

Abstract
Complete congenital stationary night blindness (cCSNB) is a clinically and genetically heterogeneous group of retinal disorders characterized by nonprogressive impairment of night vision, absence of the electroretinogram (ERG) b-wave, and variable degrees of involvement of other visual functions. We report here that mutations in GPR179, encoding an orphan G protein receptor, underlie a form of autosomal-recessive cCSNB. The Gpr179nob5/nob5 mouse model was initially discovered by the absence of the ERG b-wave, a component that reflects depolarizing bipolar cell (DBC) function. We performed genetic mapping, followed by next-generation sequencing of the critical region and detected a large transposon-like DNA insertion in Gpr179. The involvement of GPR179 in DBC function was confirmed in zebrafish and humans. Functional knockdown of gpr179 in zebrafish led to a marked reduction in the amplitude of the ERG b-wave. Candidate gene analysis of GPR179 in DNA extracted from patients with cCSNB identified GPR179-inactivating mutations in two patients. We developed an antibody against mouse GPR179, which robustly labeled DBC dendritic terminals in wild-type mice. This labeling colocalized with the expression of GRM6 and was absent in Gpr179nob5/nob5 mutant mice. Our results demonstrate that GPR179 plays a critical role in DBC signal transduction and expands our understanding of the mechanisms that mediate normal rod vision.
Main Text
Congenital stationary night blindness (CSNB) is a severe disability that impairs night vision. Complete CSNB (cCSNB) is a genetically heterogeneous form of the disorder that is caused by mutations in genes that are required for signal transduction through retinal depolarizing bipolar cells (DBCs).1–8 The function of photoreceptors and DBCs can be assessed noninvasively with the electroretinogram (ERG), and their light-induced activities are reflected in the a-wave and b-wave, respectively.9 Individuals with cCSNB and animal models of the disorder have an ERG waveform that lacks the b-wave because of a failure to transmit the photoreceptor signal through the DBCs. Depolarization of the DBCs is initiated by a metabotropic glutamate receptor-mediated (GRM6)10 modulation of a transient receptor potential melastatin 1 cation channel (TRPM1).11–13 This G protein signal transduction cascade utilizes Gαo1,14 Gβ5,15 and depends on the auxiliary protein nyctalopin.16,17 Mutations in GRM6 (MIM 604096), TRPM1 (MIM 613216), or NYX (MIM 300278), which encodes nyctalopin, all can cause cCSNB in humans.1–8 Mice with mutations in Grm6, Trpm1, Gna0, and Gnb5 or Nyx also have a no b-wave (nob) ERG phenotype.10–16,18–20 In this report, we define a critical role for GPR179, a previously uncharacterized orphan G protein receptor, in the DBC signal transduction cascade and in human cCSNB. Specifically, mutations in GPR179 in humans are responsible for a form of cCSNB. Consistent with this result, nob5 mice have a mutation in Gpr179 (Gpr179nob5/nob5) and a nob ERG phenotype. Finally, zebrafish, whose gpr179 expression is knocked down via morpholino injection, have a reduced ERG b-wave amplitude.
The nob5 mouse arose as a spontaneous mutation in a colony of C3H mice and was identified via ERG when this line was crossed to a line of C3H mice lacking the rd1 mutation (C3H-f+/+). To identify the causative mutation, we crossed affected nob5 mice to wild-type (WT) C57BL/6J mice and the resulting F1 mice were intercrossed to generate a segregating mapping cross. We identified F2 progeny homozygous for the nob5 locus by ERG and used them to map the phenotype by using a genome-wide screen with 103 simple sequence length polymorphic markers distributed throughout the genome.21 Initial mapping localized the gene to chromosome 11. Subsequently, > 600 additional informative meioses refined the map location of the nob5 locus to between D11Mit54 and D11Mit67. This 1.3 Mb region contains over 90 genes, none of which were known to be involved in DBC signal transduction or had been identified in molecular analyses of enriched pools of DBCs.22–24 To identify the mutation underlying the nob5 phenotype we used genome capture and high-throughput sequencing. Comparison of the sequence encompassing the critical region in nob5/nob5 and WT C3H mice revealed the presence of an insertion in intron 1 of Gpr179 (Figure 1A). The next-generation sequence data provided only 10 bp of sequence on either side of the insertion, but these data suggested the insertion was a transposable element. To examine this directly, we used PCR to amplify the insertion and its flanking intronic DNA. This revealed the presence of the predicted 1.3 kb fragment in WT mice and a 7.8 kb fragment in homozygous affected littermates, indicating the insertion is ∼6.5 kb (Figure 1A). Both bands were seen in heterozygotes. Henceforth, the mutant nob5 allele will be referred to as Gpr179nob5. Sequence analyses of the ends of the insertion indicated it was an endogenous retroviral element of the ERV2 class (Figure 1B). To evaluate the impact of this insertion on Gpr179 expression, we used a quantitative intron-spanning Taqman RT-PCR assay to determine Gpr179 mRNA levels of WT and Gpr179nob5/nob5 retinas (Figure 1C). The expression of mRNA representing Gpr179 in the Gpr179nob5/nob5 retina was decreased more than 800-fold compared to the expression of mRNA in the WT retina. These data indicate that the Gpr179nob5/nob5 phenotype is caused by a large insertion mutation in intron 1 of Gpr179; this insertion dramatically reduces gene expression and probably represents a null allele.
