

Abstract
Dopamine release in the striatum has been implicated in various forms of reward dependent learning. Dopamine leads to production of cAMP and activation of protein kinase A (PKA), which are involved in striatal synaptic plasticity and learning. PKA and its protein targets are not diffusely located throughout the neuron, but are confined to various subcellular compartments by anchoring molecules such as A-Kinase Anchoring Proteins (AKAPs). Experiments have shown that blocking the interaction of PKA with AKAPs disrupts its subcellular location and prevents LTP in the hippocampus and striatum; however, these experiments have not revealed whether the critical function of anchoring is to locate PKA near the cAMP that activates it or near its targets, such as AMPA receptors located in the post-synaptic density. We have developed a large scale stochastic reaction-diffusion model of signaling pathways in a medium spiny projection neuron dendrite with spines, based on published biochemical measurements, to investigate this question and to evaluate whether dopamine signaling exhibits spatial specificity post-synaptically. The model was stimulated with dopamine pulses mimicking those recorded in response to reward. Simulations show that PKA colocalization with adenylate cyclase, either in the spine head or in the dendrite, leads to greater phosphorylation of DARPP-32 Thr34 and AMPA receptor GluA1 Ser845 than when PKA is anchored away from adenylate cyclase. Simulations further demonstrate that though cAMP exhibits a strong spatial gradient, diffusible DARPP-32 facilitates the spread of PKA activity, suggesting that additional inactivation mechanisms are required to produce spatial specificity of PKA activity.
Author Summary
The striatum is a part of the basal ganglia which plays a role in addiction and reward learning. Its importance is underscored by pathologies such as Parkinson's disease and Huntington's disease in which degeneration of the dopamine inputs to the striatum or degeneration of neurons in the striatum, respectively, produces motor dysfunction. Dopamine in the striatum activates cascades of signaling molecules, ultimately producing an activity dependent change in the strength of connections between neurons. However, the dispersive movement of signaling molecules seems incompatible with the strengthening of specific subsets of connections, which is required for formation of distinct memories. Anchoring proteins, which restrict molecules to particular compartments within the neuron, are proposed to achieve specificity. We develop a reaction-diffusion model of dopamine activated signaling pathways to explore mechanisms whereby anchoring proteins can produce specificity. We use an efficient Monte-Carlo simulator to implement the cascades of signaling molecules in a neuronal dendrite with multiple dendritic spines. Simulations demonstrate that spatial specificity requires both anchoring proteins and inactivation mechanisms that limit the diffusion of signaling molecules.
Introduction
The striatum is the primary input nucleus of the basal ganglia, a set of forebrain nuclei implicated in addiction [1], motor control [2], and reinforcement learning [3], [4]. The function of the striatum is attributed to integration of glutamatergic inputs from cortex and thalamus [5] with dopaminergic afferents from midbrain [6]. In particular, dopamine signaling plays a preeminent role in striatal dependent learning [2] and in synaptic plasticity of the medium spiny projection neurons (MSPN) [7], [8]. The importance of dopamine is underscored by Parkinson's disease [9], which is caused by degeneration of dopamine producing neurons. Therefore understanding intracellular signaling produced by dopamine receptor activation is paramount for understanding the role of the striatum in learning and motor function.
Dopamine binding to the D1 receptor leads to production of cAMP which activates protein kinase A (PKA). PKA phosphorylation of DARPP-32 (Dopamine- and cAMP-regulated phosphoprotein with molecular weight 32 kD) and Ser845 of the AMPA receptor GluA1 subunit have been implicated in learning [10], addiction [1] and synaptic plasticity [7], [11]. The spatial specificity of synaptic plasticity [12]–[14] seems incompatible with diffusion of signaling molecules such as cAMP; thus, mechanisms are required to produce local activation, i.e. microdomains.
Creation of microdomains depends on several factors such as diffusional barriers, embodied by dendritic spines [15], [16], or inactivation mechanisms [17], exemplified by phosphodiesterases [18]. Development of microdomains is facilitated by multi-protein complexes, such as produced by the A-kinase-anchoring-protein AKAP5 [19], which is highly expressed in the striatum [20], [21]. AKAPs bind not only PKA but diverse molecules such as adenylate cyclase [22], and sodium channels [23]. These multi-protein complexes promote efficient PKA activation and keep its activity spatially constrained [24]. Consistent with this role, blocking anchoring of PKA to the AKAP disrupts hippocampal late-phase long term potentiation (LTP) [25], sodium channel phosphorylation [23] and striatal LTP [26]. Under conditions of synaptic activation, it is unclear whether the crucial role of AKAPs is to anchor PKA near adenylate cyclase, the cAMP source, or close to the target substrate.
Several computational modeling studies [27]–[29] have investigated the control of PKA activity and DARPP-32 phosphorylation by dopamine receptor activation, but the formation of microdomains and the role played by the subcellular location of molecule species remains largely unexplored. To resolve these important issues, we investigate the role of PKA anchoring using a spatial, Monte Carlo model of dopamine activated post-synaptic signaling pathways in a MSPN dendrite with multiple spines. Simulations demonstrate that colocalization of PKA with the source of cAMP is more important than colocalization with target molecules, and demonstrate the role of inactivation mechanisms for the generation of microdomains.
Methods
Signaling Pathways
The biochemical signaling network of direct pathway MSPNs (Fig. 1A, Table 1) was adapted from [27]. Gαolf coupled D1 type receptors connect dopamine stimulation to activation of adenylate cyclase [30], [31], with type 5 being the major type in the striatum [32]. cAMP produced by adenylate cyclase binds to the PKA holoenzyme, causing the two inherently active, catalytic subunits (PKAc) [33] to dissociate and diffuse away from the regulatory subunit dimer (PKAr). Alternatively, the cAMP saturated holoenzyme can bind to targets, which stabilizes dissociation of the catalytic subunits [34]. Targets of PKA include Thr34 of DARPP-32, Ser845 of the AMPA receptor GluA1 subunit [35], and phosphodiesterase type 10, which is the major phosphodiesterase in MSPN [36], [37]. Similar to the effect of PKA phosphorylation on phosphodiesterase type 4 [38], the activity of phosphodiesterase type 10 is enhanced by PKA phosphorylation. PhosphoThr34 DARPP-32 binds to and inhibits protein phosphatase 1 (PP1) [39]. DARPP-32 also can be phosphorylated on Thr75 by cdk5, and this form of DARPP-32 inhibits PKA [40].
Figure 1. Model of striatal medium spiny projection neuron dendrite with spines.

A. Diagram of biochemical signaling pathways. Each arrow is modeled with one or more bimolecular or enzyme reactions. See text and Table 1 for details. B. Morphology of dendrite with four spines, and location of calcium influx in the model. Subvolumes of height 0.12 µm adjacent to the top and bottom surface of the dendrite are considered submembrane subvolumes. Other dendritic subvolumes are part of the cytosol. Diffusion is two-dimensional in the dendrite and one-dimensional in the spine. C. Experimental Design: The role of anchoring is evaluated using four spatial variations in the location of adenylate cyclase and PKA. The adenylate cyclase-D1R complex (AC) is located either in the spine head or a focal dendritic submembrane area. Similarly, the PKA holoenzyme is located either in the spine head or the focal dendritic submembrane area. AMPA receptors containing GluA1 subunits are in the PSD compartment of the spine head for all cases.
Table 1. Reactions and rate constants of signaling pathways in the model.
| Reaction Equation | kf (nM−1 s−1) | kb (s−1) | kcat (s−1) | Ref |
| Da⇌Da_Ext | 2 | 2.00E-05 | Adj | |
| Da+R⇌DaR | 0.00111 | 10 | [27] | |
| DaR+Gαβγ⇌DaRGαβγ⇀DaRGβγ+GαGTP | 6.00E-04 | 0.001 | 20 | [27] |
| Gαβγ+R⇌GαβγR | 6.00E-05 | 0.0003 | [27] | |
| GαβγR+Da⇌DaRGαβγ⇀DaRGβγ+GαGTP | 0.00333 | 10 | 20 | PMR |
| DaRGβγ⇀DaR+Gβγ | 80 | # | ||
| GαGTP⇀GaGDP | 10 | [27] | ||
| GαGDP+Gβγ⇀Gαβγ | 100 | [27] | ||
| GαβγGTP+AC⇌ACGαGTP | 0.0385 | 50 | [27] | |
| ACGαGTP+ATP⇌ACGαGTP−ATP | 0.000128 | 0.2612 | [27] | |
| ACGαGTP−ATP⇌ACGαGTP+cAMP | 28.46 | 0.000259 | [27] | |
| AC+Ca⇌ACCa | 0.001 | 0.9 | [27] | |
| GαGTP+ACCa⇌ACGαGTPCa | 0.01923 | 25 | [27] | |
| ACGαGTPCa+ATP⇌ACGαGTPCa−ATP | 6.38E-05 | 0.1306 | [27] | |
| ACGαGTPCa−ATP⇌cAMP+ACGαGTPCa | 14.23 | 0.00013 | [27] | |
| PDE1+CamCa4⇌PDE1CamCa4 | 0.1 | 1 | [27] | |
| PDE1CamCa4+cAMP⇌PDE1CamCa4cAMP⇀PDE1CamCa4+AMP | 0.0046 | 44 | 11 | [27] |
