

Abstract
Phosphatidylinositol glycan class A (PIGA) is involved in the first step of glycosylphosphatidylinositol (GPI) biosynthesis. Many proteins, including CD55 and CD59, are anchored to the cell by GPI. Loss of CD55 and CD59 on erythrocytes causes complement-mediated lysis in paroxysmal nocturnal hemoglobinuria (PNH), a disease that manifests after clonal expansion of hematopoietic cells with somatic PIGA mutations. Although somatic PIGA mutations have been identified in many PNH patients, it has been proposed that germline mutations are lethal. We report a family with an X-linked lethal disorder involving cleft palate, neonatal seizures, contractures, central nervous system (CNS) structural malformations, and other anomalies. An X chromosome exome next-generation sequencing screen identified a single nonsense PIGA mutation, c.1234C>T, which predicts p.Arg412∗. This variant segregated with disease and carrier status in the family, is similar to mutations known to cause PNH as a result of PIGA dysfunction, and was absent in 409 controls. PIGA-null mutations are thought to be embryonic lethal, suggesting that p.Arg412∗ PIGA has residual function. Transfection of a mutant p.Arg412∗ PIGA construct into PIGA-null cells showed partial restoration of GPI-anchored proteins. The genetic data show that the c.1234C>T (p.Arg412∗) mutation is present in an affected child, is linked to the affected chromosome in this family, is rare in the population, and results in reduced, but not absent, biosynthesis of GPI anchors. We conclude that c.1234C>T in PIGA results in the lethal X-linked phenotype recognized in the reported family.
Main Text
Phosphatidylinositol glycan class A (PIGA [MIM 311770]) is one of seven proteins involved in the first step of GPI anchor biosynthesis: transferring N-acetylglucosamine (GlcNAc) from uridine 5′-diphospho N-acetylglucosamide (UDP-GlcNAc) to PI to form GlcNac-PI.1 Many proteins are anchored to cells by GPI, and in the absence of GPI anchors, these proteins are degraded. The loss of CD55 and CD59 on erythrocytes causes complement-mediated lysis in paroxysmal nocturnal hemoglobinuria (PNH [MIM 300818]), a disease that manifests after clonal expansion of hematopoietic cells with somatic PIGA mutations.2–5 Although somatic PIGA mutations have been identified in many patients with PNH, it has been proposed that germline PIGA mutations are lethal.6,7
Germline mutations in PIGA have never been identified, but mutations in genes encoding PIGM,8 PIGV,9 and PIGN,10 proteins involved in GPI-anchor biosynthesis, have been reported. A promoter mutation in PIGM (MIM 610273) was identified in two families affected by inherited glycosylphophatidylinosital deficiency (MIM 610293) that resulted in thrombosis and seizures. Mutations in PIGV (MIM 610274) were identified in four families affected by hyperphosphatasia mental retardation syndrome (HPMR), also known as Mabry syndrome (MIM 239300). Individuals with Mabry syndrome present with elevated alkaline phosphatase levels, mental retardation, seizures, hypotonia, and facial dysmorphism. Lastly, a missense mutation was identified in PIGN (MIM 606097) in a large consanguineous family affected by multiple-congenital-anomalies-hypotonia-seizures syndrome (MCAHS [MIM 614080]). We identified a nonsense mutation in PIGA by using X chromosome exome sequencing in a family with a male-lethal X-linked disorder. This report both expands the phenotype for deficiencies of the GPI biosynthesis pathway and more specifically identifies the phenotype for germline mutations in PIGA.
The family described here presented with three deceased male children and two obligate carrier females. The proband (IV-2, Figures 1 and 2) was a male infant and the second pregnancy of a 33-year-old woman. He was delivered by Cesarean section secondary to his having been in the breech position. Prenatal ultrasound showed a right pelviectasis. Apgar scores were 11 and 7.5 Birth weight was 3540 g (∼75th centile), length was 53.5 cm (>90th centile), and occipital frontal circumference (OFC) was 37 cm (>90th centile). He had Pierre Robin sequence; a prominent occiput; an enlarged fontanelle; a depressed nasal bridge; a short, anteverted nose; malar flattening; upslanted palpebral fissures; an overfolded helix; gingival overgrowth; a small mouth with downturned corners and a triangular shape; a short neck (Figure 1A); a globulous chest; and small nails. There was a systolic II–III/VI murmur with a fixed split S2. He had hip, knee, and elbow contractures, broad palms with short fingers, and poor central tone with brisk reflexes.
Figure 1.