Figure 1.
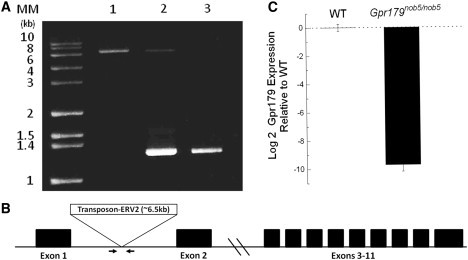
Transposable Element Disrupts Gpr179 Expression in nob5 Mouse
(A) PCR fragments from Gpr179nob5/nob5 (lane 1), Gpr179nob5/+ (lane 2), and WT C3H (lane 3). The insertion is ∼6.5 kb.
(B) Schematic exon map of Gpr179 indicating location of nob5 insertion mutation. The arrows indicate location of PCR primers used in (A).
(C) Quantitative PCR of Gpr179 cDNA generated from mRNA isolated from retinas of WT and Gpr179nob5/nob5 mice. Expression of Gpr179 was normalized to that of 18S RNA and is relative to the Gpr179 expression in WT. The error bars indicate mean ± standard deviation for three mice. Gpr179 expression in Gpr179nob5/nob5 retina is significantly reduced (p < 0.0005). All animal studies were approved by the local institutional animal care and use committees and conformed to all regulatory standards.
A series of dark-adapted ERGs obtained from representative WT, Gpr179nob5/+, and Gpr179nob5/nob5 mice are shown in (Figure 2A). Throughout the stimulus range examined, WT ERGs are dominated by a positive polarity b-wave, which increases in amplitude with increasing flash luminance and reflects the light-induced activity of DBCs.25 At higher flash luminance, the b-wave was preceded by a negative polarity a-wave, reflecting the light-induced closure of cation channels along rod photoreceptor outer segments.26 ERG responses in heterozygous Gpr179nob5/+ mice resembled the responses of WT mice, consistent with autosomal-recessive inheritance. In contrast, whereas large a-waves are obtained from homozygous Gpr179nob5/nob5 mice, these responses lack the b-wave component, revealing slow PIII, an ERG component generated by the radial Müller glial cells.27 Summary plots for the major components of the dark-adapted ERGs are shown in Figure 2B. ERG a-wave amplitudes were comparable across the three genotypes and the b-waves of Gpr179nob5/+ and WT mice were indistinguishable. The b-wave component is absent in Gpr179nob5/nob5 mice, and therefore, these data are not plotted. This ERG phenotype, in which the b-wave is absent while the a-wave is preserved, indicates that rod phototransduction is unaffected in Gpr179nob5/nob5 mice, whereas synaptic transmission between photoreceptors and DBCs, or DBC activity itself, is grossly abnormal.28 Light-adapted ERGs obtained from representative WT, Gpr179nob5/+, and Gpr179nob5/nob5 mice are shown in Figure 2C. In WT mice, the cone ERG was dominated by the positive polarity b-wave and higher frequency oscillatory potentials, which reflect activity through the DBC pathway.29 In contrast, cone ERGs of Gpr179nob5/nob5 mice are electronegative. Summary plots for cone ERGs recorded from all three genotypes are shown in Figure 2D. When cone ERG amplitude is measured from the negative trough to the following positive peak, the Gpr179nob5/nob5 response is reduced in amplitude, whereas those from WT and Gpr179nob5/+ heterozygotes are comparable. The Gpr179nob5/nob5 ERG phenotype is essentially indistinguishable from those of mouse mutants for other proteins involved in DBC signal transduction, including GRM6,10,19,20 TRPM1,11–13 and NYX,16,18 a protein required for correct localization of TRPM1 channels in DBC dendrites.17
Figure 2.
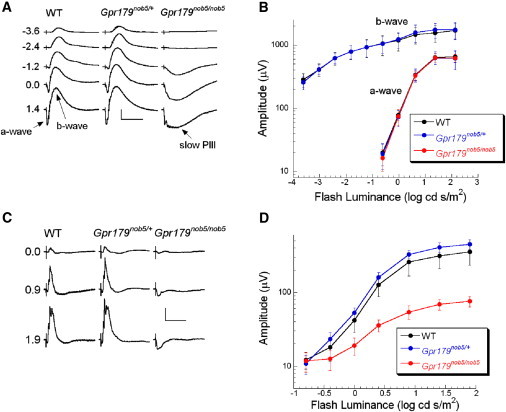
ERG Phenotype of Gpr179nob5/nob5 Mice
ERGs were recorded from mice anesthetized with ketamine (80 mg/kg) and xylazine (16 mg/kg) after overnight dark adaptation via a published procedure.16
(A) Dark-adapted ERG series obtained from representative WT (left), Gpr179nob5/+ (middle), and Gpr179nob5/nob5 (right) littermates at 6 months of age. The scale bars indicates 100 ms and 500 μV. Values to the left of each row of waveforms indicate flash luminance in log cd s/m2. (B) Luminance-response functions for the major components of the dark-adapted ERG. The b-wave component is absent in Gpr179nob5/nob5 mice and therefore these data are not plotted. (C) Cone-mediated ERG series obtained from WT (left), Gpr179nob5/+ (middle), and Gpr179nob5/nob5 (right) littermates at 6 months of age. Scale bar indicates 100 ms and 100 μV. (D) Luminance-response functions for the cone ERG b-wave. Values to the left of each row of waveforms indicate flash luminance in log cd s/m2.