| PKAc+PDE10⇌PKAcPDE10⇀pPDE10+PKAc | 6.25E-04 | 0.6022 | 0.1506 | adj |
| PKAcAMP4+PDE10⇌PKAcAMP4PDE10 | 6.25E-05 | 0.6022 | * | |
| PKAcAMP4PDE10⇌PKAcPDE10+R2C_cAMP4 | 0.38 | 0.016 | PMR | |
| PKAc+PDE10cAMP⇌PKAcPDE10cAMP⇀pPDE10cAMP+PKAc | 6.25E-04 | 0.6022 | 0.1506 | adj |
| PKAcAMP4+PDE10cAMP⇌PKAcAMP4PDE10cAMP | 6.25E-05 | 0.6022 | * | |
| PKAcAMP4PDE10cAMP⇌PKAcPDE10cAMP+R2C_cAMP4 | 0.38 | 0.016 | PMR | |
| pPDE10⇀PDE10 | 0.01036 | Adj | ||
| PDE10+cAMP⇌PDE10cAMP⇀PDE10+AMP | 0.084 | 16.8 | 4.2 | [36] |
| pPDE10+cAMP⇌pPDE10cAMP ⇀ pPDE10+AMP | 0.1008 | 16.8 | 8.4 | adj |
| AMP⇀ATP | 1 | adj | ||
| Ca+pmca⇌pmcaCa⇀pmca+CaOut | 0.05 | 7 | 3.5 | [53] |
| Ca+ncx⇌ncxCa⇀ncx+CaOut | 0.0168 | 11.2 | 5.6 | [52] |
| CaOut⇌Ca | 0.0017 | adj | ||
| Ca+Calbindin⇌CalbindinCa | 0.028 | 19.6 | [50] | |
| Cam+Ca2⇌CamCa2 | 0.006 | 9.1 | [27] | |
| CamCa2+Ca2⇌CamCa4 | 0.1 | 1000 | [27] | |
| Cam+PP2B⇌PP2BCam | 0.0046 | 0.0012 | PMR | |
| CamCa2+PP2B⇌PP2BCamCa2 | 0.046 | 0.0012 | [44] | |
| PP2BCam+Ca2⇌PP2BCamCa2 | 0.006 | 0.91 | [27] | |
| CamCa4+PP2B⇌PP2BCamCa4 | 0.046 | 0.0012 | [44] | |
| PP2BCamCa2+Ca2⇌PP2BCamCa4 | 0.1 | 1000 | PMR | |
| CamCa4+CaMKII⇌CaMKIICamCa4 | 0.01 | 3 | [118] | |
| CaMKIICamCa4+CaMKIICamCa4⇌Complex | 0.0001 | 10 | & | |
| CaMKIIpCamCa4+CaMKIICamCa4⇌pComplex | 0.0001 | 10 | & | |
| CaMKIIpCamCa4+Complex⇀CaMKIIpCamCa4+pComplex | 0.0001 | & | ||
| CaMKIICamCa4+Complex⇀CamKIICamCa4+pComplex | 0.0001 | & | ||
| Complex+Complex⇀Complex+pComplex | 0.01 | & | ||
| Complex+pComplex⇀pComplex+pComplex | 0.03 | & | ||
| CaMKIIpCamCa4⇌CamCa4+CaMKIIp | 0.0008 | 0.01 | [118] | |
| CaMKIIp+PP1⇌CaMKIIpPP1⇀PP1+CaMKII | 1.00E-05 | 0.085 | 0.025 | [119] |
| PKA+cAMP2⇌PKAcAMP2 | 8.70E-05 | 0.02 | [33] | |
| PKAcAMP2+cAMP2⇌PKAcAMP4 | 1.15E-04 | 0.2 | [33] | |
| PKAcAMP4⇌R2C_cAMP4+PKAc | 0.038 | 0.016 | [33] | |
| R2C_cAMP4⇌PKAr+PKAc | 0.152 | 0.004 | adj | |
| DARPP32+PKAc⇌DARPP32-PKA⇀PKAc+p34-DARPP32 | 0.0027 | 8 | 2 | [27] |
| p34-DARPP32+PKAcAMP4⇌p34-DARPP32−PKAcAMP4 | 0.00027 | 8 | * | |
| p34-DARPP32−PKAcAMP4⇌R2C_cAMP4+DARPP32-PKA | 0.38 | 0.016 | PMR | |
| p34-DARPP32+PP1⇌p34-DARPP32−PP1 | 0.4 | 0.58 | [27] | |
| p34-DARPP32+PP2BCamCa4⇌p34-DARPP32−PP2BCamCa4⇀PP2BCamCa4+DARPP32 | 0.000179 | 2 | 0.5 | [46] |
| p34-DARPP32-PP1+PP2BCamCa4⇌p34-DARPP32-PP1-PP2BCamCa4⇀PP1-PP2BCamCa4+p34-DARPP32 | 2.98E-05 | 0.333 | 0.0833 | [46] |
| PP1-PP2BCamCa4⇀PP2BCamCa4+PP1 | 5 | # | ||
| p34-DARPP32+PP2A_X⇌p34-DARPP32-PP2A_X⇀PP2A_X+DARPP32 | 0.00152 | 56 | 14 | [46], [56] |
| p34-DARPP32-PP1+PP2A_X⇌p34-DARPP32-PP1-PP2A_X⇀PP1-PP2A_X+DARPP32 | 0.000253 | 9.33 | 2.33 | [46], [56] |
| PP1-PP2A_X ⇀ PP2A_X+PP1 | 50 | # | ||
| Cdk5+DARPP32⇌CDK5D32⇀p75-DARPP32+Cdk5 | 0.0045 | 40 | 10 | adj |
| p75-DARPP32+PKAc⇌p75-DARPP32−PKAc | 0.00037 | 1 | [27] | |
| PP2A_B56d+PKAc⇌PKAcPP2A_B56d⇀PKAc+pPP2A | 0.0025 | 0.3 | 0.1 | [54] |
| PP2A_B56d+PKAcAMP4⇌PKAcAMP4PP2A_B56d | 0.00025 | 0.3 | * | |
| PKAcAMP4PP2A_B56d⇌R2C_cAMP4+PKAcPP2A_B56d | 0.38 | 0.016 | PMR | |
| PP2Ap⇌PP2A_B56d | 0.004 | adj | ||
| PP2A_BPR72+Ca⇌CaPP2A | 3.33E-05 | 0.1 | [55] | |
| p75-DARPP32+pPP2A⇌p75-DARPP32-pPP2A⇀DARPP32+pPP2A | 0.03 | 336 | 84 | [54] |
| p75-DARPP32+PP2A_X⇌p75-DARPP32-PP2A_X⇀DARPP32+PP2A_X | 0.0046 | 168 | 42 | [55], [56] |
| p75-DARPP32+CaPP2A⇌p75-DARPP32-CaPP2A⇀CaPP2A+DARPP32 | 0.03 | 336 | 84 | [55] |
| GluA1+PKAc⇌GluA1-PKAc⇀pS845-GluA1+PKAc | 0.00402 | 24 | 6 | [120] |
| PKAcAMP4+GluA1⇌GluA1−PKAcAMP4 | 0.000402 | 24 | * | |
| GluA1−PKAcAMP4⇌R2C_cAMP4+GluA1−PKAc | 0.38 | 0.016 | PMR | |
| GluA1+CaMKIICamCa4⇌GluA1-CaMKIICamCa4⇀pS831-GluA1+CaMKIICaMCa4 | 2.22E-05 | 1.6 | 0.4 | [120] |
| GluA1+CaMKIIpCamCa4⇌GluA1-CaMKIIpCamCa4⇀pS831-GluA1+CaMKIIpCamCa4 | 2.78E-05 | 2 | 0.5 | [120] |
| GluA1+CaMKIIp⇌GluA1-CaMKIIp⇀pS831-GluA1+CaMKIIp | 2.22E-05 | 1.6 | 0.4 | [120] |
| pS845-GluA1+PP1⇌pS845-GluA1-PP1⇀GluA1+PP1 | 0.000218 | 0.17 | 0.0425 | [120] |
| pS845pS831-GluA1+PP1⇌pS845pS831-GluA1-PP1⇀pS831-GluA1+PP1 | 0.000219 | 0.35 | 0.0875 | [120] |
| pS831-GluA1+PP1⇌pS831-GluA1-PP1⇀GluA1+PP1 | 0.000219 | 0.35 | 0.0875 | [120] |
| pS845-GluA1+PP2BCamCa4⇌pS845-GluA1 -PP2B⇀GluA1+PP2BCamCa4 | 0.00201 | 8 | 2 | [120] |
Rates were either obtained from the biochemical measurements, adjusted (adj) to reproduce other data, such as basal phosphoThr75-DARPP-32, or constrained by the thermodynamic principle of microscopic reversibility (PMR). R2C_cAMP4 is comprised of 2 regulatory PKA subunits and a single catalytic subunit.
*The cAMP saturated PKA holoenzyme likely binds to various targets at a lower rate than binding by the catalytic subunit [34]. Two types of reactions were added because NeuroRD is restricted to first or second order reactions:
CaMKII phosphorylation reactions involving “Complex” are required to produce the observed calcium sensitivity [45], and capture the probability that two calmodulin bound CaMKII subunits are adjacent in the holoenzyme;
Rapid dissociation after enzyme reaction prevents accumulation of these intermediate forms. PP2A_X indicate either BPR72 or B56d regulatory subunit. Phosphorylation of Ser845 and Ser831 are independent of each other, thus only one of two possible reactions are listed. Similarly, dephosphorylation of Ser831 is independent of whether Ser845 is phosphorylated or not, and dephosphorylation of Ser845 by calcineurin is independent of Ser831 phosphorylation.
Calcium either binds to and inhibits adenylate cyclase [41], or binds to calmodulin, which has four calcium-binding sites: one pair of fast, high affinity sites, and one pair of slow, low affinity sites [42], [43]. Calcium-bound-calmodulin binds to and activates several molecules in the striatum, including phosphodiesterase 1B, calcineurin [44], and calcium-calmodulin-dependent protein kinase type II (CaMKII) [45]. Activated calcineurin dephosphorylates phosphoThr34 DARPP-32 [46]; whereas activated CaMKII phosphorylates Ser 831 of GluA1 [47]. PP1 dephosphorylates both phosphoSer845 GluA1 and phosphoSer831 GluA1, whereas calcineurin dephosphorylates only phosphoSer845 GluA1 [48]. In addition to these molecules, to accurately simulate the calcium dynamics caused by cortical stimulation, calcium binds to the endogenous buffer calbindin [49], [50] and is extruded through the low affinity sodium calcium exchanger [51], [52], or the high affinity plasma membrane calcium ATPase pump [53].
Protein phosphatase 2A (PP2A) is a constitutively active phosphatase comprised of three subunits: catalytic, structural, and regulatory. In the model, half of the PP2A has the B56δ regulatory subunit and thus its activity is enhanced by PKA phosphorylation [54]. The remainder of the PP2A has the B″/PR72 regulatory subunit, and thus its activity is enhanced by binding a single calcium ion [55]. PP2A dephosphorylates phosphoThr75 DARPP-32 and also phosphoThr34 DARPP-32 [56].
The diffusion constant for each diffusible molecule was estimated as previously [57] but decreased due to cytosolic viscosity [58] to produce a calcium gradient from spine to dendrite similar to that measured experimentally [59]. The diffusible molecules included cAMP, ATP, all forms of calmodulin, CaMKII, DARPP-32 and the catalytic subunit of PKA (Table 2). To simulate anchoring, the diffusion coefficient of anchored molecules was set to 0. The molecules were anchored in specific regions by initializing the concentration to zero in all but the anchored regions in the morphology (Table 3,4). The anchored molecules included the dopamine receptor, G protein, adenylate cyclase, PKA (holoenzyme and regulatory subunits), phosphodiesterase and AMPA receptors. G proteins were colocalized with both the D1 receptor and adenylate cyclase for all simulations due to their limited mobility in the membrane [60] and as suggested experimentally [30], [61]. Anchored molecules can interact with either diffusible or anchored molecules that are in the same subvolume (see Morphology). To evaluate the role of colocalization, D1R, G proteins, adenylate cyclase, and PKA were anchored either in the spine head, or in a small dendritic region which had a volume equal to that of the spine head. Note that in neurons receptors are not completely immobilized in the membrane [62]; in addition, pools of PKA are anchored in multiple regions within the neuron. Therefore, non-diffusion of anchored molecules in our model serves to enhance the effect of colocalization, which facilitates evaluation of distinct functions of anchoring.
Table 2. Diffusion constants for diffusible molecules in the model.
| Molecule Name | Diffusion Constant (µm2/sec) |
| Ca | 174.3 |
| Calbindin | 9.3 |
| CalbindinCa | 9.3 |
| Da | 150 |
| ATP | 74.7 |
| AMP | 85.5 |
| cAMP | 86.4 |
| Cam | 11 |
| CamCa2 | 11 |
| CamCa4 | 11 |
| CaMKIICamCa4 | 3.6 |
| pCaMKIICamCa4 | 3.6 |
| CaMKII | 3.6 |
| pCaMKII | 3.6 |
| PKAc | 8.1 |
| DARPP32 | 10.6 |
| DARPP32-PKAc | 10.6 |
| pT34-DARPP32 | 10.6 |
| pT75-DARPP32 | 10.6 |
| pT75-DARPP32-PKAc | 10.6 |
Molecules not listed do not diffuse; thus, their diffusion constants are zero. To calculate diffusion coefficients as in [57] we used a cytosolic viscosity of 4.1 for small molecules and a cytosolic viscosity of ∼8.7 for proteins . These values yielded calcium gradients similar to those measured experimentally [59], and diffusion constants similar to those measured experimentally [50].