Phenotype of IV-2 and IV-4
(A–C) Individual IV-2. (A) Craniofacial features including micrognathia, a prominent occiput, a depressed nasal bridge, a short, anteverted nose, malar flattening, upslanted palpebral fissures, a small, triangular mouth, downturned corners of the mouth, and a short neck. (B) Sagittal T1-weighted cranial MRI demonstrates thinning of the genu of the corpus callosum, mild sulcal prominence, and cerebellar hypoplasia. (C) Axial T1-weighted MRI demonstrates white-matter or myelination immaturity for the individual's age.
(D–F) Individual IV-4. (D) Facial image showing similar facial features to those of IV-2. (E) Sagittal T1 cranial MRI demonstrates a thin corpus callosum, sulcal prominence, and a small cerebellum. (F) Axial T1-weighted cranial MRI demonstrates white-matter immaturity.
Figure 2.
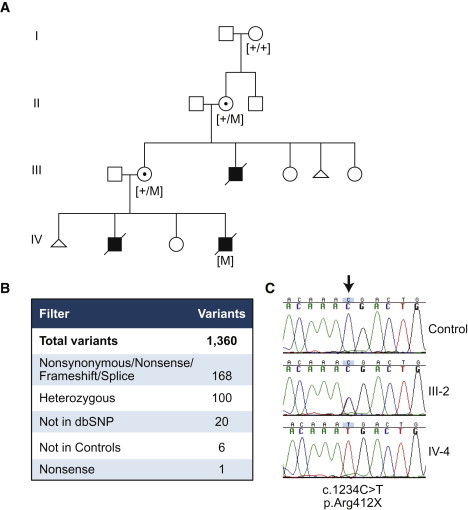
Identification and Segregation of the PIGA Mutation
(A) Pedigree showing segregation of carrier or affected status with the presence of the mutation (+, wild-type; M, mutant).
(B) Filtering criteria used for identification of the mutation from a total of 1,360 variants identified in individual III-2.
(C) Sanger sequence verification; an arrow shows the position of the mutation.
On evaluation, he had obstructive apnea, an atrial septal defect, vesicoureteral reflux, and a duplicated collecting system. Cranial MRI showed a thin corpus callosum, white-matter immaturity, no septum pellucidum, a dilated left lateral ventricle, and a small cerebellum (Figures 1B and 1C). He had myoclonic seizures requiring multiple medications, and his EEG showed burst suppression. Ionized calcium was 0.97 mmol/liter (1.13–1.32 mmol/liter), Hgb was 12.3 g/dl (14.0–18.0 g/dl), RBC was 3.92 M (4.7–6.1 M), MCV was 91.9 fl (80–94 fl), and RDW was 14.7% (11.5%–14.5%). He lived for about 11 weeks and died from pneumonia. No autopsy was performed.
The brother (IV-4 in Figure 2) of individual IV-2 was born at 35 weeks. Prenatal ultrasound showed polyhydramnios. Delivery was by Cesarean for fetal distress and breech. Apgar scores were 1,1 6,5 and 7.10 Birth weight was 3500 g (>90th centile), length was 48 cm (75th–90th centile), and OFC was 35.5 cm (∼97th centile). His features were similar to those of his brother (Figure 1D). He also had a fused metopic suture, a high-arched palate, and underdeveloped gums. Poor central tone with brisk reflexes and contractures were noted with severe myoclonic seizures.
Additional findings included a small PDA. Calcium was normal, but alkaline phosphatase was 412 IU/liter (39–117 IU/liter). Cranial MRI showed a thin corpus callosum, sulcal prominence, a small cerebellum, and white matter immaturity (Figures 1E and 1F). An EEG showed a burst suppression pattern. Hgb was 17.3 g/dl, RBC was 4.74 M, MCV was 103.7 fl, and RDW was 16.2%.
He lived for 10 weeks and died of respiratory failure. The autopsy showed hepatic microvesicular steatosis and bronchopneumonia. Examination of the central nervous system (CNS) showed dilated superficial vasculature. The corpus callosum was thin, and the cerebellum was small. The olfactory bulb and tracts were absent. White matter appeared dark (reversed cortical ribbon sign) and was decreased. Microscopically, abnormal cortical lamination was present. There were many red, ischemic neurons. The inferior olivary nucleus was simplified. The basis pontis had multiple dysplastic changes. The diagnosis was hypoxic ischemic encephalopathy and brain malformation with arhinencephaly. Muscles and bones showed noted variation in myofibril diameter and trilineage hematopoiesis, respectively.