GPR179 encodes a predicted orphan G protein-linked receptor that has not been previously characterized in any cell or tissue. The nob5 phenotype predicts that the GPR179 gene product is required for DBC function and therefore should be present in DBCs. Because DBCs receive input from photoreceptors via ribbon synapses in the outer plexiform layer (OPL), we initially characterized gross retinal morphology and OPL ultrastructure of the Gpr179nob5/nob5 retina. However, all cellular and synaptic layers appeared normal in the Gpr179nob5/nob5 retina (Figure 3A). Furthermore, Gpr179nob5/nob5 ribbon synapses are indistinguishable from WT (Figure 3B). A normal retinal morphology is typical of all other mouse models of cCSNB.10,12,13,18,30
Figure 3.
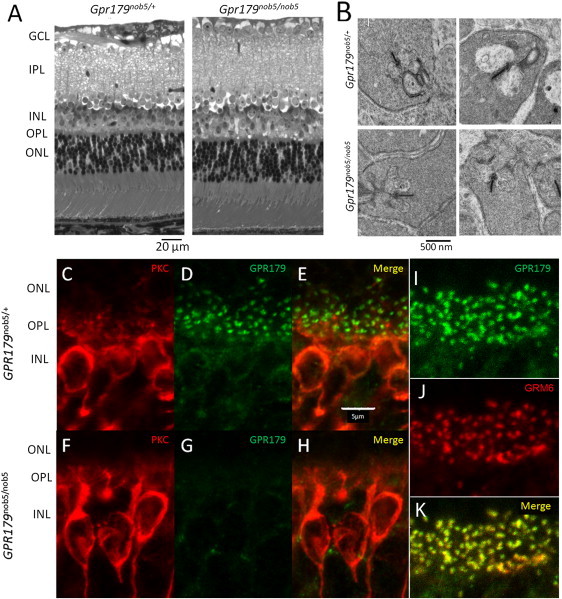
Anatomical Phenotype of Gpr179nob5/nob5 Retina
(A) Retinal cross-sections obtained from Gpr179nob5/+ and Gpr179nob5/nob5 mouse retinas fixed with 2.5% glutaraldehyde/2% paraformaldehyde and prepared according to published procedures.35
(B) Electron micrographs of ribbon structures in Gpr179nob5/+ and Gpr179nob5/nob5 mouse retinas prepared according to published procedures.
(C–K) Confocal immunohistochemistry of retinas fixed with 4% paraformaldehyde for 15 min and prepared according to published procedures.16 The scale bar indicates 5 μm. (C–E) Gpr179nob5/+ retina labeled with antibodies against (C) PKCα, (D) GPR179, and (E) merge of (C) and (D). (F–H) Gpr179nob5/nob5 retina labeled with antibodies against (F) PKCα, (G) GPR179, and (H) merge of (F) and (G). (I–K) Gpr179nob5/+ retina labeled with antibodies for (I) GPR179, (J) GRM6, and (K) merge of I–J. The following antibodies were used: GPR179; affinity purified polyclonal sheep anti-GPR179 peptide (KVQEETPGEDLDRPVLQKR), 1:1,000; mouse monoclonal anti-ctbp2/Ribeye, 1:1,000 (BD Biosciences); guinea pig anti-GRM6, 1:1000 (see Koike et al.12); rabbit anti-PKCα (1:1,000, Sigma). The following secondary antibodies were used: Alexa-488 donkey anti-sheep, Alexa-555 goat anti-rabbit, and Alexa-633 goat anti-guinea pig (all at 1:1,000; Invitrogen).
To examine the cellular localization of GPR179 in the mouse retina, we developed a polyclonal sheep antibody to a peptide (KVQEETPGEDLDRPVLQKR) located within the amino terminal extracellular domain. Retinal cryosections from Gpr179nob5/+ and Gpr179nob5/nob5 littermates were reacted with our antibody to GPR179 and an antibody against PKCα to label rod DBCs. In Gpr179nob5/+ retina, PKCα labeled the entire rod DBC (Figures 3C and 3E), whereas the GPR179 antibody produced a punctuate-labeling pattern (Figures 3D and 3E), which corresponds to the location of the OPL. In the Gpr179nob5/nob5 retina, labeling for PKCα (Figures 3F and 3H) is comparable to Gpr179nob5/+ and labeling for GPR179 is absent (Figures 3G and 3H), consistent with decreased Gpr179 mRNA expression in Gpr179nob5/nob5 retinas (Figure 1C). The punctuate-labeling pattern of GPR179 in the OPL is typical of proteins that are localized to the dendritic tips of DBCs, including GRM6, TRPM1 and NYX.12,16 To confirm this localization, we double-labeled retinal sections with antibodies to GPR179 (Figure 3I) and GRM6 (Figure 3J) The punctate labeling for GPR179 colocalizes with GRM6 (Figure 3L), the glutamate receptor known to mediate signaling in DBCs. These data show that GPR179 is expressed on the dendritic terminals of DBCs. GPR179 is not expressed elsewhere in the retina (data not shown). The combined ERG, genetic and immunohistochemical data argue strongly that the Gpr179nob5/nob5 mouse phenotype is caused by the insertion and results in a functionally null allele of GPR179.