Table 3. Initial concentrations of non-anchored molecule species in the simulation.
| Molecule | General Cytosol (nM) |
| Ca | 58.7 |
| CaOut | 2005230.1 |
| Calbindin | 145508.3 |
| CalbindinCa | 14329.0 |
| Da | 10.0 |
| Da_ext | 1000000.4 |
| ATP | 1998350.8 |
| cAMP | 35.6 |
| PDE1 | 3125.4 |
| PDE1CamCa4 | 855.5 |
| PDE10 | 800.6 |
| PDE10cAMP | 118.3 |
| pPDE10 | 364.3 |
| pPDE10cAMP | 34.0 |
| AMP | 606.0 |
| CamCa2 | 341.9 |
| Cam | 4921.5 |
| PP2BCam | 2359.2 |
| PP2BCamCa2 | 1218.4 |
| PP2BCamCa4 | 8.1 |
| CaMKII | 11318.4 |
| CaMKIIpCamCa4 | 140.0 |
| CaMKIIp | 500.0 |
| CaMKIIpPP1 | 242.7 |
| CaMKIIpCamCa4PP1 | 8.1 |
| PP1 | 3750.0 |
| PKA | 1000.0 |
| PKAcAMP2 | 160.0 |
| PKAcAMP4 | 25.0 |
| PKAc | 50.0 |
| PKAr | 25.0 |
| PP2A_BPR72 | 800.0 |
| PP2A_B56d | 630.0 |
| DARPP32 | 36400.0 |
| p34-DARPP32-PP1 | 650.0 |
| p34-DARPP32-PP1-PP2A_B56d | 11.3 |
| Cdk5 | 1088.6 |
| CDK5-DARPP32 | 2057.7 |
| p75-DARPP32 | 10370.9 |
| p75-DARPP32-PP2A_BPR72 | 200.0 |
| p75-DARPP32-PP2A_B56d | 200.0 |
| p75-DARPP32-CaPP2A | 25.0 |
| p75-DARPP32-pPP2A | 120.0 |
| pPP2A | 140.0 |
| CaPP2A | 33.0 |
Molecules not listed have initial concentrations of 0. A single molecule produces a concentration of 28 nM in the dendrite subvolumes of the morphology; thus molecule concentrations less than 28 nM indicate that some subvolumes contained a single molecule and some did not, to produce the indicated concentration averaged over the entire morphology. General cytosol means that molecules populated the entire morphology.
*Molecules initialized in the dendrite submembrane are specified in picoMoles per µm2 (picoSD).
Molecules initialized in the spine cytosol were excluded from the PSD.
Table 4. Initial concentrations of anchored molecule species in the simulation.
| Anchored Molecules | Spine Cytosol (nM)# | Focal Dendrite Submembrane (picoSD)* |
| R | 153.9 | 20.3 |
| G | 8158.3 | 1012.5 |
| GR | 1255.5 | 160.8 |
| GαGTP | 8.1 | 0.0 |
| GαGDP | 90.0 | 12.0 |
| Gβγ | 43.2 | 0.0 |
| AC | 7428.0 | 887.6 |
| ACGαGTPCa-ATP | 14.0 | 0.0 |
| ACGαGTP-ATP | 114.5 | 19.0 |
| ACCa | 2376.0 | 259.9 |
| PKA | 7050.0 | 1130.0 |
| PKAcAMP2 | 925.0 | 155.0 |
| PKAcAMP4 | 137.5 | 30.0 |
| PKAc | 300.0 | 60.0 |
| PKAr | 150.0 | 30.0 |
Only one of these concentrations applied, depending on whether molecules were anchored in the spine, or in the dendrite.
*Molecules initialized in the dendrite submembrane are specified in picoMoles per µm2 (picoSD).
Molecules initialized in the spine cytosol were excluded from the PSD, except for PKA species.
Morphology
The biochemical network was simulated in a multi-compartment morphology comprised of a 7.75 µm long segment of dendrite (0.6 µm wide by 0.4 µm depth) with four spines (Fig. 1B). Each spine was comprised of a spine head (0.6 µm diameter), a neck (0.2 µm diameter and 0.3 µm long) and a post-synaptic density (PSD) [63]. The morphology was subdivided into multiple compartments in order to simulate the reactions and diffusion mesoscopically. The dendrite was subdivided into 310 cuboidal subvolumes of dimension 0.12×0.125×0.4 µm3, allowing 2-dimensional diffusion. One layer of dendritic subvolumes on either edge was considered as the submembrane region. The approximation of a cylindrical dendrite as a rectangular cuboid with a 0.12 µm width submembrane region produced the same ratio of submembrane to cytosol volume as a cylinder with a 70 nm submembrane region. Each spine was subdivided into 0.1 µm cylindrical slices, yielding 3 spine neck subvolumes, 2 spine head subvolumes and 1 PSD subvolume, permitting 1-dimensional diffusion. One set of simulations addressed the formation of microdomains using a 23 µm long dendrite with 12 spines. For these simulations, no molecules were anchored in the dendrite; thus it was subdivided into 342 subvolumes of dimension 0.2×0.2×0.4 µm 3 to improve simulation speed. Empirically, these subvolume sizes were both large enough to meet the well-stirred criterion [64] and smaller than the length scale of observed concentration gradients.
Stimulation
The dopamine D1 receptors in the model were activated by 1 sec duration, 1 µM dopamine elevations similar to those measured using voltammetry during learning tasks in behaving animals [65]. This dopamine elevation was repeated 5 times with a 20 sec interval, similar to the intertrial interval used in behavioral tasks. In most simulations, the diffusion coefficient of dopamine was sufficiently fast to prevent any gradients, as proposed by the volume transmission hypothesis. In a subset of simulations, the diffusion coefficient was decreased to produce a small gradient in dopamine concentration, as suggested experimentally [66]. During one set of simulations dopamine and calcium were applied together. In these simulations, 100 pulses of calcium influx to the PSD region of two of the spines (3 msec duration, 10 msec inter pulse interval, 125 molecules/msec influx) approximates the calcium influx through NMDA receptors during 100 Hz, 1 sec stimulation [59] used for LTP protocols.
Numerical Methods
NeuroRD [57] is used for computationally efficient, Monte Carlo (stochastic) simulation of reaction and diffusion of these signaling pathways. The stochastic algorithm is essential because many of the molecular populations are small, especially in the spine, invalidating the assumption of continuous concentration of molecules. Furthermore, the computational properties (i.e. bistability, threshold) of synaptic signaling pathways that are exhibited in deterministic models of spines are not necessarily maintained in stochastic models of spines [67]. NeuroRD is used because the total number of molecules in the morphology described (Fig. 1B, Table 3,4) is large enough to make simulation of individual molecules in microscopic stochastic simulators computationally prohibitive. NeuroRD achieves its efficiency by integrating the tau-leap stochastic reaction algorithm [68] with a computationally-efficient, tau-leap style of stochastic diffusion algorithm [69]. The leaping approach maintains accuracy while dramatically reducing the number of time-steps required for a simulation [70] as compared to spatial extensions of the Gillespie exact stochastic simulation algorithm [71]. Both the simulation software and the files used for the model simulations are freely available from modelDB (http://senselab.med.yale.edu/ModelDB/)and the authors website (http://krasnow.gmu.edu/CENlab/). Even with this accelerated algorithm, 500 sec of simulation time (using a simulation time step of 5 µsec) required 4 days for the 7.75 µm long dendrite and 7 days for the 23 µm long dendrite. Simulations in the 7.75 µm long dendrite were repeated 4 times using a different random number seed and simulations in the 23 µm long dendrite were repeated 6 times, analogous to repeated experimental trials. Numbers of molecules were converted into concentration, either in particular microdomains or in the entire morphology by dividing by the appropriate volume, to facilitate comparison between microdomains of different sizes. Because GluA1 is limited to the PSD, concentration is less meaningful; thus, the percent of receptors phosphorylated on Ser845 (both phosphorylated and unphosphorylated on Ser831) is reported.
Results
Previous results have shown that PKA anchoring is important for striatal LTP [26] and striatal dependent behavior [72], but these experiments did not delineate whether PKA needs to be anchored near its target molecules, such as DARPP-32 and the AMPA receptor GluA1 subunit, or near a source of activator molecules, such as adenylate cyclase. Though disruption of PKA anchoring prevents phosphorylation of targets such as sodium channels [23] or adenylate cyclase 5 [73] in response to a general elevation in cAMP, the role of anchoring in response to spatially constrained cAMP has not been investigated. In order to clarify which of these two associations is critical to PKA function, a model of the signaling pathways implicated in striatal synaptic plasticity (Fig. 1A) was implemented using NeuroRD [57], in a 7.75 µm long dendrite with four spines (Fig. 1B).
Validation of the Model
Prior to evaluating the role of PKA and adenylate cyclase location, the model was validated by comparison with experiments that measured phosphorylation of PKA targets in response to bath application of agonists. For this validation, both PKA and adenylate cyclase were located in the spine and dendrite in equal amounts.
The first validation simulates the response to bath application of 10 µM dopamine. Fig. 2A shows that a simulated increase in dopamine from 10 nM (basal level) to 10 µM leads to a 6-fold increase in phosphoThr34 DARPP-32 within 2 min, and a decrease of phosphoThr75 DARPP-32 to 60% of basal (Fig. 2B) comparable to the changes measured experimentally using dopamine D1R agonists [74]. In addition, simulated phosphoSer845 GluA1 increases by 8 fold within 5 to 10 minutes (Fig. 2C), comparable to measurements performed in the absence of D2 receptor stimulation [48].
Figure 2. Validation of the model via simulation of agonist bath application.

(A) Change in phosphoThr34 DARPP-32 is similar to that observed experimentally in response to 10 µM dopamine alone, 600 nM calcium alone (inset), and the combination of dopamine with calcium. (B) Decrease in phosphoThr75 DARPP-32 for same three conditions as (A). (C) Change in phosphoSer845 GluA1 for same three conditions as (A). All responses are similar to experimental measurements.
The second validation simulates the response to a sustained increase in intracellular calcium concentration, as occurs due to experimental activation of NMDA receptors and voltage dependent calcium channels [75]. In response to this calcium increase, simulated phosphorylation of both DARPP-32 residues decreases, to half (Thr34) or two-thirds (Thr75) the basal level (Fig. 2A and 2B), in agreement with experimental data [56], [76]. Furthermore, the simulated calcium elevation leads to dephosphorylation of phosphoSer845 GluA1 (Fig. 2C) as previously reported [75].
The last model validation evaluates the change in phosphorylation of Thr34 and Thr75 in response to paired dopamine and calcium elevations. As demonstrated experimentally [75], [76], the addition of calcium reduces the increase in phosphoThr34 caused by dopamine (Fig. 2A), and enhances the decrease in phosphoThr75 (Fig. 2B). Furthermore, paired elevation of dopamine and calcium completely blocks the simulated phosphorylation of GluA1 Ser845 (Fig. 2C).
