, 12% of these E13.5 birthdated TBR1+ cells were labeled by CT-β-thal injections (Fig. 7E–H,H′). These results demonstrated that some layer 5 neurons in Fezf2−/− mice switched their identity to become corticothalamic neurons, and suggested that Fezf2 represses corticothalamic identity in layer 5 neurons by preventing TBR1 expression.
CTIP2 expression and CST development are partially rescued in Fezf2−/−; Tbr1−/− double mutant mice
The specification of deep-layer cortical neuronal identity into thalamic projecting requires Tbr1 and subcerebral projecting requires Fezf2. To gain insight into the regulatory relationship between these genes in controlling this cell fate decision, we generated mice deficient in both Fezf2 and Tbr1, and investigated CTIP2 expression and axonal projections.
As demonstrated in Figure 3, layer 5 neurons expressed CTIP2high in control Tbr1+/−; Fezf2+/− mice, and layer 6 neurons expressed CTIP2low (Fig. 8E,I). Approximately twice as many CTIP2high cells were present in Tbr1−/−; Fezf2+/− mice as in controls (Figs. 3Y, 8F,J). As reported previously (Molyneaux et al., 2005; Chen et al., 2008), very few neurons expressed CTIP2high in Tbr1+/−; Fezf2−/− mutants (Fig. 8G,K). Importantly, compared to Tbr1+/−; Fezf2−/−, there were threefold more CTIP2high cells in Tbr1−/−; Fezf2−/− brains (Fig. 8H,L); although, in comparison to control Tbr1+/−; Fezf2+/− mice, the CTIP2high number in double mutant mice was still reduced (Fig. 8E,H,I,L). Thus, deleting Tbr1 function in Fezf2−/− mice partially rescued the CTIP2high expression, which marked subcerebral projection neurons.
Figure 8.
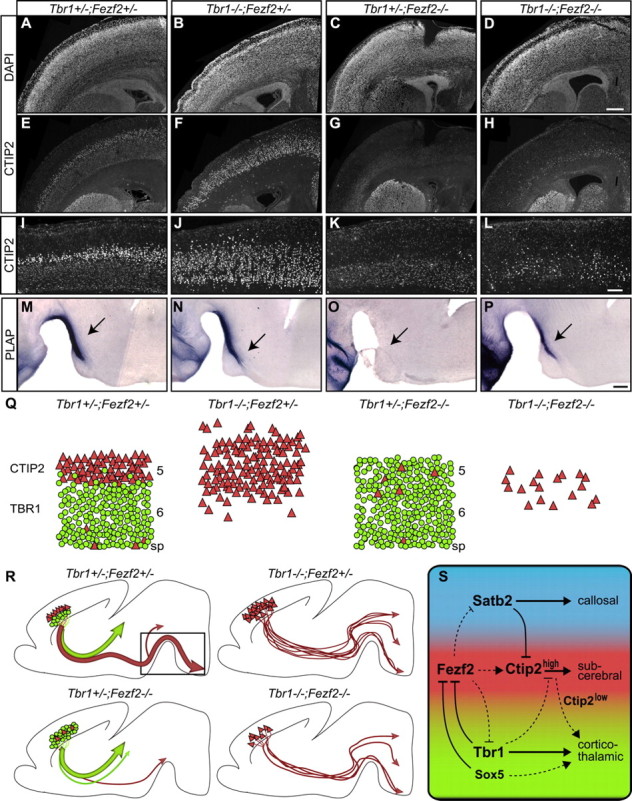
CTIP2high expression and subcerebral projections were partially rescued in Tbr1−/−; Fezf2−/− mice. Analysis in P0 mice. A–D, DAPI staining. E–H, CTIP2 staining for sections shown in A–D. I–L, Higher-magnification images of CTIP2 staining. M–P, PLAP-labeled subcerebral axons showing the boxed area in R. Q, R, Summary of phenotypes in control, single mutant, and compound mutant mice. Green represents TBR1+ corticothalamic neurons and their axons. Red represents CTIP2high subcerebral neurons and their axons. S, Proposed mechanisms for establishing cortical projection neuron identities. The dashed arrow between Fezf2 and Ctip2: an indirect positive regulation. The dashed, bar-end lines from Fezf2 to Satb2, Fezf2 to Tbr1, and Tbr1 to Ctip2: direct inhibition has not been established. The dashed arrows from Sox5 and Ctip2low demonstrate that they are important for corticothalamic axon development, although corticothalamic axons are still present in Sox5−/− and Ctip2−/− mice (Arlotta et al., 2005; Lai et al., 2008). Scale bars: A–H, 200 μm, I–L; 100 μm; M–P, 200 μm.