To directly determine whether reduced GPR179 expression could recapitulate the reduced b-wave phenotype, we used morpholino knockdown in zebrafish, which have a single copy of gpr179 in their genome. We injected 1 cell stage zebrafish embryos with morpholinos (MOs) targeted against the Gpr179 translation start site (MO-Gpr179 5′-GCCCATACTTTTAGCAACTGCTTCT-3′), and recorded ERGs at 4-6 days post fertilization. As comparisons, MOs against either the nyx translation start site (MO-Nyx 5′-GATGAAACACATCACTGGCTTC-3′)31 or control (C-MO 5′-CCTCTTACCTCAGTTACAATTTATA-3′) were injected. Embryos injected with MO-Gpr179 had a significantly reduced ERG b-wave amplitude, similar to embryos injected with MO-Nyx (positive control) (Figures 4A and 4B). The b-wave was unaffected following injection of the control MO (Figure 4B). The b-wave/a-wave ratio decreased from 3.79 ± 0.46 (n = 4) in control to 0.75 ± 0.11 (n = 7) in Gpr179-MO (p = 0.005). These results indicate that gpr179 expression is required for normal DBC function in zebrafish.
Figure 4.
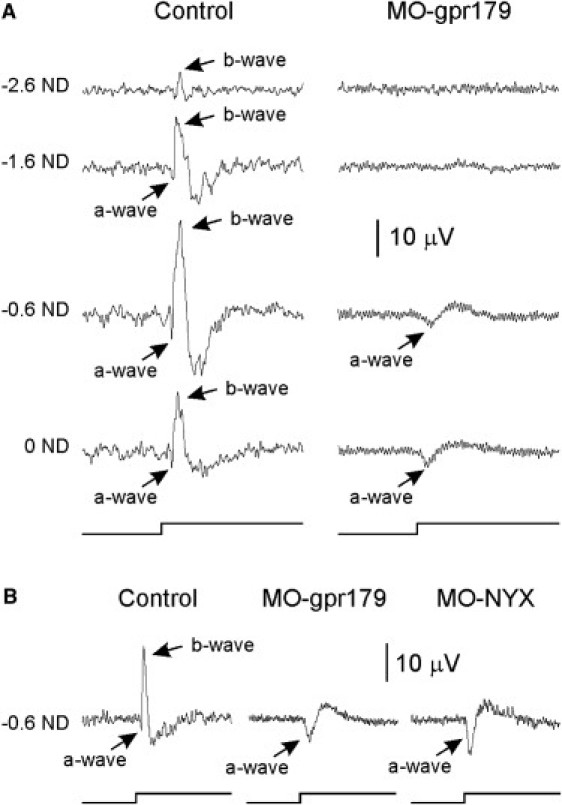
GPR179 Knockdown in Zebrafish Decreases ERG b-Wave
Injected MOs (Gene Tools) were designed against the gpr179 translation site (MO 5′-GCCCATACTTTTAGCAACTGCTTCT-3′), which occurs as a single copy in the zebrafish genome, or the nyx translation site (MO 5′-GATGAAACACATCACTGGCTTC-3′) or were a standard control (MO 5′-CCTCTTACCTCAGTTACAATTTATA-3′). In each case, 30 ng of MO was injected into the chorions of one-cell-stage zebrafish embryos. ERGs were recorded from larvae at 4–6 days post fertilization with a 1 s stimulus after 30 min of dark adaption, as previously described.36
(A) ERGs from embryos injected with control (left column) or MO-gpr179 (right column) MOs and tested at four flash intensities. Flash intensity at 0 log is 9.3 W/m2. Neutral density (ND) filters reduced the intensity by the indicated amount in log units. The b-wave amplitudes obtained from MO-gpr179 injected embryos were strongly reduced at all intensities. The b-wave/a-wave ratio decreased from 3.79 ± 0.46 (n = 4) in control to 0.75 ± 0.11 (n = 7) in MO-gpr179 (p = 0.005), showing that a specific knockdown of gpr179 reduces DBC function.
(B) The magnitude of b-wave reduction for MO-gpr179 and MO-nyx were comparable. Injections of MO-control did not affect the ERG (data not shown).
Mutations in NYX, GRM6 or TRPM1 have been identified in patients with cCSNB.1–8 However, a small number of individuals with cCSNB did not bear mutations in any of these genes.8 To evaluate the potential involvement of GPR179 in cCSNB patients lacking DBC function, we sequenced the 11 exons and flanking splice sites of the human gene in 44 patients (see Table 1 for primers). All human studies were undertaken with the approval of the appropriate institutional review board. We identified two probands with inactivating mutations in the GPR179 gene. Proband 1 had no family history of night blindness or consanguinity and was 10 years old at the time of diagnosis. He presented with 20/70 best corrected visual acuities, mild myopic refractive error, congenital nystagmus, a history of early onset nightblindness, a normal retinal appearance and full Goldmann visual fields. ERGs obtained under ISCEV standard conditions from proband 1 are shown in Figure 5A. Under dark-adapted conditions (upper traces), the ERG b-wave recorded to a low luminance stimulus was markedly reduced in amplitude, whereas the ERG obtained to a high flash luminance had a robust a-wave without the subsequent b-wave seen in controls (middle traces). Under light-adapted conditions (lower pair of traces), the ERG waveform showed a square a-wave but retained a late positive ERG component. ERGs of proband 2 showed a similar selective absence of the dark-adapted b-wave and a square light-adapted ERG a-wave (data not shown). These ERG abnormalities have been uniquely associated with human cases of DBC dysfunction32–34 and readily distinguish cCSNB from other retinal disorders with a reduced b-wave, such as incomplete CSNB (MIM 300071 and 610427).33
Table 1.