In addition to individuals IV-2 and IV-4, an uncle (III-3 in Figure 2) was reported to have had similar characteristics. The uncle died at one month due to “stroke.” There are no medical or autopsy records available. Neither obligate carrier female had any of the clinical manifestations that were seen in the affected males.
The pedigree suggested X-linked inheritance in this family, and X-chromosome-inactivation studies (Greenwood Genetics Center) showed the pattern of X inactivation to be 100% skewed in both obligate carrier females, supporting X linkage. On the basis of the evidence for X-linked inheritance, one sample from an obligate carrier female was included in a sequencing screen of all X exons. For the purpose of this project, “X exons” were defined as all exons between positions 2,675,000 bp and 154,500,000 bp of known coding and noncoding genes in build 36 of the UCSC Genome Browser. We used solution hybridization for X exons (Sure Select, Agilent, Santa Clara, CA) to generate a single-end sequencing platform library (Illumina, San Diego, CA). The X exon oligonucleotide library was designed to select 81.3% of the target. The library was sequenced on one lane of an Illumina GAII in single-end 36 bp configuration. The sequencing yielded 18,509,569 reads. Of these, 52.1% could be uniquely aligned to the X exome target. This yielded an overall coverage of 107×. Coverage was ≥1× for 80.4% of the X exome, and 72.9% had ≥20× coverage. The MPG genotype-calling software11 made calls on 69.2% of the X exon sequence. Coding changes were predicted with custom-designed software.12 We found 1,271 substitutions and 89 insertions or deletions.
The genotypes were filtered for several attributes (Figure 2). We reasoned that the variant should be severe because it caused a lethal phenotype in hemizygous males, so we filtered for nonsynonymous, splice-site, frame-shift, and stop alleles. We used heterozygosity because the test subject was an unaffected female carrier for an X-linked trait. We sequenced eight control samples by using the same methodology and excluded variants that were present in multiple samples. Additionally, we filtered against variants present in dbSNP or the ClinSeq cohort (572 control individuals).13 This left six variants, including a single nonsense mutation, c.1234C>T in PIGA (RefSeq accession number NM_002641.2), which predicts p.Arg412∗. This variant was confirmed in the other individuals by Sanger sequencing (Figure 2), and mutation status segregated with disease and carrier status. The maternal grandmother (II-2 in Figure 2) was a carrier of c.1234C>T, but the great-grandmother (I-2) was not. Analyses of marker GATA175D03, 14 kb telomeric to PIGA, showed that I-2 did not share an allele with her affected great-grandchild (IV-4). The great-grandfather (I-1) was not available for testing. We conclude that the mutation most likely arose in II-2 on the X chromosome inherited from her father. Additionally, no frameshift, nonsense, or splice-site mutations in PIGA were identified in the eight independent samples sequenced with the same methodology, 572 samples from the ClinSeq exome cohort13 (the whole-exome capture included PIGA), or a survey of 208 patients with X-linked mental retardation.14 The c.1234C>T p.Arg412∗ variant found in the family was not identified in the 1000 Genomes data set.15 Human studies were performed according to an approved human subjects research protocol.
p.Arg412∗ occurs in the last exon of PIGA and is predicted to result in a truncated protein missing the final C-terminal 109 amino acids. Similar mutations are known to cause dysfunction of PIGA16 in PNH, and both frame-shift and nonsense mutations have been identified in the 3′ region of the gene. Together, the genetic data show that the c.1234C>T p.Arg412∗ variant is a severe change in the gene, is rare in the population, is present in an affected child, and is linked to the affected chromosome in this family. We conclude that it caused the phenotype in this family.
In addition to the genetic data, we studied the possible functional consequences of this genetic variant. This was challenging because of the X inactivation status of the heterozygous carriers (see above) and the fact that the affected males were deceased. We tested the hematopoietic cells of the mother (III-2) of the two affected individuals for evidence of a small proportion of cells displaying absent GPI-anchored proteins. We tested her erythrocytes for evidence of CD59-negative, glycophorin-A-positive cells but could find none (data not shown). We also attempted to deplete her GPI-positive cells by exposing them to aerolysin, but this did not allow detection of any glycophorin-A-positive, CD59-negative cells (data not shown).