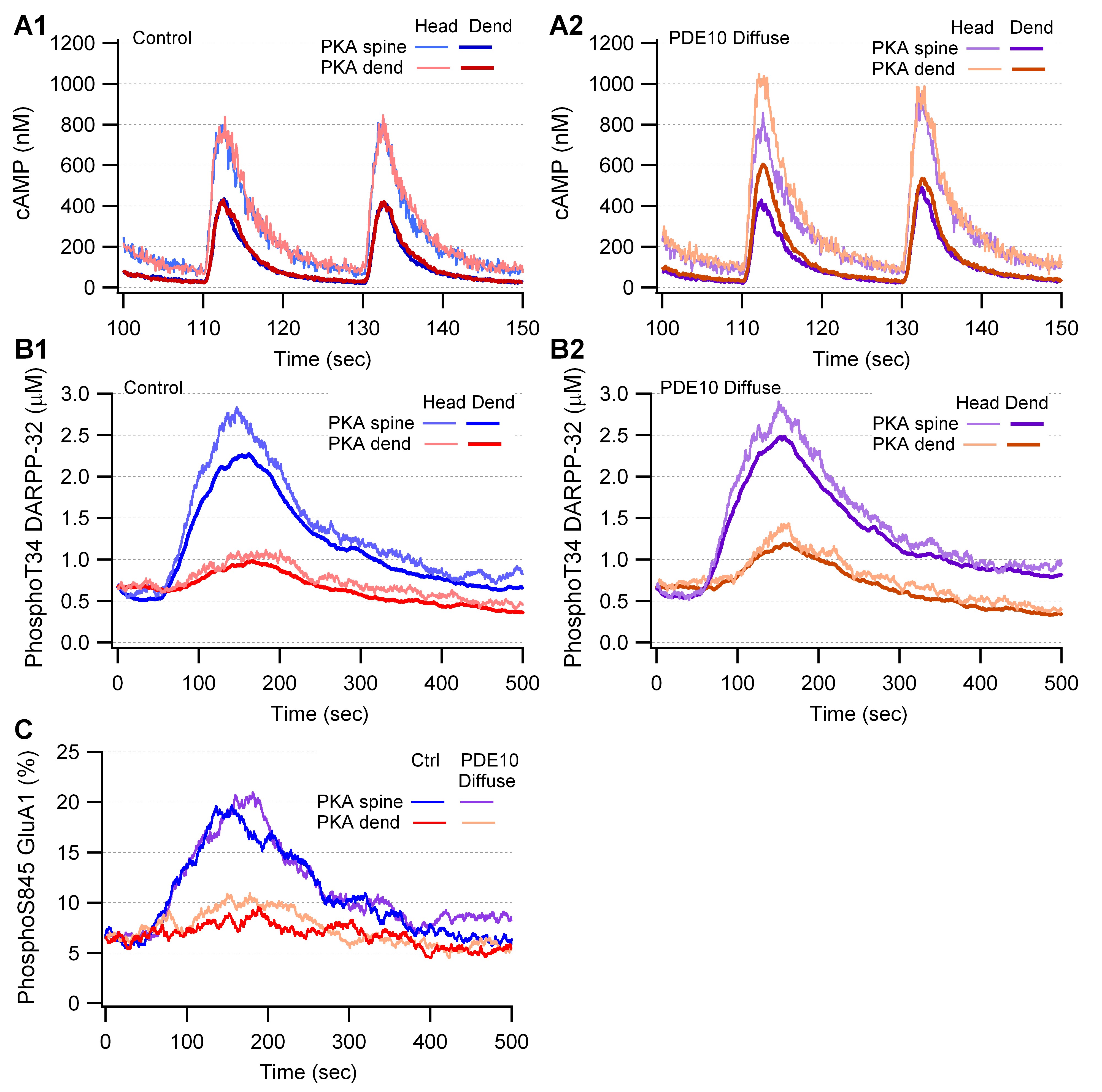
PKA Activity is Greater when Anchored near Adenylate Cyclase
Using this validated model, we explore the function of PKA anchoring relative to the location of both adenylate cyclase that activates it and PKA targets such as AMPA receptors (Fig. 1C). The D1 receptor is a metabotropic G protein coupled receptor functionally associated with adenylate cyclase [30], [31]. Immunohistochemical studies have shown D1 receptors located in spine heads and necks [77]. On the other hand, studies using immunogold labeling suggest D1 receptors are localized in extra synaptic sites homogeneously distributed in dendritic shafts and spines [78]. Therefore, prior to evaluating the effect of colocalization, we first examine the cAMP distribution when the adenylate cyclase/G protein/D1 receptor complex is located either in the spine head or in the submembrane region of the dendritic shaft.
Simulations show that when the adenylate cyclase-D1 receptor complex is located in the spine head, cAMP concentration is significantly higher in the spine head compared to the dendrite (Fig. 3A). This gradient is independent of PDE10 distribution (Suppl Fig S1). Conversely, when the complex is located in the submembrane compartment of the dendrite, cAMP concentration is higher in the dendrite than in the spine head (Fig. 3B). In summary, regardless of adenylate cyclase location, a cAMP gradient develops, with the highest concentration near the source (adenylate cyclase-D1 receptor complex); therefore, subsequent evaluations of the role of colocalization take into account the local cAMP concentration.
Figure 3. The gradient in cAMP concentration depends on the location of adenylate cyclase.

(A) When adenylate cyclase (AC) is in the spine head, there is a large difference (gradient) between spine cAMP and dendrite cAMP. (B) When adenylate cyclase is in the dendrite, there is a small gradient from dendrite to spine.
PKA location is regulated by interaction with A-Kinase anchoring proteins [19]. Various AKAPs anchor PKA to different locations, such as to the spine (e.g. AKAP5) [79], [80] or the dendritic shaft (e.g. MAP2) [81]. Recent evidence suggests that AKAP5 also anchors adenylate cyclase [22], [24], which would colocalize adenylate cyclase, PKA and GluA1 receptors. Alternatively, a pool of adenylate cyclase may be in the dendrite together with the dopamine D1 receptors found there [78]. In order to evaluate whether the critical function of anchoring is to place PKA near adenylate cyclase or near phosphoprotein targets, PKA is localized either in the spine heads or in a small submembrane region of the dendrite. For both of these spatial variations of PKA, the adenylate cyclase/D1 receptor complex is placed either in the spine heads or in the small submembrane region of the dendrite. Thus, four different spatial configurations are simulated (Fig. 1C). Note that the volume of the small dendrite region equals the volume of the spine head; thus, both global and local concentrations of these molecules are equal for all simulations. This 2×2 experimental design (Fig. 1C) allows assessment of the role of PKA location relative to adenylate cyclase - the source of cAMP, or a non-diffusible target - the AMPA receptor GluA1 subunit. Note that membrane associated molecules have limited mobility in the membrane [62] and pools of PKA are anchored in multiple regions within the neuron; nonetheless, our approach serves to enhance the effect of colocalization and facilitates evaluation of distinct functions of anchoring.
Simulations show that PKA colocalization with adenylate cyclase, either in the dendrite or the spine, produces a higher quantity of the active catalytic subunit than when PKA is apart from the adenylate cyclase (Fig. 4A, B). The large fluctuations in catalytic subunit are caused by its low quantity, which is due to a high affinity for the regulatory subunit and DARPP-32. Both the large fluctuations and the slow dynamics of PKA activation obscure the individual pulses that are visible in the cAMP traces. Colocalization in the spine produces greater PKA activity than colocalization in the dendrite because local spine cAMP is greater when adenylate cyclase is in the spine due to impeded cAMP diffusion. The effect of local cAMP (measured at the site of PKA anchoring) and colocalization (whether PKA was colocalized with adenylate cyclase or not) were evaluated using the SAS procedure GLM. Analysis revealed that local cAMP concentration predicted PKA activity (P<0.0001, R2 = 0.91), whereas colocalization did not reach significance (P = 0.083); thus the role of colocalization is primarily to place PKA in a microdomain of high cAMP concentration.
Figure 4. Colocalization of PKA with adenylate cyclase enhances PKA activity.

(A, B) Concentration of free catalytic subunit (PKAc) is greater for the two colocalized cases. (A) Traces are averaged over 4 trials; nonetheless the stochastic fluctuations are so large that the traces overlay each other and are difficult to distinguish. (B) Mean and S.E.M. of the total PKA activity (PKAc summed between 50 and 350 s). (C,D) Concentration of phosphoThr34 DARPP-32 is greater for the two colocalized cases. (C) Traces are the average over four trials. (D) Mean and S.E.M. of the concentration of phosphoThr34 DARPP-32 averaged between 50 and 350 s. (E, F) Percent of phosphoSer845 GluA1 is greater for the two colocalized cases. (E) Traces are the average over four trials. (F) Mean and S.E.M. of the percent of phosphoSer845 GluA1 averaged between 50 and 300 s.
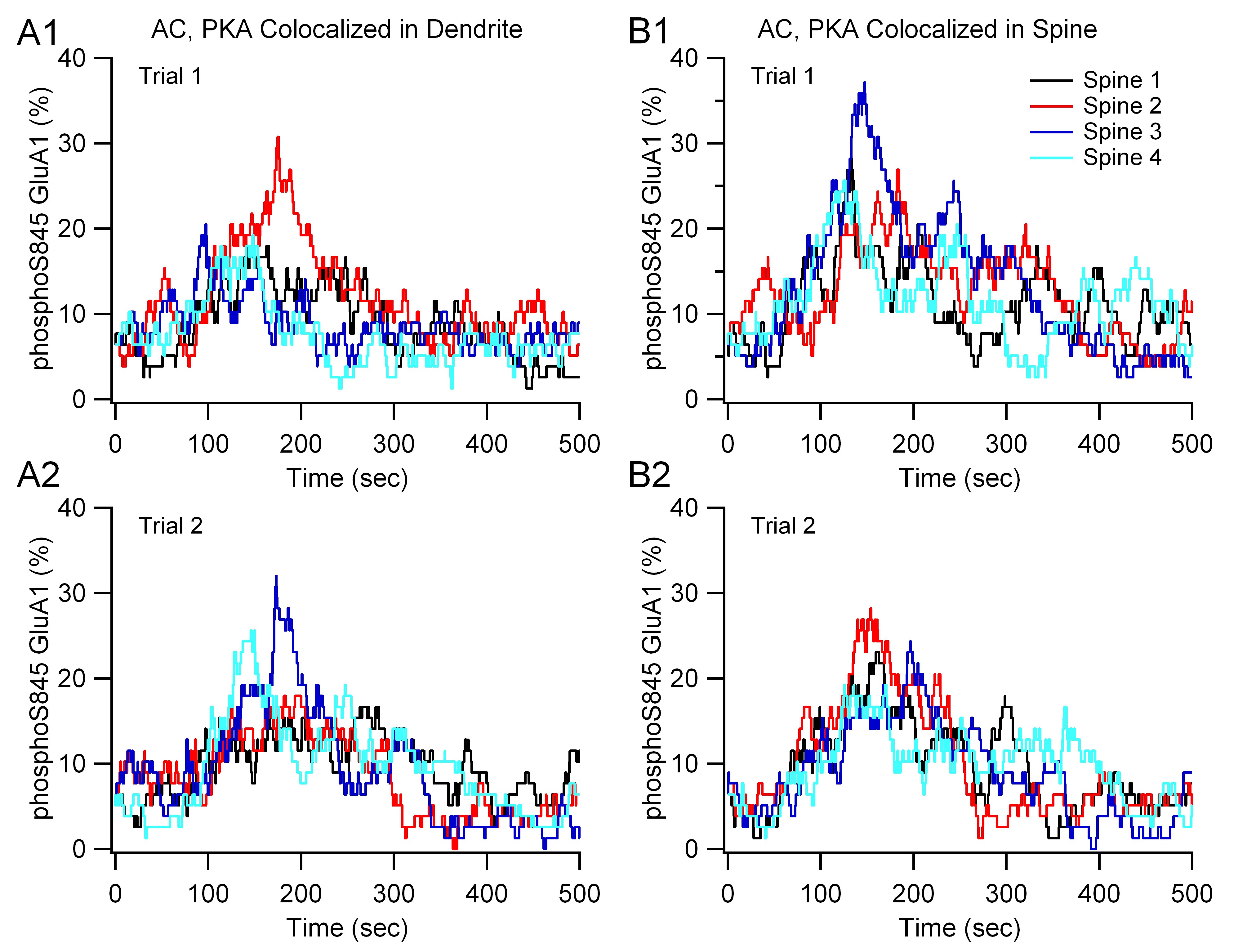
The effect of colocalization is propagated to downstream targets: Colocalization of PKA and adenylate cyclase (in spine or dendrite) leads to increased phosphoThr34 DARPP-32 (Fig. 4C,D) and phosphoSer845 GluA1 (Fig. 4E,F) compared to non-colocalized cases. In fact, both phosphoThr34 DARPP-32 and phosphoSer845 GluA1 are completely predicted by PKA activity (P = 0.0001, R2 = 0.999 and P = 0.0001, R2 = 0.93, respectively). The concentration of phosphoThr34 DARPP-32 (total quantity divided by volume of entire morphology) is illustrated because the gradients are much smaller than those of cAMP (Suppl Fig. S1). Evaluation of GluA1 in single spines on individual trials (Suppl Fig. S2) reveals that GluA1 phosphorylation varies considerably, exceeding 20% more often when PKA and adenylate cyclase are colocalized in the spine. This variability would not be evident in deterministic simulations.
Previous experiments have shown that disruption of PKA anchoring using Ht31 peptide blocks L-LTP induction in hippocampus [25] and LTP induction in the striatum [26]. To evaluate this experimental observation, we performed simulations with PKA uniformly distributed in the morphology, mimicking the disruption of anchoring by Ht31 peptide [23], [25]. The simulation shows that phosphoThr34 DARPP-32 and phosphoSer845 GluA1 are reduced (ratio<1) when PKA is uniformly distributed (Fig. 5), especially for the case of adenylate cyclase located in the spine. This decreased PKA activity supports experimental studies showing that PKA anchoring is required for LTP induction.
Figure 5. Disruption of PKA anchoring, as caused by Ht31 peptide, decreases PKA phosphorylation of downstream targets.

