

Abstract
Exome sequencing of an individual with congenital cataracts, hypertrophic cardiomyopathy, skeletal myopathy, and lactic acidosis, all typical symptoms of Sengers syndrome, discovered two nonsense mutations in the gene encoding mitochondrial acylglycerol kinase (AGK). Mutation screening of AGK in further individuals with congenital cataracts and cardiomyopathy identified numerous loss-of-function mutations in an additional eight families, confirming the causal nature of AGK deficiency in Sengers syndrome. The loss of AGK led to a decrease of the adenine nucleotide translocator in the inner mitochondrial membrane in muscle, consistent with a role of AGK in driving the assembly of the translocator as a result of its effects on phospholipid metabolism in mitochondria.
Main Text
Sengers syndrome (MIM 212350) is an autosomal-recessive disorder characterized by congenital cataracts, hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance, and lactic acidosis but normal mental development. Since the first report in 1975 by Sengers et al.,1 about 40 individuals have been described as having this unique mitochondrial disease.2–8 The clinical course varies from severe forms that cause death in infancy to more benign forms that allow survival into the fourth decade. Cause of death is invariably heart failure due to a hypertrophic form of cardiomyopathy. Histopathological investigations have shown abnormal structure of mitochondria and storage of lipid and glycogen in both skeletal and heart muscle. In 2002, Jordens et al. described a decrease in both the amount of skeletal muscle and the activity of adenine nucleotide translocator 1 (ANT1 [SLC25A4]) in two families with Sengers syndrome.9 Because mutations in SLC25A4 (MIM 103220) were excluded by linkage analysis, Jordens et al. proposed that transcriptional, translational, or posttranslational events might cause the carrier deficiency. The phenotype of hypertrophic cardiomyopathy, exercise intolerance, and lactic acidemia of Slc25a4-knockout mice shows a broad overlap with the clinical findings seen in Sengers syndrome.10 However, the molecular basis underpinning the defect has remained elusive.
To identify the disease-causing sequence variation, we performed exome sequencing in a single German index case (54027 in Table 1) with Sengers syndrome. Written informed consent was obtained from all participants or their guardians at the recruiting center, and the study was approved by the ethical committee of the Technical University of Munich. The boy was born a dizygotic twin from unrelated parents of Italian origin. The primary adaptation was uneventful. Bilateral central cataracts were noticed in a routine physical examination performed at day 5. On the boy's 13th day of life, the family noticed that the child was very fatigued and had muscular hypotonia. The next day, he arrived at the hospital with decompensated cardiomyopathy and tachydyspnea. Echocardiography revealed massive hypertrophy of both ventricles. Lactate was 7.3 mmol/liter in plasma (normal is 0.5–2.2 mmol/L). The boy died on the 18th day of life as a result of cardiac failure. A muscle biopsy was taken on the 15th day of life and revealed mild myopathic abnormalities and deposition of fine lipid droplets in a histological investigation. Activity staining of cytochrome c oxidase and succinate dehydrogenase was normal.
Table 1.
Genetic and Clinical Findings in Sengers Individuals with AGK Mutations
| ID | Sex |
AGK Mutations Identified |
Biochemical Investigations |
Clinical Features |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA (NM_018238.3) | Protein (NP_060708) | OXPHOS Defect | ATP Synthesis | Substrate Oxidation | AO | Course | CM | Cataract | Other Clinical Features | Literature | ||
| 54027a | male | c.306C>T c.841C>T |
p.Tyr102∗ p.Arg281∗ |
I, II+III, and V | impaired | impaired | 1 weeks | death at 18 days | yes | congenital | floppy infant, tachydyspnoea, lactic acidosis plasma, and CSF | this study |
| 60453 | male | c.3G>C c.517C>T |
p.Met1Ile p.Gln173∗ |
normal | ND | ND | 3 months | alive for 36 years | yes | 3 months | motor developmental delay, muscular hypotonia, exercise intolerance, normal mental development, and lactic acidosis | Lalive d'Epinay et al. (“fall 1”)3 |
| 60455 b | male | c.412C>T c.1137_1143del |
p.Arg138∗ p.Gly380Leufs∗16 |
normal | ND | ND | 1 week | death at 11 months | yes | congenital | muscular hypotonia, moderate motor retardation, and lactic acidosis depending on exercise | this study |