PLAP staining revealed the axonal projections of the deep-layer neurons. Consistent with the partial rescue of CTIP2high expression, there was a restoration of the subcerebral axon development in the Tbr1−/−; Fezf2−/− mice when compared to the Tbr1+/−; Fezf2−/− brains. PLAP-labeled CST axons, which were missing in Tbr1+/−; Fezf2−/− mice, were detected in Tbr1−/−; Fezf2−/− mice (Fig. 8O,P). Furthermore, CT-β injected into the cerebral peduncle of Tbr1−/−; Fezf2−/− embryos, retrogradely labeled CTIP2high neurons (supplemental Fig. S7E–G′, available at www.jneurosci.org as supplemental material). Despite the rescue of CST, development of the corticothalamic axons in the Tbr1−/− mutants was not restored by deletion of Fezf2 (supplemental Fig. S7A–D, available at www.jneurosci.org as supplemental material). The partial rescue of CTIP2high expression and CST development in Tbr1−/−; Fezf2−/− mice supports the hypothesis that Fezf2 regulates the specification of layer 5 subcerebral projection neurons in part by inhibiting TBR1 expression.
Discussion
In this study, we investigate the molecular mechanisms regulating fate divergence of closely related, but distinct, layer 6 corticothalamic and layer 5 subcerebral projection neurons. First, we investigated the function of TBR1 in regulating the identity of layer 6 neurons. Previously, it was shown that corticothalamic axons were absent in Tbr1−/− mice (supplemental Fig. S1, available at www.jneurosci.org as supplemental material and Hevner et al., 2001). We found that in Tbr1−/− mice, early-born neurons failed to express many layer 6 neuron markers. Instead they expressed markers associated with layer 5 neurons, and extended their axons into subcerebral targets. Furthermore, overexpressing TBR1 in layer 5 neurons prevented them from extending axons into the brainstem. Expression of Fezf2 and CTIP2, both of which are required for subcerebral neuron development, were increased in Tbr1−/− mice. ChIP analysis showed that TBR1 bound to a conserved region in the Fezf2 gene. In vitro, overexpressing TBR1 in cortical cells inhibits a luciferase reporter, which contained the TBR1 binding region of Fezf2. Thus, TBR1 promotes the identity of corticothalamic neurons and represses subcerebral fates, likely through reducing expression of Fezf2 and CTIP2. In the Fezf2−/− mice, TBR1 expression was increased in layer 5 neurons; these ectopic TBR1+ layer 5 neurons projected their axons into thalamus, a normal target for layer 6 neurons. These results suggest that Fezf2 blocks corticothalamic fate in layer 5 by reducing Tbr1 expression. This conclusion was supported by analysis of Fezf2 mutant and Tbr1−/−; Fezf2−/− compound mutant mice. Thus we have uncovered that Tbr1 and Fezf2 participate in central transcriptional mechanisms that regulate fate specification of corticothalamic (layer 6) and subcerebral (layer 5) neocortical projection neurons.