When adenylate cyclase (AC) is in the spine, disruption of PKA anchoring reduces by 30% both phosphoThr34 DARPP-32 (p = 0.0006) and phosphoSer845 GluA1 (p = 0.0036). When adenylate cyclase is in the dendrite, disruption of PKA anchoring produces a significant decrease in phosphoThr34 DARPP-32 (p = 0.0005), but not phosphoSer845 GluA1 (p = 0.16). Most of the diffusely distributed PKA is in the dendrite; thus, cAMP diffusion out of the spine (when adenylate cyclase is in the spine) to reach the PKA is more difficult than cAMP diffusion within the dendrite (when adenylate cyclase is in the dendrite). Consequently, disruption of PKA anchoring has a larger effect when adenylate cyclase is in the spine. PhosphoThr34 DARPP-32 is averaged between 50 and 350 s and phosphoSer845 GluA1 is averaged between 50 and 300 s.
Spine Neck Length Enhances the Effect of Colocalization
Recent studies have demonstrated that synaptic function and plasticity are correlated with synaptic structure and spine morphology [82], [83]. In particular, spine-neck geometry is an important determinant of NMDA receptor-dependent calcium signaling in the dendrite [84], [85]. To evaluate how spine morphology affects the interaction between PKA and cAMP, simulations are repeated using a spine with either a longer (1.0 µm) or shorter (0 µm) spine neck, representing the range of experimentally measured values in striatal dendrites [86]. Keeping the spine head the same size and shape maintains both the same quantity and concentration of molecules localized in the spine head. For these simulations, the four configurations of PKA and adenylate cyclase introduced previously are used.
Simulations show that spine neck enhances the effect of colocalization through control of cAMP concentration. An increase in spine neck length increases the cAMP gradient due to reduced diffusional coupling. Thus, an increase in neck length produces a larger cAMP in the spine when adenylate cyclase is in the spine, and a smaller cAMP in the spine when adenylate cyclase is in the dendrite. The effect of neck length on cAMP concentration gradient propagates downstream to PKA targets (Fig. 6A,B), yielding a larger effect of colocalization for the longer spine neck. Furthermore, local cAMP concentration predicts PKA activity regardless of neck length (P<0.0001, R2 = 0.93), and PKA activity predicts both phosphoThr34 DARPP-32 (P<0.0001, R2 = 0.99) and phosphoSer845 GluA1 (P<0.0001, R2 = 0.93). These results reinforce the observation that colocalization of PKA with its source of cAMP functions to locate PKA in a microdomain of high cAMP concentration, and also show that the effect of colocalization is robust to variation in spine neck length.
Figure 6. Results are robust to variation in parameters, though spine neck length enhances the effect of colocalization.

(A) The difference in phosphoThr DARPP-32 between colocalized and non-colocalized cases increases with a longer spine neck. (B) The difference in phosphoSer845 GluA1 between colocalized and non-colocalized cases increases with spine neck length. (C) Increase in diffusion constant decreases the activity of PKA when colocalized in the spine with adenylate cyclase (AC), but maintains enhancement over the non-colocalized case. (D) Changes in rate of GluA1 phosphorylation changes the level of phosphoSer845 GluA1, but maintains the difference between colocalized and non-colocalized cases. PhosphoThr34 DARPP-32 is averaged between 50 and 350 s and phosphoSer845 GluA1 is averaged between 50 and 300 s.
Several other simulations were performed to demonstrate that the results were robust to variations in parameters. The effect of colocalization was evaluated for different values of diffusion constants, rate of GluA1 dephosphorylation, and PDE10 location. For all these parameter variations, simulations demonstrate that colocalization of PKA with its activator still yields the greatest PKA activity. GluA1 dephosphorylation rate had no effect on PKA activity (results not shown), whereas an increase in diffusion constant (Fig. 6C) predictably decreased, but did not eliminate the effect of colocalization on PKA activity. Similarly, the GluA1 dephosphorylation rate changed the level of GluA1 Ser845 (Fig. 6D), but maintained the enhancement in phosphoSer845 GluA1 due to colocalization. Fig. S1 illustrates the effect of distributing PDE10 throughout the morphology (instead of locating it in the spine and submembrane region) for the two cases with adenylate cyclase in the spine. Though cAMP concentration and phosphoThr34 DARPP-32 are slightly increased when PKA is in the dendrite, PKA colocalization with adenylate cyclase still produces a much greater activity.
Effect of Calcium on Dopamine Response
Experimental evidence demonstrates that calcium influx through NMDA receptors is required for plasticity [87], [88]; however the molecular mechanisms of calcium action are less clear. One possibility suggested by a previous model [27] is that transient calcium elevation (in contrast with prolonged calcium elevation) paired with dopamine enhances phosphoThr34 DARPP-32 compared with dopamine alone. Another possibility is that multiple kinases are required for synaptic plasticity, and, in particular, that calcium is required for CaMKII activation [45]. Therefore to explore the interaction between dopaminergic and glutamatergic transient signals in the medium spiny projection neuron, the model was stimulated with simultaneous dopamine and calcium pulses.
Transient calcium influx to spines was applied at 100 Hz for 1 sec, which approximates the calcium influx through NMDA receptors during LTP protocols [59]. This calcium influx generates a transient increase in calcium concentration that is constrained to the stimulated spines, as observed experimentally. Peak calcium levels in the stimulated spines (1500–2000 nM) are much higher than both the non-stimulated spines (500–600 nM) and the dendrite (∼400 nM), regardless of PKA and adenylate cyclase location.
Independent of whether PKA and adenylate cyclase were colocalized in the spine or dendrite, the PKA activity, phosphoThr34 DARPP-32, and phosphoSer845 were 10% smaller in response to calcium plus dopamine than in response to dopamine alone (Fig. 7). The inhibition in PKA activity is caused by three pathways: inhibition of adenylate cyclase 5, activation of phosphodiesterase 1B and activation of calcineurin that dephosphorylates phosphoThr34 DARPP-32. The small increase in PKA activity in response to calcium alone is due to enhanced dephosphorylation of phosphoThr75 DARPP-32 by calcium bound PP2A (Fig. 7B) which cannot compensate for the other inhibitory actions of calcium, regardless of PP2A distribution or affinity for calcium (results not shown).
Figure 7. Effect of calcium on kinase activity.

(A) Calcium alone produces a large increase in CaMKII phosphorylation and a small increase in PKA activity. Calcium together with dopamine leads to CaMKII phosphorylation only 10% lower than that observed with calcium alone, and PKA activity only 10% lower than that observed with dopamine alone. Both PKA and CaMKII activity are summed between 50 and 350 sec. (B) The calcium induced increase in PKA activity is caused by a small activation in PP2A, leading to a small decrease in phosphoThr75 (not shown).
To evaluate the possibility that calcium is needed for activation of other kinases, we measured the quantity of phosphorylated CaMKII, implicated in striatal LTP [89], in response to calcium, dopamine, and calcium plus dopamine. Fig. 7A shows that calcium stimulation alone causes a tremendous increase in phosphoCaMKII when compared with dopamine stimulation alone, while the combination of dopamine and calcium produces only a 10% reduction compared to calcium alone. This suggests that the increase in phosphoCaMKII more than compensates for the small decrease in PKA activity due to calcium. Though PKA and CaMKII have distinct targets in the model, extracellular signal-regulated kinase type II, important for dopamine signaling in the striatum [90], is phosphorylated by several kinases [91], and other points of PKA and CaMKII convergence are possible. In summary, these results suggest that both dopamine and calcium are required for LTP due to the need for activation of multiple kinases.
Colocalization of Dopamine Terminals and Receptors
There are two conflicting hypotheses on the relationship between the extracellular concentration of dopamine and the activation of dopamine receptors [92]: either dopamine concentration exhibits a spatial gradient and preferentially activates receptors near terminals, or dopamine concentration does not exhibit a spatial gradient and activates receptors homogenously through volume transmission. The latter hypothesis is supported by the microscopic anatomy of dopamine receptors and terminals [78], [93]. On the other hand, voltammetry experiments coupled with modeling demonstrate a spatial gradient of dopamine concentration, suggesting that nearby low affinity receptors (e.g. D1R) are activated preferentially [66], [94], [95]. Therefore the next set of simulations evaluates the extent to which an extracellular microdomain in dopamine would produce an intracellular microdomain of D1R activated second messengers.
To approximate the spatio-temporal profile of dopamine observed in voltammetry experiments [66], dopamine diffusion was decreased by a factor of 3. This approach avoided the need for dopamine uptake via the dopamine transporter and subsequent degradation, which was beyond the scope of the current paper that focuses on post-synaptic mechanisms. The quantity of dopamine released at the spine was adjusted so that dopamine concentration at the release site remained the same, which yielded no change in PKA activity when PKA, D1R and adenylate cyclase were colocalized with the dopamine terminals in the spine. The effect of the resulting dopamine spatial gradient was evaluated by measuring the change in PKA activity when PKA, D1R and adenylate cyclase were colocalized in the dendrite, but 3 µm from the dopamine terminals.
Simulations show that the dopamine spatial gradient (Fig. 8B1 inset) leads to a small but significant decrease in amplitude of cAMP concentration (p = 0.0012) and phosphoThr34 DARPP-32 (p = 0.046), compared to no dopamine gradient (Fig. 8). In contrast, the time of the peak cAMP concentration (Fig. 8A1), phosphoThr34 DARPP-32 (Fig. 8B1) and phosphoSer845 GluA1 (Fig. 8C1) were not altered. This small change in intracellular signaling molecules caused by the dopamine spatial gradient suggests that small distances between receptor and terminal may not be important, especially if the intracellular molecules do not exhibit a spatial gradient. This idea was further evaluated with the next simulation, in which spatial gradients of intracellular signaling molecules were evaluated in a longer dendrite.
Figure 8. Locating dopamine receptors several microns away from the dopamine release site produces only a small change in cAMP signaling.

A spatial gradient of dopamine decreases (A) cAMP and PKA activity as measured by (B) phosphoThr34 DARPP-32 and (C) phosphoSer845 GluA1, but no delay in time course. A1, B1 and C1 show the time course of a single simulation, A2 show mean and stdev (n = 4) of cAMP averaged between 50 and 200 s; B2 and C2 show mean and stdev (n = 4) of phosphoThr34 DARPP-32 averaged between 50 and 350 s, and phosphoSer845 GluA1 averaged between 50 and 300 s, respectively. The inset of B1 shows the gradient in dopamine concentration from the PSD to the dendrite. Traces are the average of four simulations.
Microdomains and Spatial Specificity
The effect of close proximity between dopamine terminals and receptors can only be important when accompanied by mechanisms for limiting the spread of intracellular signaling molecules. Spatial specificity of intracellular signaling is critical for information processing, in particular for a neuron to discriminate between different patterns of input. To evaluate if spatial specificity of dopamine signaling would propagate to downstream targets, we performed simulations using a longer dendrite with an average spine density of 1/µm, as experimentally measured [86]. These simulations use the slow dopamine diffusion which produces a gradient in dopamine to ensure that only the dopamine receptors at one end are activated.
Fig. 9A shows that dopamine stimulation of one end of the dendrite produces a spatial gradient in cAMP concentration, reaching ∼400 nM at the stimulated end, and decaying to basal concentration within 13 µm of the stimulation site. The spatial profile of cAMP is well described by a single spatial decay constant of 4.69±0.14 µm (Fig. 9B). This gradient does not propagate to PKA activity, as measured by phosphoThr34 DARPP-32 activity (Fig. 9C), or phosphoSer845 GluA1 (Fig. 9E). The lack of intracellular gradient of PKA activity supports the ineffectiveness of extracellular gradients in dopamine. This result further suggests that PKA anchoring is not sufficient by itself to produce PKA microdomains, in part because when PKA is activated, the catalytic subunit diffuses.
Figure 9. Dopamine gradients produce intracellular gradients of cAMP, but not PKA activity.