| 62014 b | female | NA | NA | ND | ND | ND | 1 week | death at 7 months | ND | congenital | muscular hypotonia and motor retardation | this study |
| 60456 c | male | c.3G>C c.672C>A |
p.Met1Ile p.Tyr224∗ |
normal | ND | ND | 3 months | alive for 35 years | yes | 3 months | motor developmental delay, exercise intolerance, normal mental development, and lactic acidosis depending on exercise | Lalive d'Epinay et al. (“fall 3”)3 |
| 62013 c | female | NA | NA | normal | ND | ND | 10 weeks | death at 19 years | yes | 10 weeks | motor developmental delay, exercise intolerance, normal mental development, esotropia, and nystagmus | Lalive d'Epinay et al. (“fall 2”)3 |
| 60182 d | male | c.1131+5G>A c.1131+5G>A |
splicing defect | I, II+III, IV, and PDHc | impaired | impaired | 1 year | death at 12 years | yes | 18 months | muscular hypotonia, muscle weakness, and exercise intolerance | Morava et al. (case 1)6 |
| 60183 d | female | c.1131+5G>A c.1131+5G>A |
splicing defect | I, II+III, IV, and PDHc | impaired | impaired | birth | alive for 10 years | yes | 5 months | lactic acidosis and exercise intolerance | Morava et al. (case 2)6 |
| 60186 | female | c.1131+5G>A c.1131+5G>A |
splicing defect | normal | impaired | impaired | 3.5 years | alive for 41 years | yes | congenital | lactic acidosis and cerebrovascular accident | van Ekeren et al. (case 12)4 |
| 62216 | female | c.672C>A c.870del |
p.Tyr224∗ p.Gln291Argfs∗8 |
I, II, III, IV, and very high CS | impaired | ND | birth | death at 10 months | yes | 4 months | lactic acidosis | this study |
| 62217 | male | c.101+?_222-?del c.101+?_222-?del |
ND ND |
I, II, III, IV, and very high CS | impaired | ND | 4 months | death at 8 months | yes | congenital | lactic acidosis, seizures, paresis of upper-left limb, dilation of brain ventricles, and axial hypotonia | this study |
| 62218 | female | c.221+1G>A c.1213C>T |
splicing defect p.Gln405∗ |
I, II, III, IV, and very high CS | ND | ND | 10 months | alive for 12 years | yes | congenital | lactic acidosis and severe muscle weakness | Di Rosa et al., (case 3);7 this study |
Abbreviations are as follows: OXPHOS, oxidative phosphorylation; AO, age of onset; CM, cardiomyopathy; CSF, cerebrospinal fluid; NA, no material available; ND, not determined; I, complex I; II+III, succinate cytochrome c oxidoreductase; IV, cytochrome c oxidase; V, oligomycin-sensitive ATPase; PDHc, pyruvate dehydrogenase complex; and CS, citrate synthase.
Investigated by exome sequencing.
Individuals 60455 and 62014 are siblings.
Individuals 60456 and 62013 are siblings.
Individuals 60182 and 60183 are siblings.
We performed in-solution targeted enrichment of exonic sequences from index case 54027 by using the 50 Mb SureSelect Human All Exon kit from Agilent. The library was subsequently sequenced as 76 paired-end runs on the GAIIx from Illumina. Read alignment to the human genome assembly hg19 was done with Burrows-Wheeler Aligner (BWA, version 0.5.8) and yielded a total of 9.6 Gb of sequence data corresponding to an average coverage of 113× (Table S1, available online). Single-nucleotide variants and small insertions and deletions were detected with SAMtools (version 0.1.7). Because Sengers syndrome is known to be a rare condition, we first excluded all variants present in 666 control exomes. We then filtered for compound heterozygous or homozygous nonsynonymous variants affecting genes that encode mitochondrial proteins11 (Table S2). This approach identified a single gene harboring the compound heterozygous nonsense mutations c.306T>G (p.Tyr102∗) and c.841C>T (p.Arg281∗) in AGK (RefSeq NM_018238.3), the gene coding for mitochondrial acylglycerol kinase (AGK). The parents and the healthy brother carried a single heterozygous mutation each, as confirmed by Sanger sequencing.
Mutation screening of AGK (MIM 610345) in 13 individuals with congenital cataracts and cardiomyopathy discovered that there were 12 alleles with predicted loss of function in ten affected individuals, confirming the causal nature of AGK variants in Sengers syndrome (Figure 1). All affected individuals displayed the clinical signs of Sengers syndrome, but they experienced a varying course of the disease and had different biochemical alterations; five individuals had a combined respiratory-chain-complex deficiency in muscle tissue. Clinical phenotypes of most individuals have been reported previously (Table 1). For the remaining individuals, the clinical manifestation and course of the disease are described in the following five paragraphs.
Figure 1.