PCR Primers Used to Generate Products for Sequence Analysis
| Name | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Amplicon Size (bp) |
|---|---|---|---|
| GPR179-EXON1_1 | GACTGCAGCCAGCCTCTG | GCATGTTGAGAAAATTGGCG | 402 |
| GPR179-EXON1_2 | AGATGCCCAGCAGCTATCAC | TCAACACTCGCTTCTTCAGG | 410 |
| GPR179-EXON1_3 | ACCTTTAACCCTCCACCAGG | GGACCTGTACTTCCTTCCCC | 410 |
| GPR179-EXON2 | AAAGTGCCATAATTCCGAGC | GAGATGCTGGGGTAGCTTTG | 330 |
| GPR179-EXON3 | GACCCAAGTCCCATGGATAG | GTACCCTCCCAACCATCCTC | 261 |
| GPR179-EXON4 | GGCTGTGGTGACTATCAGGG | TAGTCTCAGGCCTCCAGTTG | 386 |
| GPR179-EXON5 | ACAGGGTGAAGCCTTAGCTC | GTGCCAGGTTTTGGCTATG | 241 |
| GPR179-EXON6 | TGGTGATGGCTCTATTGCAG | ATGAGTGAGCTGGGGTGG | 284 |
| GPR179-EXON7 | CAGCAATGGACAGAGACCAG | CCAGAACAAGAGGAACCCTG | 401 |
| GPR179-EXON8 | TCTTTCAGAGAAAGGGGTGG | GTAGCAGTGTTGCCAGGATG | 313 |
| GPR179-EXON9 | CTAAGCGTATCAGGGTTGGG | CTCCACAGTGGCCCTGAC | 274 |
| GPR179-EXON10 | TGAGTGCAGGAGTGGACAAG | GACTTGCAGTGGCACATAGC | 311 |
| GPR179-EXON11-1 | AAGGGTGGAGGAACAGACAG | CGCTGGCTGACCTGAAG | 497 |
| GPR179-EXON11-2 | GACCCGCCTCTTCTTGACTC | TAGAAACCTGGTGCCTGATG | 472 |
| GPR179-EXON11-3 | AGGCCATCAGCCAGGAG | GCCCTGCTTTTCTCTACTGC | 473 |
| GPR179-EXON11-4 | CAGCCAAGCAAGAAAATGTG | TCTGTTGGTTCTGGAGGGAC | 471 |
| GPR179-EXON11-5 | CGTTCTCGGAGCACCTACAG | GCTGACGCGTTTCTGATTC | 481 |
| GPR179-EXON11-6 | ATCAAAAGAAACCCCTGTCG | TCAGGGGTATGAGCTTCTGG | 480 |
| GPR179-EXON11-7 | CTGGGAGACGAGTGAAGGAG | AAGTCTCCTCCCGGTTTCTG | 474 |
| GPR179-EXON11-8 | AGTGAAGGAGGCGAGGATG | TCTCCCTGGGACAAACTGAC | 463 |
| GPR179-EXON11-9 | AATGCTGAGTCTGGGGACAG | GTCTTGAGGACGTGGTTGTG | 529 |
| GPR179-EXON11-10 | TGGAAAATCGGAAATCGAAG | ACCTGGATGCTGAGAACAGG | 460 |
| GPR179-EXON11-11 | CCCAGGGAGAGAGCTGTTAC | GCTGCTGGGCAAGTCTGAG | 474 |
| GPR179-EXON11-12 | GCCTGTTCTCAGCATCCAG | TGGGTTATGTGTTCTGGGAAG | 482 |
| GPR179-EXON11-13 | GAGTAGTGAAGTGGCAGAGGG | CTGAACTGGCACACCTTTTG | 487 |
| GPR179-EXON11-14 | AATCTGACAGCAGTGCCAAG | AGGAACATCTCTCCTTGCCC | 445 |
| GPR179-EXON11-15 | CTGTGGGAAGTGGTAGAGGC | TTTTCAGGAGCTGTGGGG | 473 |
| GPR179-EXON11-16 | AGGGTACCATGGCAGACATC | TTCAGCTCTTTGGGAACACC | 486 |
Figure 5.