(A) cAMP concentration versus time and distance from dopamine release site. (B) cAMP concentration, averaged from 50 to 150 sec, is well fit by single exponential decay. (C) phosphoThr34 DARPP-32 concentration versus time and distance from dopamine release site exhibits minimal spatial gradient. (D) Concentration of phosphoThr34 DARPP-32, averaged between 100 and 250 sec, exhibits a spatial gradient when diffusion of all DARPP-32 forms is zero (red), or diffusion of PKA bound DARPP-32 is zero (blue). Blocking the phosphoThr75-PKA interaction does not change the gradient that appears when diffusion of all DARPP-32 forms is zero (black). All three cases overlap and have the same decay space constant; thus, they are difficult to distinguish in the figure. The inset shows the fits alone, which also overlap. (E) Percent of GluA1 phosphorylated on Ser845, averaged between 100 and 250 sec, versus distance from dopamine release site. (F) Percent of GluA1 phosphorylated on Ser845, averaged between 100 and 250 sec, exhibits a spatial gradient when diffusion of the DARPP-32 forms is zero (red), or diffusion of PKA bound DARPP-32 is zero (blue). Blocking the phosphoThr75-PKA interaction does not change the gradient that appears when diffusion of all DARPP-32 forms is zero (black).
The decay length of a molecule's concentration gradient is governed not only by its diffusion constant but also by its inactivation rate [17], [96]. Though PKA is inactivated by the large quantity (∼11 µM) of phosphoThr75 DARPP-32, this inactivation is reversible, similar to calcium binding to calcium buffers [97]. In addition, diffusion of phosphoThr34 DARPP-32 could obscure the gradient of PKA activity. Therefore, to evaluate the role of DARPP-32, simulations were repeated with diffusion constants for various forms of DARPP-32 set to zero.
Fig. 9D and 9F show that a gradient in phosphoThr34 DARPP-32 and phosphoSer845 GluA1 is observed in the absence of diffusion of any form of DARPP-32 (red traces). The gradient has a spatial decay constant of 14.7±8.5 µm for phosphoThr34 DARPP-32, which does not differ significantly from the spatial decay constant of 16.1±12.2 µm for phosphoSer845 GluA1 (P = 0.84). To demonstrate that DARPP-32 acts similar to calcium buffers in spreading PKA activity, simulations were repeated with diffusion for only the PKA-bound forms set to zero (both the PKA-DARPP-32 complex and the inhibited PKA-phosphoThr75 DARPP-32 complex), but diffusion of phosphoThr34 DARPP-32 enabled. Figs. 9D and 9E show that a gradient in phosphoThr34 DARPP-32 and phosphoSer845 GluA1 are observed in these conditions (blue traces). More importantly, the spatial decay constants of the gradient do not differ between these two cases (P = 0.8 for phosphoThr34 DARPP-32 and P = 0.4 for phosphoSer845 GluA1). As a further demonstration that PKA binding to phosphoThr75 DARPP-32 does not act as an inactivation mechanism, simulations were repeated with this reaction blocked, in the absence of DARPP-32 diffusion. Fig. 9D and 9F (black traces) show that blocking this reaction does not decrease the gradient, and thus the reaction does not act as an inactivation mechanism at this spatial scale.
Discussion
The requirement of PKA activity for striatal synaptic plasticity in both D1R and D2R containing medium spiny projection neurons is well known [98], [99], but this research demonstrates that the subcellular location of PKA is critical, a prediction that has been confirmed experimentally [26]. AKAPs spatially constrain PKA through the organization of macromolecular complexes that effectively colocalize activators and effectors of enzymes. We investigated whether the critical function of AKAPs is to localize PKA near target proteins or near the source of cAMP, using a multi-compartmental stochastic reaction-diffusion model of the signaling pathways leading to PKA activation in medium spiny projection neurons. Simulations show that anchoring PKA near adenylate cyclase, the source of cAMP, is crucial because it places PKA in a high cAMP concentration microdomain. Such microdomains are created when inactivation, i.e., by phosphodiesterases, is strong enough to prevent the widespread elevation of cAMP elsewhere in the cell [18], [57]. Anchoring of PKA near its phosphoprotein targets is less important because PKA activity is less constrained due to the weak inactivation mechanisms and the spread of PKA bound to DARPP-32. This result leads to the experimental prediction that disruption of LTP by Ht31 peptide [24] will be rescued by phosphodiesterase inhibitors, which increase the diffusional distance of cAMP.
These simulations have behavioral implications because striatal dependent reward learning is associated with cortical glutamatergic stimulation followed by dopamine release in the striatum. One mechanism to ensure enhancement of only those synapses that assist in obtaining the reward is for dopamine to interact preferentially with a subset of synapses to create intracellular microdomains of cAMP. Though evidence suggests that dopamine is spatially unconstrained [78], [93], our simulations investigated whether an extracellular spatial gradient of dopamine could support an intracellular, spatial gradient of PKA activity. The results showed that the extracellular dopamine gradient produces a cAMP gradient, but not a gradient of phosphorylated PKA targets: neither phosphoThr34 DARPP-32 nor phosphoSer845 GluA1. Had the extracellular dopamine gradient been steeper, as in [94], the intracellular cAMP gradient may have been steeper. Nonetheless, the lack of inactivation mechanisms for PKA, and the diffusion of PKA bound forms of DARPP-32 would still minimize, if not eliminate, the gradient in PKA activity. On the other hand, though a gradient of PKA activity was not observed, the time of peak phosphoThr34 DARPP-32 was delayed in the part of the dendrite furthest from the dopamine release site. This result suggests that molecules sensitive to the temporal interval between PKA activity and calcium elevation could provide spatial specificity, and supports the hypothesis that dopamine provides synaptic specificity through temporal association with glutamatergic inputs [92].
The lack of gradient in PKA activity is due to weak inactivation mechanisms of PKA, which is surprising given the 11 µM concentration of phosphoT75-DARPP-32. It also seems inconsistent with prior demonstrations that disruption of PKA anchoring blocks phosphorylation of sodium channels [23] and calcium channels [100] in response to a general cAMP elevation. It is important to note that our results do not demonstrate that anchoring near targets is unimportant, just that anchoring near cyclase is more important in response to synaptic activation. Indeed, anchoring of PKA and adenylate cyclase in the spine produced greater phosphorylation of GluA1 than when PKA and adenylate cyclase were anchored in the dendrite. Enriching phosphatases in the PSD near the GluA1 receptor did not enhance the effect of colocalizing PKA with GluA1 (results not shown), suggesting the need to identify specific inactivation mechanisms for PKA to exhibit spatial specificity.
Neurons may have other inactivation mechanisms for PKA which are not included in our model. For example, multiple pools of anchored PKA coexist in a single neuron and distributing PKA throughout the neuron may have accelerated PKA inactivation. These distinct pools of anchored PKA probably phosphorylate unique post-synaptic effectors that are important for learning, addiction and synaptic plasticity. For example, PKA anchored in the spine may phosphorylate proteins in the spine head, of which GluA1 Ser845 is the example used in our simulations because it is upregulated in LTP [11]. Other pools of PKA, either in the dendrite or in the soma, may phosphorylate DARPP-32, CREB and molecules of the ERK pathway [101], which are implicated in synaptic plasticity [102] and the nuclear response to psychostimulants [103]. Nonetheless, the purpose of simulating the anchoring of PKA in a single location was to evaluate whether individual pools of PKA are spatially constrained.
Another possible PKA inactivation mechanism is excess quantity of regulatory subunit, as suggested in recent studies of cardiac myocytes [104], which might increase the importance of anchoring near targets and reveal a gradient even in the presence of DARPP-32 diffusion. A third possibility is a dynamic increased in phosphoThr75-DARPP-32 which requires modulation of cdk5 activity or location [105]. As more information about these signaling pathways becomes available, they can be evaluated by incorporation into the model. Alternatively, a gradient is likely to appear with focal dopamine release onto a much longer dendrite, given that a spatial gradient of PKA activity was observed in a model of hippocampal dendrites [106]. Nevertheless, the diffuse distribution of dopamine terminals makes it unlikely for a spatial gradient to develop under physiological conditions without stronger inactivation mechanisms. Ultimately, neuronal imaging using the AKAR sensor [107] of the spatial extent of PKA activity in response to focal application of dopamine is required to validate this model result.
Though inactivation mechanisms are clearly important for generation of spatial microdomains, morphology also plays an important role. Though qualitatively the results were robust to the length of the spine neck, quantitatively a longer spine neck enhanced the effect of colocalization, and a shorter spine neck decreased the effect of colocalization. This effect of spine neck may contribute to a positive feedback loop of synaptic regulation. A number of imaging studies have demonstrated that structural plasticity is correlated with functional synaptic plasticity [82]. In particular, LTP induction leads to longer spines [15], which would enhance the activation of PKA and possibly other molecules anchored in the spine head, such as Rap1GAP [108], which in turn would further enhance spine size.
The reduction in PKA activity caused by transient elevation in calcium contrasts with the results of previous models that show a calcium induced enhancement of either phosphoThr34 DARPP-32 [27] or phosphorylated AMPA receptors [29]. In the striatum, calcium inhibits PKA activity through its direct inhibition of adenylate cyclase 5, and calcium-calmodulin dependent activation of phosphodiesterase type 1B. The only mechanism for calcium enhancement of PKA activity in our model is calcium activation of protein phosphatase 2A, which enhances dephosphorylation of phosphoThr75 DARPP-32. In contrast to the Lindskog model, dependence of protein phosphatase 2A on calcium is first order, consistent with recent experiments [55], [109]. Therefore, the small amplitude of calcium bound PP2A (individual pulses clearly discernable in Fig. 7B) and inhibition of phosphoThr75 DARPP-32 is insufficient to compensate for other inhibitory actions of calcium.
This result is puzzling because experiments clearly demonstrate that both NMDA receptors and dopamine are required for synaptic plasticity [87], [88]. One possible post-synaptic action of calcium, supported by this model, is activation of CaMKII [89]; however, other calcium activated molecules, such as PKC [7], may be important. In addition, calcium activates calDAG GEF1 [110] and RasGRF [111] both of which lead to ERK activation, which modulates striatal LTP [102]. Another pathway is activation of casein kinase 1 by calcineurin, which then phosphorylates Ser137 of DARPP-32, thereby impairing the ability of calcineurin to dephosphorylate phosphoThr34 DARPP-32. Though this pathway was included in prior computational models [28], [29], it did not lead to synergistic phosphorylation of Thr34 DARPP-32. Nonetheless, colocalizing casein kinase 1 near calcineurin may produce such synergistic activation and should be explored in future studies. In addition to these interactions between calcium and dopamine pathways, direct interactions between dopamine and NMDA receptors may be important. Pre-synaptic glutamate receptors may modulate dopamine release [112], though NMDA receptors in dopamine neurons do not appear necessary for classical conditioning [113]. Alternatively, PKA phosphorylation of striatal enriched phosphatase (STEP) inactivates STEP, decreasing NMDA dephosphorylation and endocytosis [114].
Dynamic anchoring of several other molecules may be important for synaptic plasticity. CaMKII anchors at the PSD in an activity dependent manner, enhancing its ability to phosphorylate GluA1 receptors and protecting them from dephosphorylation [115]. Phosphodiesterase type 10 is primarily membrane associated, but moves to the cytosol when phosphorylated by PKA [116]. This translocation could accelerate degradation of cytosolic cAMP while allowing greater elevations in submembrane cAMP. Calcineurin also anchors to AKAP5 [117], which would enhance its activation by calcium in the spine, and make it less sensitive to calcium influx in the dendrite. The control of molecule anchoring by binding reactions or post-translational modification adds yet another mechanism for flexible regulation of signaling pathways.
Supporting Information
Diffuse distribution of PDE10 does not change the effect of colocalization. (A) cAMP: diffuse distribution of PDE10 (A2) enhances the cAMP concentration compared to control (A1) when PKA is in the dendrite, but not when PKA is in the spine. There is no change in the spine to dendrite gradient. (B) phosphoT34 DARPP-32: diffuse distribution of PDE10 (B2) increases phosphoT34 DARPP-32 slightlycompared to control (B1) when PKA is in the dendrite, probably due to the increased cAMP concentration. (C) phosphoS845 Glur1: No effect of PDE10 location is observed.