TBR1 regulates the identities of layer 6 neurons
TBR1 promotes the identity of corticothalamic neurons and represses subcerebral fates, possibly through repressing Fezf2 expression. In the Tbr1−/− mice, the early-born neurons switched their molecular and cellular identities from corticothalamic neurons to subcerebral neurons. This fate switch resulted in significantly more subcerebral neurons and projections in Tbr1−/− mice. ChIP analysis showed that TBR1 binds to a conservative non-protein-coding region in the Fezf2 gene. In vitro, overexpressing TBR1 inhibits a luciferase reporter, which contained the TBR1 binding region of Fezf2. Ectopic TBR1 expression in subcerebral neurons prevented them from sending axons to subcerebral targets.
In addition to deep-layer neurons, TBR1 is expressed in upper layers. We investigated whether upper-layer (later-born) neurons in Tbr1−/− mice upregulate subcerebral markers by combining immunohistochemistry and BrdU birthdating. We found that later-born cells in Tbr1−/− mice do not show increased expression of CTIP2high (Fig. 4A,B). Thus, the ectopic subcerebral axons in P1 Tbr1−/− mice do not originate from upper-layer neurons. TBR1 is also expressed in a few neurons in layer 5 (Fig. 1). However, since only 1% of the retrogradely labeled neurons after CT-β-Cpd injection express TBR1 protein (Fig. 1), if the ectopic subcerebral projections in the P1 Tbr1−/− mice were due to “genuine” layer 5 subcerebral axons extending prematurely toward their targets, the effect of Tbr1 mutation would likely be non-cell-autonomous.
Our results demonstrate that TBR1 regulates the development of layer 6 neurons by preventing them from adopting layer 5 neuronal fates. It is intriguing that, even though ectopic expression of TBR1 in layer 5 neurons prevented them from sending axons to subcerebral targets, the electroporated layer 5 neurons still expressed CTIP2high (Fig. 6N,O). However, this result is consistent with our previous observations (Chen et al., 2008): (1) restoring Fezf2 expression in Fezf2−/− neurons restored CST development, but CTIP2high was not detected in the electroporated neurons; (2) ectopic expression of Fezf2 in layer 2/3 neurons redirected the corticocortical axons into corticofugal targets without inducing CTIP2 expression. These results suggest that besides CTIP2, other gene(s) can activate the cellular program to direct cortical axons toward subcerebral targets. However, it is also possible that high levels of TBR1 may affect axons of the electroporated cells at a level that is downstream of Fezf2 and/or Ctip2 transcriptional regulation.
The upregulation of Fezf2 and Ctip2 expression in early-born neurons of Tbr1−/− mice suggests that TBR1 represses the expression of these genes, directly or indirectly (Fig. 8Q–S). Indeed we found that TBR1 protein binds to a region in the 3′ of Fezf2, and this region confers repression by TBR1 in a luciferase assay, suggesting a possible direct inhibition. It should be emphasized that TBR1 only partially represses Fezf2 and Ctip2 expression, since low levels of Fezf2 and Ctip2 expression normally persist in developing layer 6 neurons. In fact, these low levels of Fezf2 and Ctip2 expression are important for normal differentiation of layer 6 neurons. In Ctip2−/− mice, even though there were many more corticothalamic axons reaching the thalamus than subcerebral axons reaching the pontine nuclei, many axons were “lost” in the forebrain and did not even enter the internal capsule (Arlotta et al., 2005). In Fezf2−/− mice, expression of many layer 6 (and subplate) neuronal markers, such as TLE4 and FOXP2, was greatly reduced (B. Chen et al., 2005). In addition, the increased SATB2 expression in both layer 5 and 6 neurons in Fezf2−/− mice showed that Fezf2 represses callosal neuron identity in both layers (Alcamo et al., 2008; Britanova et al., 2008; Chen et al., 2008).