To assess the pathogenicity of the c.1234C>T (p.Arg412∗) mutant, we performed transient transfection experiments in two previously characterized PIGA-deficient cell lines (LD17 and TF118) and compared vectors containing wild-type or mutant PIGA to empty vectors (Figure 3). PIGA mRNA was amplified from lymphoblast RNA (One-Step RT-PCR, QIAGEN), and products were cloned into a PCR4-TOPO vector (Invitrogen, Carlsbad, CA). A single full-length cDNA clone was identified, and sequence confirmation identified three nonsynonymous alterations that were corrected by site-directed mutagenesis (Quick Change, Agilent). Additionally, we removed an EcoRI site for cloning purposes without altering the predicted amino acid sequence. For the mutant cDNA, the c.1234C>T mutation was introduced. Wild-type and mutant cDNAs were subcloned into the EcoRI site of the pPB-Ubc plasmid (Dr. Kosuke Yusa, Sanger Institute, Cambridge, UK). The resultant pPB-wtPIGA and pPB-mutPIGA vectors express wild-type and mutant PIGA, respectively, under the control of human Ubc promoter. All plasmid constructs were confirmed by restriction digestion and sequencing.
Figure 3.
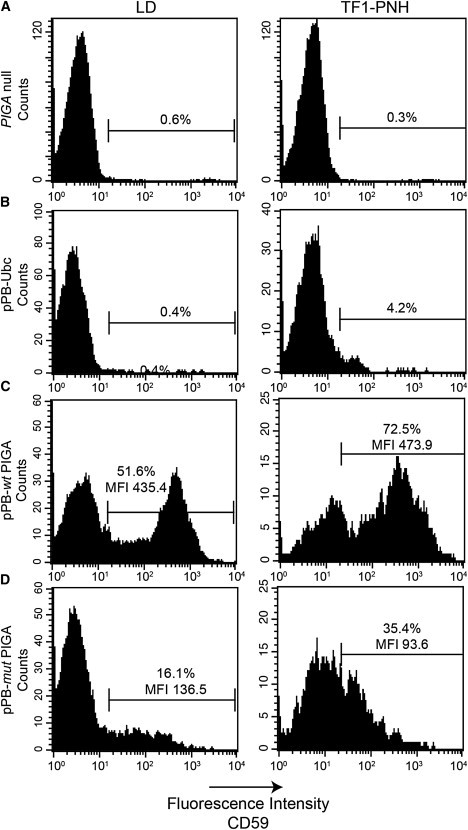
Reduced Activity of the p.Arg412∗ PIGA Mutant in Restoring Surface Expression of GPI-Anchored Protein after Transfection into PIGA-Null Cell Lines
Flow-cytometry analysis showing that cell-surface expression of CD59, a GPI-anchored protein, is absent in both LD cells (A, left panel) and TF1-PNH cells (A, right panel). Seventy-two hours after transfection, the cells that received vector expressing wild-type PIGA (pPB-wtPIGA) significantly upregulated surface CD59 expression (C) in comparison to those that received empty vector controls (B). The expression of mutant PIGA (pPB-mutPIGA) partially restored the surface expression of CD59 (D).
The LD17 and TF1-PNH cells18 were cultured in RPMI 1640 with 10% fetal calf serum (FCS). For TF1 culture, 1 ng/ml GM-SCF was added to the medium. Transfection of LD and TF1-PNH cells with 1 μg of PIGA-expression plasmid or control empty vector was performed with the Nucleofection Kit V (Lonza, Walkersville, MD). Twenty-four hours after transfection, the cells were washed with PBS and cultured in fresh RPMI supplemented with 1 ng/ml GM-SCF. Seventy-two hours after transfection, the cells were collected and washed with PBS before being stained with CD59 monoclonal antibody (BD Biosciences, Sparks, MD) for analysis of restoration of surface CD59 expression in a FACScan cytometer (BD Biosciences). Both wild-type and mutant PIGA restored cell-surface expression of CD59, a GPI-anchored protein. However, the percentages of CD59+ cells and the mean fluorescence intensity (MFI) of both PIGA-null cell lines were significantly lower when they were transfected with the c.1234C>T PIGA mutant than when they were transfected with wild-type PIGA.
Heretofore, germline mutations in PIGA have not been described in humans and were presumed to be embryonic lethal on the basis of experiments in mice3 and in both murine19 and human embryonic stem cells.20 Attempts to create knock-out mice by traditional methods resulted in low levels of chimerism and embryonic lethality.21 Pluripotent human embryonic stem cells (hES) deficient in PIGA are defective in trophoblast formation because they lack GPI-anchored BMP4 coreceptors.20 The data presented here are consistent with the hypothesized embryonic lethality of null mutations in PIGA and suggest that residual function of the protein is necessary for fetal development. The neonatal lethality seen in this family suggests that even residual function of PIGA is insufficient for normal development and survival.