(JPG)
{kind=link}
Variability in the phosphorylation of S845 GluR1. (A) AC, PKA colocalized in the Dendrite; (B) AC, PKA colocalized in the spine. Top row shows results for one random seed; bottom row shows results for a different random seed. The percent phosphorylation, and the spine with greatest GluR1 phosphorylation differs between trials.
(JPG)
{kind=link}
Footnotes
The authors have declared that no competing interests exist.
This work was supported by the joint NSF-NIH Collaborative Research in Computational Neuroscience program, through NIH grant RO1AA-16022, and ONR grant MURI N00014-10-1-0198. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hiroi N, Fienberg AA, Haile CN, Alburges M, Hanson GR, et al. Neuronal and behavioural abnormalities in striatal function in DARPP-32-mutant mice. Eur J Neurosci. 1999;11:1114–1118. doi: 10.1046/j.1460-9568.1999.00570.x. [DOI] [PubMed] [Google Scholar]
- 2.Willuhn I, Steiner H. Motor-skill learning in a novel running-wheel task is dependent on D1 dopamine receptors in the striatum. Neuroscience. 2008;153:249–258. doi: 10.1016/j.neuroscience.2008.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin HH, Ostlund SB, Knowlton BJ, Balleine BW. The role of the dorsomedial striatum in instrumental conditioning. Eur J Neurosci. 2005;22:513–523. doi: 10.1111/j.1460-9568.2005.04218.x. [DOI] [PubMed] [Google Scholar]
- 4.Cromwell HC, Hassani OK, Schultz W. Relative reward processing in primate striatum. Exp Brain Res. 2005;162:520–525. doi: 10.1007/s00221-005-2223-z. [DOI] [PubMed] [Google Scholar]
- 5.Doig NM, Moss J, Bolam JP. Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J Neurosci. 2010;30:14610–14618. doi: 10.1523/JNEUROSCI.1623-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayer HM, Glimcher PW. Midbrain dopamine neurons encode a quantitative reward prediction error signal. Neuron. 2005;47:129–141. doi: 10.1016/j.neuron.2005.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, et al. Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J Neuroscience. 2000;20:8443–8451. doi: 10.1523/JNEUROSCI.20-22-08443.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendez I, Vinuela A, Astradsson A, Mukhida K, Hallett P, et al. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat Med. 2008;14:507–509. doi: 10.1038/nm1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heyser CJ, Fienberg AA, Greengard P, Gold LH. DARPP-32 knockout mice exhibit impaired reversal learning in a discriminated operant task. Brain Res. 2000;867:122–130. doi: 10.1016/s0006-8993(00)02272-1. [DOI] [PubMed] [Google Scholar]
- 11.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 12.Govindarajan A, Israely I, Huang SY, Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron. 2011;69:132–146. doi: 10.1016/j.neuron.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloodgood BL, Sabatini BL. Neuronal activity regulates diffusion across the neck of dendritic spines. Science. 2005;310:866–869. doi: 10.1126/science.1114816. [DOI] [PubMed] [Google Scholar]
- 16.Yuste R, Majewska A, Holthoff K. From form to function: calcium compartmentalization in dendritic spines. Nat Neurosci. 2000;3:653–659. doi: 10.1038/76609. [DOI] [PubMed] [Google Scholar]
- 17.Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–176. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaccolo M. Phosphodiesterases and compartmentalized cAMP signalling in the heart. Eur J Cell Biol. 2006;85:693–697. doi: 10.1016/j.ejcb.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 20.Glantz SB, Amat JA, Rubin CS. cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell. 1992;3:1215–1228. doi: 10.1091/mbc.3.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostroveanu A, Van der Zee EA, Dolga AM, Luiten PG, Eisel UL, et al. A-kinase anchoring protein 150 in the mouse brain is concentrated in areas involved in learning and memory. Brain Res. 2007;1145:97–107. doi: 10.1016/j.brainres.2007.01.117. [DOI] [PubMed] [Google Scholar]
- 22.Dessauer CW. Adenylyl cyclase–A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA. Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci. 1999;19:RC21. doi: 10.1523/JNEUROSCI.19-17-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, et al. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem. 2010;285:14450–14458. doi: 10.1074/jbc.M110.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie T, McDonough CB, Huang T, Nguyen PV, Abel T. Genetic disruption of protein kinase A anchoring reveals a role for compartmentalized kinase signaling in theta-burst long-term potentiation and spatial memory. J Neurosci. 2007;27:10278–10288. doi: 10.1523/JNEUROSCI.1602-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans RC, Blackwell KT. The role of PKA anchoring in striatal synaptic plasticity. 2010. Soc Neurosci Abstract Viewer/Itinerary Planner 452.17.
- 27.Lindskog M, Kim M, Wikstrom MA, Blackwell KT, Kotaleski JH. Transient calcium and dopamine increase PKA activity and DARPP-32 phosphorylation. PLoS Comput Biol. 2006;2:e119. doi: 10.1371/journal.pcbi.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez E, Schiappa R, Girault JA, Le Novere N. DARPP-32 is a robust integrator of dopamine and glutamate signals. PLoS Comput Biol. 2006;2:e176. doi: 10.1371/journal.pcbi.0020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano T, Doi T, Yoshimoto J, Doya K. A kinetic model of dopamine- and calcium-dependent striatal synaptic plasticity. PLoS Comput Biol. 2010;6:e1000670. doi: 10.1371/journal.pcbi.1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herve D, Levi-Strauss M, Marey-Semper I, Verney C, Tassin JP, et al. G(olf) and Gs in rat basal ganglia: possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J Neurosci. 1993;13:2237–2248. doi: 10.1523/JNEUROSCI.13-05-02237.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhuang X, Belluscio L, Hen R. GOLFalpha Mediates Dopamine D1 Receptor Signaling. J Neuroscience. 2000;20:RC91. doi: 10.1523/JNEUROSCI.20-16-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwamoto T, Okumura S, Iwatsubo K, Kawabe J, Ohtsu K, et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem. 2003;278:16936–16940. doi: 10.1074/jbc.C300075200. [DOI] [PubMed] [Google Scholar]
- 33.Zawadzki KM, Taylor SS. cAMP-dependent protein kinase regulatory subunit type IIbeta: active site mutations define an isoform-specific network for allosteric signaling by cAMP. J Biol Chem. 2004;279:7029–7036. doi: 10.1074/jbc.M310804200. [DOI] [PubMed] [Google Scholar]
- 34.Johnson DA, Leathers VL, Martinez AM, Walsh DA, Fletcher WH. Fluorescence resonance energy transfer within a heterochromatic cAMP-dependent protein kinase holoenzyme under equilibrium conditions: new insights into the conformational changes that result in cAMP-dependent activation. Biochemistry. 1993;32:6402–6410. doi: 10.1021/bi00076a013. [DOI] [PubMed] [Google Scholar]
- 35.Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 36.Fujishige K, Kotera J, Omori K. Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur J Biochem. 1999;266:1118–1127. doi: 10.1046/j.1432-1327.1999.00963.x. [DOI] [PubMed] [Google Scholar]
- 37.Nishi A, Kuroiwa M, Miller DB, O'Callaghan JP, Bateup HS, et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br J Pharmacol. 2002;136:421–433. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hemmings HC, Jr, Greengard P, Tung HY, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984;310:503–505. doi: 10.1038/310503a0. [DOI] [PubMed] [Google Scholar]
- 40.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 41.Guillou JL, Nakata H, Cooper DM. Inhibition by calcium of mammalian adenylyl cyclases. J Biol Chem. 1999;274:35539–35545. doi: 10.1074/jbc.274.50.35539. [DOI] [PubMed] [Google Scholar]
- 42.Gaertner TR, Putkey JA, Waxham MN. RC3/Neurogranin and Ca2+/calmodulin-dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium. J Biol Chem. 2004;279:39374–39382. doi: 10.1074/jbc.M405352200. [DOI] [PubMed] [Google Scholar]
- 43.Faas GC, Raghavachari S, Lisman JE, Mody I. Calmodulin as a direct detector of Ca(2+) signals. Nat Neurosci. 2011;14:301–304. doi: 10.1038/nn.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quintana AR, Wang D, Forbes JE, Waxham MN. Kinetics of calmodulin binding to calcineurin. Biochem Biophys Res Commun. 2005;334:674–680. doi: 10.1016/j.bbrc.2005.06.152. [DOI] [PubMed] [Google Scholar]
- 45.De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 46.Hemmings HC, Jr, Nairn AC, Elliott JI, Greengard P. Synthetic peptide analogs of DARPP-32 (Mr 32,000 dopamine- and cAMP-regulated phosphoprotein), an inhibitor of protein phosphatase-1. Phosphorylation, dephosphorylation, and inhibitory activity. J Biol Chem. 1990;265:20369–20376. [PubMed] [Google Scholar]
- 47.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 48.Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, et al. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parent A, Fortin M, Cote PY, Cicchetti F. Calcium-binding proteins in primate basal ganglia. Neurosci Res. 1996;25:309–334. doi: 10.1016/0168-0102(96)01065-6. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt H, Kunerth S, Wilms C, Strotmann R, Eilers J. Spino-dendritic cross-talk in rodent Purkinje neurons mediated by endogenous Ca2+-binding proteins. J Physiol. 2007;581:619–629. doi: 10.1113/jphysiol.2007.127860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lorincz A, Rozsa B, Katona G, Vizi ES, Tamas G. Differential distribution of NCX1 contributes to spine-dendrite compartmentalization in CA1 pyramidal cells. Proc Natl Acad Sci U S A. 2007;104:1033–1038. doi: 10.1073/pnas.0605412104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gall D, Gromada J, Susa I, Rorsman P, Herchuelz A, et al. Significance of Na/Ca exchange for Ca2+ buffering and electrical activity in mouse pancreatic beta-cells. Biophys J. 1999;76:2018–2028. doi: 10.1016/S0006-3495(99)77359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sedova M, Blatter LA. Dynamic regulation of [Ca2+]i by plasma membrane Ca(2+)-ATPase and Na+/Ca2+ exchange during capacitative Ca2+ entry in bovine vascular endothelial cells. Cell Calcium. 1999;25:333–343. doi: 10.1054/ceca.1999.0036. [DOI] [PubMed] [Google Scholar]
- 54.Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, et al. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci U S A. 2007;104:2979–2984. doi: 10.1073/pnas.0611532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahn JH, Sung JY, McAvoy T, Nishi A, Janssens V, et al. The B″/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc Natl Acad Sci U S A. 2007;104:9876–9881. doi: 10.1073/pnas.0703589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishi A, Bibb JA, Matsuyama S, Hamada M, Higashi H, et al. Regulation of DARPP-32 dephosphorylation at PKA- and Cdk5-sites by NMDA and AMPA receptors: distinct roles of calcineurin and protein phosphatase-2A. J Neurochem. 2002;81:832–841. doi: 10.1046/j.1471-4159.2002.00876.x. [DOI] [PubMed] [Google Scholar]
- 57.Oliveira RF, Terrin A, Di Benedetto G, Cannon RC, Koh W, et al. The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PLoS One. 2010;5:e11725. doi: 10.1371/journal.pone.0011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swaminathan R, Hoang CP, Verkman AS. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J. 1997;72:1900–1907. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheuss V, Yasuda R, Sobczyk A, Svoboda K. Nonlinear [Ca2+] signaling in dendrites and spines caused by activity-dependent depression of Ca2+ extrusion. J Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee GM, Zhang F, Ishihara A, McNeil CL, Jacobson KA. Unconfined lateral diffusion and an estimate of pericellular matrix viscosity revealed by measuring the mobility of gold-tagged lipids. J Cell Biol. 1993;120:25–35. doi: 10.1083/jcb.120.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Borgdorff AJ, Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- 63.Harris KM, Stevens JK. Dendritic spines of CA1 pryramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Isaacson SA. The Reaction-Diffusion Master Equation as an Asymptotic Approximation of Diffusion to a Small Target. SIAM J Appl Math. 2009;70:77. [Google Scholar]