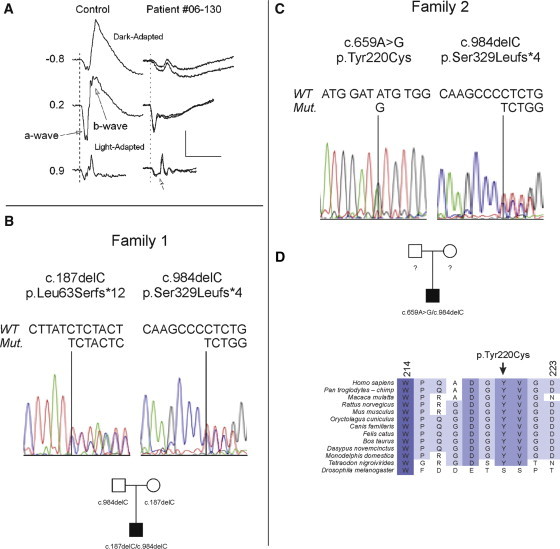
GPR179 Mutations Are Present in Two Probands with Autosomal-Recessive cCSNB
(A) ERGs obtained from a control subject and proband 1 for a standard series of stimulus conditions that allow rod- and cone-mediated responses to be evaluated. For proband 1 (patient 06-130), the two records indicate ERGs obtained from the two eyes. Under dark-adapted conditions, ERGs obtained from the proband had markedly reduced b-waves. Under light-adapted conditions (30 cd/m2), the cone ERG had a squared a-wave (arrow). Values indicate flash luminance in log cd s/m2.
(B and C) GPR179 (accession number NM_001004334.2) exons were sequenced from DNA samples isolated from 44 patients with cCSNB. Chromatograms containing the mutant sequences found in probands 1 (B) and 2 (C). In addition to the chromatogram, each subsection shows the mutation, the predicted impact on the amino acid sequence, and the segregation pattern. The pedigree for proband 1 shows that he inherited one mutant GPR179 allele from each parent (c.187delC and c.984delC), who had normal vision (data not shown). The parents of proband 2 were not available for analyses.
(D) Comparison of the region of GPR179 containing the amino acid substitution (p.Tyr220Cys) identified in proband 2 across phyla. With the exception of Drosophila melanogaster, Tyr220 is conserved for every species for which data were available. In general, this region of the protein is highly conserved (the shade of blue indicates the amount of conservation; dark blue indicates the most conserved). The study followed the tenets of the Declaration of Helsinki and was approved by the ethics committee of the Academic Medical Centre, Amsterdam. All participants provided signed informed consent for participation in the study.
Proband 1 was a compound heterozygote for two frameshift mutations in GPR179, c.187delC and c.984delC (NM_001004334.2) resulting in predicted protein truncations p.Leu63Serfs∗12 and p.Ser329Leufs∗4, respectively (Figure 5B). The premature chain termination is expected to result in functional null alleles. The probands' unaffected parents were each heterozygous for one of the mutations (Figure 5B).
Proband 2 was 20 years old at the time of diagnosis. She is of Norwegian descent and presented with rotatory nystagmus, a very unusual blond fundus, and congenital nightblindness and was able to see 20/30 with a −12.00 D prescription. Although not known to be related, proband 2 also carried the c.984delC frameshift mutation identified in proband 1, suggesting that this might be a founder mutation. Proband 2 carried a second mutation, c.659A>G, that would result in missense mutation, p.Tyr220Cys. (Figure 5C). The three variants in probands 1 and 2 were not present in 210 healthy control chromosomes. Two out of three Alamut analyses, which predict mutation impact on function, classified the p.Tyr220Cys as potentially pathogenic. Moreover, by introducing a new cysteine into the GRP179 protein, the mutation is likely to impact its structure. The functional importance of Tyr220 is supported by its conservation across species ranging from human to Tetraodon (Figure 5D). DNA of family members of proband 2 was not available for segregation analysis.
The combined data indicate that GPR179 is required for DBC signal transduction and that mutations that disrupt the function of GPR179 cause a recessive form of cCSNB. Although Gpr179nob5/nob5 mice lack expression of GPR179, we have not detected any anatomical defect in the retina, and the mice have no apparent health problem. Similarly, the two human patients we have identified with cCSNB and mutations in GPR179 have no other known health problems. Given the ERG phenotype in mice, zebrafish, and humans, as well as the colocalization of GPR179 with GRM6, we postulate that GPR179 plays a critical role in DBC signal transduction, possibly by forming heterodimers with GRM6. Future studies will be required to define the specific role of GPR179 in this process. The availability of the Gpr179nob5/nob5 mouse model will be an important tool with which to address this question.
Acknowledgments
We are grateful to Mathieu Gauvin for assistance with data analyses, Wim de Graaff for the morpholino injections in zebrafish larvae, and Takahisa Furakawa for the gift of the GRM6 antibody. We also thank the patients for their participation. This research was supported by grants from the National Institutes of Health (R21EY021852 to N.S.P. and R.G.G.; R01EY12354 to R.G.G.; R01EY014701 to M.A.M.; RR017890 to A.F.X.G.; R01EY016501 and P30CA34196 to P.M.N.; R01EY004864 and P30EY006360 to P.M.I.; and R01NS43459 and R01EY020821 to G.T.); the Veterans Administration Medical Research Service; the Netherlands Organization for Scientific Research (Zon-MW NWO to M.K.); ODAS and European Commission FP7 Grant RETICIRC HEALTH-F2-2009-223156 (to M.K.); Algemene Nederlandse Vereniging ter Voorkoming van Blindheid (to A.A.B.B.); the Foundation Fighting Blindness Canada (to R.K.K.); the Canadian Institutes for Health Research (to R.K.K.); Reseau Vision (to R.K.K.); Fonds de recherche du Québec–Santé (to R.K.K.), a Foundation Fighting Blindness Center Grant to the Cole Eye Institute, Cleveland Clinic; and unrestricted awards from Research to Prevent Blindness to the Department of Ophthalmology, Cleveland Clinic Lerner College of Medicine; to the Department of Ophthalmology, Emory University; and to the Department of Ophthalmology and Visual Sciences, University of Louisville. P.M.I. is a recipient of an RPB Senior Scientific Investigator Award.