Germline mutations have been described in mannosyltransferases in the GPI pathway, supporting an important role for GPI anchor biosynthesis in development and homeostasis. A PIGM promoter mutation causing reduced GPI-linked protein expression was described in two families affected by portal- and hepatic-vein thrombosis and persistent absence seizures.8 Missense PIGV mutations cause hyperphosphatasia mental retardation syndrome (HPMR [MIM 239300]).9 Individuals with HPMR have elevated alkaline phosphatase, a distinct facial gestalt, and neurological features including seizures, muscular hypotonia, and intellectual disability. A germline missense mutation has also been reported in PIGN, encoding GPI ethanolamine phosphate transferase 1.10 Studies in a knockout mouse cell line suggest that PIGN is not essential for surface expression of GPI-anchored proteins22 although surface expression of CD59 in patient fibroblasts was reduced. The resulting phenotype includes hypotonia, seizures, and multiple congenital anomalies. There is overlap among the phenotypes associated with these disorders and those of affected members of the family discussed here. Cases 1 and 2 had hypotonia, seizures, and elevated alkaline phosphatase. No thromboses were recognized, although their uncle, case 3, reportedly died from a perinatal stroke. Similar to PIGA p.Arg412∗, reported PIGM, PIGV, and PIGN alterations are hypomorphic and lead to partial loss of GPI anchor biosynthesis. Interestingly, hemolytic anemia, the primary characteristic of PNH, is not reported in patients with PIGM, PIGV, or PIGN mutations and was not found in the patients reported here. Specifically, the affected patients reported here did not manifest clinical hemoglobinuria. Unfortunately, the affected individuals in this report died prior to identification of the causative mutation, and the presence of CD59-negative erythrocytes could not be assessed. However, autopsies did not reveal any abnormalities in the hematopoietic system.
A further understanding of the role of GPI anchors in development comes from studies of female mice obtained via a Cre/loxP system targeting Piga in preimplantation embryos.6 These mice have mosaicism for cells expressing GPI-anchored proteins due to X-inactivation. The level of GPI-negative cells in different tissues indicates the importance of GPI-anchored proteins in those tissues. Heart, lung, brain, and kidney tissues showed high levels of GPI-positive cells, suggesting that normal development of these tissues depends on GPI anchors. The CNS includes many GPI-anchored proteins, including neural-cell-adhesion molecules (neural CAMs)23 and glypicans24 important for neuronal migration. Defects in neuronal migration cause CNS dysfunction, specifically seizures. We hypothesize that the CNS structural anomalies and dysfunction observed here were caused by reduced function of GPI-anchored CNS proteins.
The advent of next-generation sequencing technologies paired with exon capture is allowing rapid discovery in the area of the genetic etiology of human disease. This study demonstrates that a single family can be used to identify the causative mutation for a disease. In a disease in which the identified alteration is not obviously deleterious, or in which the function of the affected gene is not well defined, additional cases might be necessary to allow one to conclude causation. In the case reported here, a nonsense mutation was identified in a well-described gene with a function that is clearly important to normal hematopoiesis and important to both development and survival in a mouse model.21 This study shows that humans with severe PIGA mutations can survive to birth, although development is aberrant. Understanding the germline phenotype of PIGA mutations will allow identification of other affected families and facilitate better understanding of the role of PIGA in development and hematopoiesis.