- 65.Sunsay C, Rebec GV. Real-time dopamine efflux in the nucleus accumbens core during Pavlovian conditioning. Behav Neurosci. 2008;122:358–367. doi: 10.1037/0735-7044.122.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venton BJ, Zhang H, Garris PA, Phillips PE, Sulzer D, et al. Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. J Neurochem. 2003;87:1284–1295. doi: 10.1046/j.1471-4159.2003.02109.x. [DOI] [PubMed] [Google Scholar]
- 67.Bhalla US. Signaling in small subcellular volumes. II. Stochastic and diffusion effects on synaptic network properties. Biophys J. 2004;87:745–753. doi: 10.1529/biophysj.104.040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillespie DT. Approximating accelerated stochastic simulation of chemically reacting systems. J Chem Phys. 2001;115:1716–1733. [Google Scholar]
- 69.Blackwell KT. An efficient stochastic diffusion algorithm for modeling second messengers in dendrites and spines. J Neurosci Methods. 2006;157:142–153. doi: 10.1016/j.jneumeth.2006.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koh W, Blackwell KT. An accelerated algorithm for discrete stochastic simulation of reaction-diffusion systems using gradient-based diffusion and tau-leaping. J Chem Phys. 2011;134:154103. doi: 10.1063/1.3572335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hattne J, Fange D, Elf J. Stochastic reaction-diffusion simulation with MesoRD. Bioinformatics. 2005;21:2923–2924. doi: 10.1093/bioinformatics/bti431. [DOI] [PubMed] [Google Scholar]
- 72.Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, et al. Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS One. 2010;5:e10325. doi: 10.1371/journal.pone.0010325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, et al. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nishi A, Bibb JA, Snyder GL, Higashi H, Nairn AC, et al. Amplification of dopaminergic signaling by a positive feedback loop. Proc Natl Acad Sci U S A. 2000;97:12840–12845. doi: 10.1073/pnas.220410397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Snyder GL, Galdi S, Fienberg AA, Allen P, Nairn AC, et al. Regulation of AMPA receptor dephosphorylation by glutamate receptor agonists. Neuropharmacology. 2003;45:703–713. doi: 10.1016/s0028-3908(03)00319-8. [DOI] [PubMed] [Google Scholar]
- 76.Halpain S, Girault JA, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- 77.Huang Q, Zhou D, Chase K, Gusella JF, Aronin N, et al. Immunohistochemical localization of the D1 dopamine receptor in rat brain reveals its axonal transport, pre- and postsynaptic localization, and prevalence in the basal ganglia, limbic system, and thalamic reticular nucleus. Proc Natl Acad Sci U S A. 1992;89:11988–11992. doi: 10.1073/pnas.89.24.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caille I, Dumartin B, Bloch B. Ultrastructural localization of D1 dopamine receptor immunoreactivity in rat striatonigral neurons and its relation with dopaminergic innervation. Brain Res. 1996;730:17–31. doi: 10.1016/0006-8993(96)00424-6. [DOI] [PubMed] [Google Scholar]
- 79.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, et al. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 80.Swayze RD, Lise MF, Levinson JN, Phillips A, El-Husseini A. Modulation of dopamine mediated phosphorylation of AMPA receptors by PSD-95 and AKAP79/150. Neuropharmacology. 2004;47:764–778. doi: 10.1016/j.neuropharm.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 81.Zhong H, Sia GM, Sato TR, Gray NW, Mao T, et al. Subcellular dynamics of type II PKA in neurons. Neuron. 2009;62:363–374. doi: 10.1016/j.neuron.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- 83.Garcia BG, Neely MD, Deutch AY. Cortical regulation of striatal medium spiny neuron dendritic remodeling in parkinsonism: modulation of glutamate release reverses dopamine depletion-induced dendritic spine loss. Cereb Cortex. 2010;20:2423–2432. doi: 10.1093/cercor/bhp317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noguchi J, Matsuzaki M, Ellis-Davies GC, Kasai H. Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron. 2005;46:609–622. doi: 10.1016/j.neuron.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grunditz A, Holbro N, Tian L, Zuo Y, Oertner TG. Spine neck plasticity controls postsynaptic calcium signals through electrical compartmentalization. J Neurosci. 2008;28:13457–13466. doi: 10.1523/JNEUROSCI.2702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson CJ, Groves PM, Kitai ST, Linder JC. Three-dimensional structure of dendritic spines in the rat neostriatum. J Neurosci. 1983;3:383–388. doi: 10.1523/JNEUROSCI.03-02-00383.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Charpier S, Deniau JM. In vivo activity-dependent plasticity at cortico-striatal connections: evidence for physiological long-term potentiation. Proc Natl Acad Sci U S A. 1997;94:7036–7040. doi: 10.1073/pnas.94.13.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, et al. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Picconi B, Gardoni F, Centonze D, Mauceri D, Cenci MA, et al. Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J Neurosci. 2004;24:5283–5291. doi: 10.1523/JNEUROSCI.1224-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Valjent E, Bertran-Gonzalez J, Aubier B, Greengard P, Herve D, et al. Mechanisms of locomotor sensitization to drugs of abuse in a two-injection protocol. Neuropsychopharmacology. 2010;35:401–415. doi: 10.1038/npp.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 92.Arbuthnott GW, Wickens J. Space, time and dopamine. Trends Neurosci. 2007;30:62–69. doi: 10.1016/j.tins.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 93.Moss J, Bolam JP. A dopaminergic axon lattice in the striatum and its relationship with cortical and thalamic terminals. J Neurosci. 2008;28:11221–11230. doi: 10.1523/JNEUROSCI.2780-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci. 2010;30:14273–14283. doi: 10.1523/JNEUROSCI.1894-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rice ME, Cragg SJ. Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res Rev. 2008;58:303–313. doi: 10.1016/j.brainresrev.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tostevin F, ten Wolde PR, Howard M. Fundamental limits to position determination by concentration gradients. PLoS Comput Biol. 2007;3:e78. doi: 10.1371/journal.pcbi.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wagner JJ, Keizer J. Effects of rapid buffers on Ca2+ diffusion and Ca2+ oscillations. Biophys J. 1994;67:447–456. doi: 10.1016/S0006-3495(94)80500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Centonze D, Grande C, Saulle E, Martin AB, Gubellini P, et al. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci. 2003;23:8506–8512. doi: 10.1523/JNEUROSCI.23-24-08506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spencer JP, Murphy KP. Activation of cyclic AMP-dependent protein kinase is required for long-term enhancement at corticostriatal synapses in rats. Neurosci Lett. 2002;329:217–221. doi: 10.1016/s0304-3940(02)00659-6. [DOI] [PubMed] [Google Scholar]
- 100.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010;3:ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Constantinescu A, Wu M, Asher O, Diamond I. cAMP-dependent protein kinase type I regulates ethanol-induced cAMP response element-mediated gene expression via activation of CREB-binding protein and inhibition of MAPK. J Biol Chem. 2004;279:43321–43329. doi: 10.1074/jbc.M406994200. [DOI] [PubMed] [Google Scholar]
- 102.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 103.Bertran-Gonzalez J, Hakansson K, Borgkvist A, Irinopoulou T, Brami-Cherrier K, et al. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology. 2009;34:1710–1720. doi: 10.1038/npp.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aye TT, Scholten A, Taouatas N, Varro A, Van Veen TA, et al. Proteome-wide protein concentrations in the human heart. Mol Biosyst. 2010;6:1917–1927. doi: 10.1039/c004495d. [DOI] [PubMed] [Google Scholar]
- 105.Philibin SD, Hernandez A, Self DW, Bibb JA. Striatal signal transduction and drug addiction. Front Neuroanat. 2011;5:60. doi: 10.3389/fnana.2011.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Neves SR, Tsokas P, Sarkar A, Grace EA, Rangamani P, et al. Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell. 2008;133:666–680. doi: 10.1016/j.cell.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paulucci-Holthauzen AA, Vergara LA, Bellot LJ, Canton D, Scott JD, et al. Spatial distribution of protein kinase A activity during cell migration is mediated by A-kinase anchoring protein AKAP Lbc. J Biol Chem. 2009;284:5956–5967. doi: 10.1074/jbc.M805606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McAvoy T, Zhou MM, Greengard P, Nairn AC. Phosphorylation of Rap1GAP, a striatally enriched protein, by protein kinase A controls Rap1 activity and dendritic spine morphology. Proc Natl Acad Sci U S A. 2009;106:3531–3536. doi: 10.1073/pnas.0813263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Janssens V, Jordens J, Stevens I, Van Hoof C, Martens E, et al. Identification and functional analysis of two Ca2+-binding EF-hand motifs in the B″/PR72 subunit of protein phosphatase 2A. J Biol Chem. 2003;278:10697–10706. doi: 10.1074/jbc.M211717200. [DOI] [PubMed] [Google Scholar]
- 110.Crittenden JR, Cantuti-Castelvetri I, Saka E, Keller-McGandy CE, Hernandez LF, et al. Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc Natl Acad Sci U S A. 2009;106:2892–2896. doi: 10.1073/pnas.0812822106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li S, Tian X, Hartley DM, Feig LA. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26:1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zigmond MJ, Castro SL, Keefe KA, Abercrombie ED, Sved AF. Role of excitatory amino acids in the regulation of dopamine synthesis and release in the neostriatum. Amino Acids. 1998;14:57–62. doi: 10.1007/BF01345243. [DOI] [PubMed] [Google Scholar]
- 113.Parker JG, Beutler LR, Palmiter RD. The contribution of NMDA receptor signaling in the corticobasal ganglia reward network to appetitive Pavlovian learning. J Neurosci. 2011;31:11362–11369. doi: 10.1523/JNEUROSCI.2411-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Synaptic plasticity: one STEP at a time. Trends Neurosci. 2006;29:452–458. doi: 10.1016/j.tins.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mullasseril P, Dosemeci A, Lisman JE, Griffith LC. A structural mechanism for maintaining the ‘on-state’ of the CaMKII memory switch in the post-synaptic density. J Neurochem. 2007;103:357–364. doi: 10.1111/j.1471-4159.2007.04744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kotera J, Sasaki T, Kobayashi T, Fujishige K, Yamashita Y, et al. Subcellular localization of cyclic nucleotide phosphodiesterase type 10A variants, and alteration of the localization by cAMP-dependent protein kinase-dependent phosphorylation. J Biol Chem. 2004;279:4366–4375. doi: 10.1074/jbc.M308471200. [DOI] [PubMed] [Google Scholar]
- 117.Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dupont G, Goldbeter A. CaM kinase II as frequency decoder of Ca2+ oscillations. BioEssays. 1998;20:607–610. doi: 10.1002/(SICI)1521-1878(199808)20:8<607::AID-BIES2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 119.Bradshaw JM, Kubota Y, Meyer T, Schulman H. An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc Natl Acad Sci U S A. 2003;100:10512–10517. doi: 10.1073/pnas.1932759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hayer A, Bhalla US. Molecular switches at the synapse emerge from receptor and kinase traffic. PLoS Comput Biol. 2005;1:137–154. doi: 10.1371/journal.pcbi.0010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Diffuse distribution of PDE10 does not change the effect of colocalization. (A) cAMP: diffuse distribution of PDE10 (A2) enhances the cAMP concentration compared to control (A1) when PKA is in the dendrite, but not when PKA is in the spine. There is no change in the spine to dendrite gradient. (B) phosphoT34 DARPP-32: diffuse distribution of PDE10 (B2) increases phosphoT34 DARPP-32 slightlycompared to control (B1) when PKA is in the dendrite, probably due to the increased cAMP concentration. (C) phosphoS845 Glur1: No effect of PDE10 location is observed.
(JPG)
Variability in the phosphorylation of S845 GluR1. (A) AC, PKA colocalized in the Dendrite; (B) AC, PKA colocalized in the spine. Top row shows results for one random seed; bottom row shows results for a different random seed. The percent phosphorylation, and the spine with greatest GluR1 phosphorylation differs between trials.
(JPG)