Contributor Information
Maarten Kamermans, Email: m.kamermans@nin.knaw.nl.
Ronald G. Gregg, Email: ron.gregg@louisville.edu.
Web Resources
The URLs for data presented herein are as follows:
Human Genome Variation Society, http://www.hgvs.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Primer3, http://frodo.wi.mit.edu/primer3/
References
- 1.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 2.Pusch C.M., Zeitz C., Brandau O., Pesch K., Achatz H., Feil S., Scharfe C., Maurer J., Jacobi F.K., Pinckers A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000;26:324–327. doi: 10.1038/81627. [DOI] [PubMed] [Google Scholar]
- 3.Dryja T.P., McGee T.L., Berson E.L., Fishman G.A., Sandberg M.A., Alexander K.R., Derlacki D.J., Rajagopalan A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA. 2005;102:4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeitz C., van Genderen M., Neidhardt J., Luhmann U.F., Hoeben F., Forster U., Wycisk K., Mátyás G., Hoyng C.B., Riemslag F. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest. Ophthalmol. Vis. Sci. 2005;46:4328–4335. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]
- 5.Audo I., Kohl S., Leroy B.P., Munier F.L., Guillonneau X., Mohand-Saïd S., Bujakowska K., Nandrot E.F., Lorenz B., Preising M. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:720–729. doi: 10.1016/j.ajhg.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z., Sergouniotis P.I., Michaelides M., Mackay D.S., Wright G.A., Devery S., Moore A.T., Holder G.E., Robson A.G., Webster A.R. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am. J. Hum. Genet. 2009;85:711–719. doi: 10.1016/j.ajhg.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Genderen M.M., Bijveld M.M., Claassen Y.B., Florijn R.J., Pearring J.N., Meire F.M., McCall M.A., Riemslag F.C., Gregg R.G., Bergen A.A., Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:730–736. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura M., Sanuki R., Yasuma T.R., Onishi A., Nishiguchi K.M., Koike C., Kadowaki M., Kondo M., Miyake Y., Furukawa T. TRPM1 mutations are associated with the complete form of congenital stationary night blindness. Mol. Vis. 2010;16:425–437. [PMC free article] [PubMed] [Google Scholar]
- 9.Frishman L.J., Wang M.H. Electroretinogram of human, monkey and mouse. In: Levin L.A., Nilsson S.F.E., Ver Hoeve J., Wu S.M., Kaufman P.L., Alm A., editors. Adler's Physiology of the Eye. 11th ed. Saunders Elsevier; New York: 2011. pp. 480–501. [Google Scholar]
- 10.Masu M., Iwakabe H., Tagawa Y., Miyoshi T., Yamashita M., Fukuda Y., Sasaki H., Hiroi K., Nakamura Y., Shigemoto R. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995;80:757–765. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- 11.Shen Y., Heimel J.A., Kamermans M., Peachey N.S., Gregg R.G., Nawy S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J. Neurosci. 2009;29:6088–6093. doi: 10.1523/JNEUROSCI.0132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koike C., Obara T., Uriu Y., Numata T., Sanuki R., Miyata K., Koyasu T., Ueno S., Funabiki K., Tani A. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc. Natl. Acad. Sci. USA. 2010;107:332–337. doi: 10.1073/pnas.0912730107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgans C.W., Zhang J., Jeffrey B.G., Nelson S.M., Burke N.S., Duvoisin R.M., Brown R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA. 2009;106:19174–19178. doi: 10.1073/pnas.0908711106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhingra A., Jiang M., Wang T.L., Lyubarsky A., Savchenko A., Bar-Yehuda T., Sterling P., Birnbaumer L., Vardi N. Light response of retinal ON bipolar cells requires a specific splice variant of Galpha(o) J. Neurosci. 2002;22:4878–4884. doi: 10.1523/JNEUROSCI.22-12-04878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao A., Dallman R., Henderson S., Chen C.K. Gbeta5 is required for normal light responses and morphology of retinal ON-bipolar cells. J. Neurosci. 2007;27:14199–14204. doi: 10.1523/JNEUROSCI.4934-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregg R.G., Kamermans M., Klooster J., Lukasiewicz P.D., Peachey N.S., Vessey K.A., McCall M.A. Nyctalopin expression in retinal bipolar cells restores visual function in a mouse model of complete X-linked congenital stationary night blindness. J. Neurophysiol. 2007;98:3023–3033. doi: 10.1152/jn.00608.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearring J.N., Bojang P., Jr., Shen Y., Koike C., Furukawa T., Nawy S., Gregg R.G. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J. Neurosci. 2011;31:10060–10066. doi: 10.1523/JNEUROSCI.1014-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pardue M.T., McCall M.A., LaVail M.M., Gregg R.G., Peachey N.S. A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest. Ophthalmol. Vis. Sci. 1998;39:2443–2449. [PubMed] [Google Scholar]