Two groups have reported that SOX5 regulates the identity and differentiation of corticofugal neurons (Kwan et al., 2008; Lai et al., 2008). The cortices in Sox5−/− mice and Tbr1−/− mice share some similarities (Hevner et al., 2001; Kwan et al., 2008; Lai et al., 2008), including migration and subplate defects. Furthermore, subplate and layer 6 neurons in both mutants express high levels of Fezf2 and Ctip2. However, the cortical phenotypes of Tbr1−/− and Sox5−/− mutants are different in several ways. For example, an increased number of CTIP2+ neurons were detected at E12.5 in Tbr1−/− cortices (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), demonstrating a premature expression of Ctip2. On the other hand, in the Sox5−/− cortex, upregulation of Ctip2 (and Fezf2) in layer 6 neurons was not detected at E12.5 and E14.5, but only at E16.5 and later stages. Furthermore, corticothalamic axons were still present in the Sox5−/− mice (Lai et al., 2008). Thus TBR1 and SOX5 appear to regulate the properties of early born subplate and layer 6 neurons via distinct mechanisms.
Recently Bedogni et al. (2010) reported laminar defects in the Tbr1−/− mice. They proposed that Tbr1, by activating Sox5 expression either directly or indirectly, inhibits Fezf2 expression in layer 6 neurons. We disagree with this model for three reasons. First, the defects in corticothalamic axons are much more severe in Tbr1−/− mice than in Sox5−/− mice. The corticothalamic axons are completely absent in the Tbr1−/− mice [Fig. 2; supplemental Fig. S1, available at www.jneurosci.org as supplemental material; and Hevner et al. (2001)], whereas in the Sox5−/− mice, the course of corticothalamic axons was reported to appear normal (Lai et al., 2008). Second, the upregulation of subcerebral neuronal markers are much more severe in Tbr1−/− mice than in Sox5−/− mice. We found upregulation of Fezf2 mRNA and CTIP2 protein in the entire Tbr1−/− neocortex (Figs. 3, 5; supplemental Fig. S4, available at www.jneurosci.org as supplemental material; and data not shown), whereas the Sox5−/− lateral neocortex is almost devoid of CTIP2 or Fezf2 expression; in the medial neocortex, CTIP2- or Fezf2-expressing neurons are ectopically located in layer 6 (Lai et al., 2008). Third, the number of SOX5-expressing neurons is not significantly affected in the Tbr1−/− mice [supplemental Fig. S2, available at www.jneurosci.org as supplemental material; and Bedogni et al. (2010)]. Therefore, while Sox5 might mediate or facilitate a subset of Tbr1-dependent mechanisms to direct corticothalamic neuron differentiation, we deem it unlikely that Tbr1 functions solely through promoting Sox5 expression to inhibit subcerebral neuronal identity in layer 6 neurons. Instead we propose that TBR1 inhibits subcerebral neuronal identity by directly inhibiting Fezf2 expression.
Mechanism for cell fate specification in cortical projection neurons
In addition to defining the function of Tbr1 in specifying corticothalamic neuron identity, we discover that Fezf2 blocks corticothalamic fate in layer 5 by reducing Tbr1 expression in subcerebral neurons. In Fezf2−/− mice, subcerebral neurons ectopically expressed TBR1 and sent axons to the thalamus. Compared to Fezf2−/− single mutants, both subcerebral gene expression and axonal projection were partially rescued in Tbr1−/−; Fezf2−/− mice.
Ctip2 plays a critical role in regulating the development of subcerebral axons; high levels of Ctip2 (CTIP2high) are associated with neurons that project to the CST (Arlotta et al., 2005). The partial rescue of CTIP2high expression and CST development in the Tbr1−/−; Fezf2−/− mice demonstrate that in the absence of Tbr1, Fezf2 is not absolutely required for Ctip2 expression. Even though Ctip2 is a major downstream effector of Fezf2 function (Chen et al., 2008), there is no evidence that Fezf2 directly activates Ctip2 transcription. Ctip2 expression is likely under tight control by multiple transcription factors, such as Satb2, and possibly also by Tbr1. We suggest that one of Fezf2's functions is to prevent high levels of Satb2 and Tbr1 expression in subcerebral neurons, which in turn allows high Ctip2 expression. In this model, the absence of Tbr1 function in Fezf2−/− neurons leads to Ctip2 expression. The fact that there is only partial rescue of CTIP2high expression may be due to increased Satb2 in these deep-layer neurons.