Acknowledgments
The authors thank M. Abubakar, G. Bowles-Johnson, S. Covington, C. Gomez, A. Hutchins, F. Porter, M. Raygada, O. Rennert, and other clinicians at Georgetown University Hospital, Children's National Medical Center, and Johns Hopkins University for clinical investigations of this family. J. Hanover assisted with attempts to revive the frozen fibroblasts of patient IV-4. Julia Fekecs assisted with graphics. Most importantly, we gratefully acknowledge the family and dedicate this work to the memory of Walker P. Carter, Jr and Michael H. Carter III. This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Almeida A., Layton M., Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: A treatable CDG. Biochim. Biophys. Acta. 2009;1792:874–880. doi: 10.1016/j.bbadis.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Brodsky R.A. Narrative review: paroxysmal nocturnal hemoglobinuria: the physiology of complement-related hemolytic anemia. Ann. Intern. Med. 2008;148:587–595. doi: 10.7326/0003-4819-148-8-200804150-00003. [DOI] [PubMed] [Google Scholar]
- 3.Brodsky R.A. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2009;113:6522–6527. doi: 10.1182/blood-2009-03-195966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ware R.E., Rosse W.F., Howard T.A. Mutations within the Piga gene in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1994;83:2418–2422. [PubMed] [Google Scholar]
- 5.Bessler M., Mason P.J., Hillmen P., Miyata T., Yamada N., Takeda J., Luzzatto L., Kinoshita T. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13:110–117. doi: 10.1002/j.1460-2075.1994.tb06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller P., Tremml G., Rosti V., Bessler M. X inactivation and somatic cell selection rescue female mice carrying a Piga-null mutation. Proc. Natl. Acad. Sci. USA. 1999;96:7479–7483. doi: 10.1073/pnas.96.13.7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller P., Payne J.L., Tremml G., Greer P.A., Gaboli M., Pandolfi P.P., Bessler M. FES-Cre targets phosphatidylinositol glycan class A (PIGA) inactivation to hematopoietic stem cells in the bone marrow. J. Exp. Med. 2001;194:581–589. doi: 10.1084/jem.194.5.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almeida A.M., Murakami Y., Layton D.M., Hillmen P., Sellick G.S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 9.Krawitz P.M., Schweiger M.R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 10.Maydan G., Noyman I., Har-Zahav A., Neriah Z.B., Pasmanik-Chor M., Yeheskel A., Albin-Kaplanski A., Maya I., Magal N., Birk E. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011;48:383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 11.Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D., NISC Comparative Sequencing Program Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnston J.J., Teer J.K., Cherukuri P.F., Hansen N.F., Loftus S.K., Chong K., Mullikin J.C., Biesecker L.G., NIH Intramural Sequencing Center (NISC) Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am. J. Hum. Genet. 2010;86:743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: Piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nafa K., Bessler M., Castro-Malaspina H., Jhanwar S., Luzzatto L. The spectrum of somatic mutations in the PIG-A gene in paroxysmal nocturnal hemoglobinuria includes large deletions and small duplications. Blood Cells Mol. Dis. 1998;24:370–384. doi: 10.1006/bcmd.1998.0203. [DOI] [PubMed] [Google Scholar]
- 17.Nagarajan S., Brodsky R.A., Young N.S., Medof M.E. Genetic defects underlying paroxysmal nocturnal hemoglobinuria that arises out of aplastic anemia. Blood. 1995;86:4656–4661. [PubMed] [Google Scholar]
- 18.Savage W.J., Barber J.P., Mukhina G.L., Hu R., Chen G., Matsui W., Thoburn C., Hess A.D., Cheng L., Jones R.J., Brodsky R.A. Glycosylphosphatidylinositol-anchored protein deficiency confers resistance to apoptosis in PNH. Exp. Hematol. 2009;37:42–51. doi: 10.1016/j.exphem.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunn D.E., Yu J., Nagarajan S., Devetten M., Weichold F.F., Medof M.E., Young N.S., Liu J.M. A knock-out model of paroxysmal nocturnal hemoglobinuria: Pig-a(-) hematopoiesis is reconstituted following intercellular transfer of GPI-anchored proteins. Proc. Natl. Acad. Sci. USA. 1996;93:7938–7943. doi: 10.1073/pnas.93.15.7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G., Ye Z., Yu X., Zou J., Mali P., Brodsky R.A., Cheng L. Trophoblast differentiation defect in human embryonic stem cells lacking PIG-A and GPI-anchored cell-surface proteins. Cell Stem Cell. 2008;2:345–355. doi: 10.1016/j.stem.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawagoe K., Kitamura D., Okabe M., Taniuchi I., Ikawa M., Watanabe T., Kinoshita T., Takeda J. Glycosylphosphatidylinositol-anchor-deficient mice: implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:3600–3606. [PubMed] [Google Scholar]
- 22.Hong Y., Maeda Y., Watanabe R., Ohishi K., Mishkind M., Riezman H., Kinoshita T. Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. J. Biol. Chem. 1999;274:35099–35106. doi: 10.1074/jbc.274.49.35099. [DOI] [PubMed] [Google Scholar]
- 23.Karagogeos D. Neural GPI-anchored cell adhesion molecules. Front. Biosci. 2003;8:s1304–s1320. doi: 10.2741/1214. [DOI] [PubMed] [Google Scholar]
- 24.Bandtlow C.E., Zimmermann D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000;80:1267–1290. doi: 10.1152/physrev.2000.80.4.1267. [DOI] [PubMed] [Google Scholar]