- 19.Pinto L.H., Vitaterna M.H., Shimomura K., Siepka S.M., Balannik V., McDearmon E.L., Omura C., Lumayag S., Invergo B.M., Glawe B. Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis. Neurosci. 2007;24:111–123. doi: 10.1017/S0952523807070149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maddox D.M., Vessey K.A., Yarbrough G.L., Invergo B.M., Cantrell D.R., Inayat S., Balannik V., Hicks W.L., Hawes N.L., Byers S. Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J. Physiol. 2008;586:4409–4424. doi: 10.1113/jphysiol.2008.157289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor B.A., Navin A., Phillips S.J. PCR-amplification of simple sequence repeat variants from pooled DNA samples for rapidly mapping new mutations of the mouse. Genomics. 1994;21:626–632. doi: 10.1006/geno.1994.1323. [DOI] [PubMed] [Google Scholar]
- 22.Dhingra A., Sulaiman P., Xu Y., Fina M.E., Veh R.W., Vardi N. Probing neurochemical structure and function of retinal ON bipolar cells with a transgenic mouse. J. Comp. Neurol. 2008;510:484–496. doi: 10.1002/cne.21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim D.S., Ross S.E., Trimarchi J.M., Aach J., Greenberg M.E., Cepko C.L. Identification of molecular markers of bipolar cells in the murine retina. J. Comp. Neurol. 2008;507:1795–1810. doi: 10.1002/cne.21639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakajima Y., Moriyama M., Hattori M., Minato N., Nakanishi S. Isolation of ON bipolar cell genes via hrGFP-coupled cell enrichment using the mGluR6 promoter. J. Biochem. 2009;145:811–818. doi: 10.1093/jb/mvp038. [DOI] [PubMed] [Google Scholar]
- 25.Kofuji P., Ceelen P., Zahs K.R., Surbeck L.W., Lester H.A., Newman E.A. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: Phenotypic impact in retina. J. Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penn R.D., Hagins W.A. Signal transmission along retinal rods and the origin of the electroretinographic a-wave. Nature. 1969;223:201–204. doi: 10.1038/223201a0. [DOI] [PubMed] [Google Scholar]
- 27.Samuels I.S., Sturgill G.M., Grossman G.H., Rayborn M.E., Hollyfield J.G., Peachey N.S. Light-evoked responses of the retinal pigment epithelium: Changes accompanying photoreceptor loss in the mouse. J. Neurophysiol. 2010;104:391–402. doi: 10.1152/jn.00088.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCall M.A., Gregg R.G. Comparisons of structural and functional abnormalities in mouse b-wave mutants. J. Physiol. 2008;586:4385–4392. doi: 10.1113/jphysiol.2008.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma S., Ball S.L., Peachey N.S. Pharmacological studies of the mouse cone electroretinogram. Vis. Neurosci. 2005;22:631–636. doi: 10.1017/S0952523805225129. [DOI] [PubMed] [Google Scholar]
- 30.Pardue M.T., Ball S.L., Candille S.I., McCall M.A., Gregg R.G., Peachey N.S. nob: A mouse model of CSNB1. In: Hollyfield J.G., Anderson R.E., LaVail M.M., editors. New Insights into Retinal Degenerative Diseases. Kluwer/Plenum Press; New York: 2001. pp. 319–328. [Google Scholar]
- 31.Bahadori R., Biehlmaier O., Zeitz C., Labhart T., Makhankov Y.V., Forster U., Gesemann M., Berger W., Neuhauss S.C. Nyctalopin is essential for synaptic transmission in the cone dominated zebrafish retina. Eur. J. Neurosci. 2006;24:1664–1674. doi: 10.1111/j.1460-9568.2006.05053.x. [DOI] [PubMed] [Google Scholar]
- 32.Lachapelle P., Little J.M., Polomeno R.C. The photopic electroretinogram in congenital stationary night blindness with myopia. Invest. Ophthalmol. Vis. Sci. 1983;24:442–450. [PubMed] [Google Scholar]
- 33.Miyake Y., Yagasaki K., Horiguchi M., Kawase Y., Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986;104:1013–1020. doi: 10.1001/archopht.1986.01050190071042. [DOI] [PubMed] [Google Scholar]
- 34.Alexander K.R., Fishman G.A., Peachey N.S., Marchese A.L., Tso M.O.M. ‘On’ response defect in paraneoplastic night blindness with cutaneous malignant melanoma. Invest. Ophthalmol. Vis. Sci. 1992;33:477–483. [PubMed] [Google Scholar]
- 35.Goldberg A.F., Ritter L.M., Khattree N., Peachey N.S., Fariss R.N., Dang L., Yu M., Bottrell A.R. An intramembrane glutamic acid governs peripherin/rds function for photoreceptor disk morphogenesis. Invest. Ophthalmol. Vis. Sci. 2007;48:2975–2986. doi: 10.1167/iovs.07-0049. [DOI] [PubMed] [Google Scholar]
- 36.Makhankov Y.V., Rinner O., Neuhauss S.C. An inexpensive device for non-invasive electroretinography in small aquatic vertebrates. J. Neurosci. Methods. 2004;135:205–210. doi: 10.1016/j.jneumeth.2003.12.015. [DOI] [PubMed] [Google Scholar]