Based on current literature and our study, we propose the following model of the molecular mechanisms for specifying cortical projection neuronal identities (Fig. 8Q–S): Ctip2, which functions downstream of Fezf2, is essential for the differentiation of subcerebral neurons (Arlotta et al., 2005; B. Chen et al., 2005, 2008; J. G. Chen et al., 2005; Molyneaux et al., 2005). By repressing Ctip2 expression in the callosal projection neurons, Satb2 ensures the development of callosal neuron identity (Alcamo et al., 2008; Britanova et al., 2008). Our current investigation shows that Tbr1 promotes corticothalamic neuron identity by preventing the expression of subcerebral projection neuron characteristics. Our previous study on the subcerebral to callosal fate switch (Chen et al., 2008), and the current results on the subcerebral to corticothalamic fate switch in the Fezf2−/− mice, demonstrated that subcerebral neuronal identity is established by repressing both the callosal and corticothalamic neuronal fates. It remains to be determined whether FEZF2 directly inhibits the transcription of Satb2 and Tbr1. Analyses of Fezf2−/−, Satb2−/−, and Tbr1−/− mice demonstrated that these genes promote the subcerebral, callosal, and corticothalamic neuronal identities, respectively, by repressing alternate neuronal fates. It remains to be determined, however, in addition to repressing alternate identities, whether FEZF2, SATB2, and TBR1 directly activate genes required for subcerebral, callosal, and corticothalamic neuronal development.
Footnotes
This work was supported by California Institute of Regenerative Medicine Grant RN1-00530 (to B.C.), a Basil O'Connor Starter Scholar Research Award from March of Dimes Foundation (to B.C.), and NINDS Grant NS34661 (to J.L.R.R.). We thank Dr. Susan McConnell, Dr. Gord Fishell, Dr. David Feldheim, Dr. Sofie Salama, Dr. Pushkar Joshi, and members of the Chen laboratory for scientific discussion and critical reading of the manuscript. Golli-GFP mice were generously provided to us by Dr. Anthony T. Campagnoni at UCLA. We deeply appreciate Dr. Rohinton Kamakaka, Dr. Nemrita Dhillon, Jesse Raab, and Giulia Gurun Ruben for great help with ChIP. We greatly appreciate Dr. Luis Puelles's help with neural anatomy.
References
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, McConnell SK. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RA, Beyer RP, Bammler TK, Rubenstein JL, Hevner RF. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci U S A. 2010;107:13129–13134. doi: 10.1073/pnas.1002285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, Sestan N, Molnár Z, Tarabykin V. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–392. doi: 10.1016/j.neuron.2007.12.028. [DOI] [PubMed] [Google Scholar]
- Bulfone A, Wang F, Hevner R, Anderson S, Cutforth T, Chen S, Meneses J, Pedersen R, Axel R, Rubenstein JL. An olfactory sensory map develops in the absence of normal projection neurons or GABAergic interneurons. Neuron. 1998;21:1273–1282. doi: 10.1016/s0896-6273(00)80647-9. [DOI] [PubMed] [Google Scholar]
- Chen B, Schaevitz LR, McConnell SK. Fezl regulates the differentiation and axon targeting of layer 5 subcortical projection neurons in cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17184–17189. doi: 10.1073/pnas.0508732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc Natl Acad Sci U S A. 2008;105:11382–11387. doi: 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Rasin MR, Kwan KY, Sestan N. Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17792–17797. doi: 10.1073/pnas.0509032102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois C, Le Bars D, Villanueva L. Organization of cortical projections to the medullary subnucleus reticularis dorsalis: a retrograde and anterograde tracing study in the rat. J Comp Neurol. 1999;410:178–196. [PubMed] [Google Scholar]
- Dhillon N, Raab J, Guzzo J, Szyjka SJ, Gangadharan S, Aparicio OM, Andrews B, Kamakaka RT. DNA polymerase epsilon, acetylases and remodellers cooperate to form a specialized chromatin structure at a tRNA insulator. EMBO J. 2009;28:2583–2600. doi: 10.1038/emboj.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Jacobs EC, Campagnoni C, Kampf K, Reyes SD, Kalra V, Handley V, Xie YY, Hong-Hu Y, Spreur V, Fisher RS, Campagnoni AT. Visualization of corticofugal projections during early cortical development in a tau-GFP-transgenic mouse. Eur J Neurosci. 2007;25:17–30. doi: 10.1111/j.1460-9568.2006.05258.x. [DOI] [PubMed] [Google Scholar]
- Koop KE, MacDonald LM, Lobe CG. Transcripts of Grg4, a murine groucho-related gene, are detected in adjacent tissues to other murine neurogenic gene homologues during embryonic development. Mech Dev. 1996;59:73–87. doi: 10.1016/0925-4773(96)00582-5. [DOI] [PubMed] [Google Scholar]
- Kwan KY, Lam MM, Krsnik Z, Kawasawa YI, Lefebvre V, Sestan N. SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc Natl Acad Sci U S A. 2008;105:16021–16026. doi: 10.1073/pnas.0806791105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, Menezes JR, Macklis JD. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron. 2008;57:232–247. doi: 10.1016/j.neuron.2007.12.023. [DOI] [PubMed] [Google Scholar]
- McConnell SK. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–768. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- O'Leary DD, Koester SE. Development of projection neuron types, axon pathways, and patterned connections of the mammalian cortex. Neuron. 1993;10:991–1006. doi: 10.1016/0896-6273(93)90049-w. [DOI] [PubMed] [Google Scholar]
- Plachez C, Lindwall C, Sunn N, Piper M, Moldrich RX, Campbell CE, Osinski JM, Gronostajski RM, Richards LJ. Nuclear factor I gene expression in the developing forebrain. J Comp Neurol. 2008;508:385–401. doi: 10.1002/cne.21645. [DOI] [PubMed] [Google Scholar]
- Polleux F, Dehay C, Kennedy H. The timetable of laminar neurogenesis contributes to the specification of cortical areas in mouse isocortex. J Comp Neurol. 1997;385:95–116. [PubMed] [Google Scholar]
- Polleux F, Dehay C, Kennedy H. Neurogenesis and commitment of corticospinal neurons in reeler. J Neurosci. 1998;18:9910–9923. doi: 10.1523/JNEUROSCI.18-23-09910.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Armstrong DM, Fibiger HC. A comparison of the distribution of central cholinergic neurons as demonstrated by acetylcholinesterase pharmacohistochemistry and choline acetyltransferase immunohistochemistry. Brain Res Bull. 1983;11:693–720. doi: 10.1016/0361-9230(83)90013-8. [DOI] [PubMed] [Google Scholar]
- Valverde F. Reticular formation of the albino rat's brain stem cytoarchitecture and corticofugal connections. J Comp Neurol. 1962;119:25–53. doi: 10.1002/cne.901190105. [DOI] [PubMed] [Google Scholar]
- Zetterström RH, Williams R, Perlmann T, Olson L. Cellular expression of the immediate early transcription factors Nurr1 and NGFI-B suggests a gene regulatory role in several brain regions including the nigrostriatal dopamine system. Brain Res Mol Brain Res. 1996;41:111–120. doi: 10.1016/0169-328x(96)00074-5. [DOI] [PubMed] [Google Scholar]