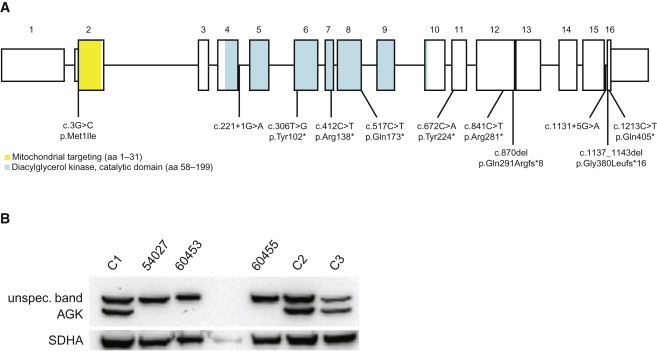
Distribution of the AGK Mutations and Their Consequences on AGK
Structure of AGK (A) and localization of identified mutations. AGK (B) was not detectable with immunoblot analysis (primary AGK antibody [1:1,000; rabbit polyclonal; GTX107413, Genetex]) in myoblasts form affected individual 54027 and in fibroblasts of individuals 60453 and 60455.
(C1) Myoblast control.
(C2 and C3) Fibroblast controls.
Individual 62014 was a girl from healthy nonconsanguineous Swiss parents. Bilateral cataracts were noticed when she was born but were interpreted as rubella embryopathy. The girl always had muscular hypotonia and failed to thrive. She died at the age of 7 months as a result of sudden infant death syndrome.
Individual 60455, the younger brother of individual 62014, was born at the 40th week of gestation after an uneventful pregnancy. He had a length of 47 cm, a body weight of 2,620 g, a head circumference of 33 cm, and Apgar scores of 9, 10, and 10. His mother first noticed a cataract in his right eye when he was three days old and another cataract in his left eye when he was two weeks old. Therefore, a cataract extraction was performed. Electro- and echocardiography were normal when the boy was 2 months of age; there was no lactate elevation in plasma and urine. When he was 4 months old, a failure to thrive was noticed. When he was 7 months old, a moderate concentric hypertrophic cardiomyopathy was first documented and was thereafter rapidly progressive. Depending on motor activity, lactate was intermittently elevated. At the age of 8.5 months, the boy presented with muscular hypotonia and moderate motor retardation. Electromyography and nerve-conduction velocity were normal. Magnetic resonance imaging (MRI) investigation of his muscle tissue showed hypotrophy without fat infiltration, and magnetic resonance spectroscopy revealed a normal 31P-spectrum of phosphate, phosphocreatine, and ATP. A muscle biopsy revealed fatty infiltrations in the muscle fibers, and electron microscopy revealed abnormal mitochondria. Lactate in the blood and urine was elevated depending on muscular activity. At 11 months old, the boy died as a result of heart failure. Autopsy was performed 15 min after his death; both muscle and heart tissue showed a normal concentration of L-carnitine. Enzymatic investigations of muscle tissue showed normal activity of creatine kinase and complexes I, II, II+III, IV, and V (ATPase). No abnormalities were found in the liver or kidneys.
Individual 62216, the only daughter of healthy nonconsanguineous Italian parents, died at 9 months of age. She was born at term by normal vaginal delivery, and her birth weight was 2,750 g. Growth delay was noted during the third trimester of pregnancy (50–25th percentile) and again postnatally (<tenth percentile). When she was 4 months of age, bilateral cataracts were noted. When she was 4.5 months of age, an ultrasound examination of the heart showed severe concentric left-cardiac hypertrophy. She then suffered from recurrent bronchitis. After vitrectomy removed the cataracts, the baby worsened and died from cardiac arrest. High lactate was only present during infections or motor activity. Biochemical analysis of bioptic and autoptic muscle fragments showed marked reduction of all respiratory-chain complexes and elevated activity of citrate synthase.
Individual 62217, a boy, was the third child of reportedly nonconsanguineous Italian parents. His older sister, who had hypertrophic cardiomyopathy and congenital cataracts, died at 4 days old, and his older brother is alive and well. At birth, bilateral cataracts and moderate hypertrophy of the interventricular septum were noted. Two surgical interventions for cataracts were performed at 2 weeks and 2 months of age, when a paresis of the upper-left limb was noted. A brain MRI showed thinning of the corpus callosum and moderate dilation of cerebral ventricles. Axial hypotonia and hypertonia of the upper limbs were present. When the boy was 4 months of age, a cardiac ultrasound examination showed severe left-ventricular hypertrophy (the posterior wall was 12 mm; the normal value is <4), modest mitral regurgitation, and preserved ejection fraction. No pulmonary arterial hypertension was detected. At the same age, he had an episode of generalized hypertonic seizures with no clonic phase. Anisocoria of the pupils and severe axial hypotonia were present. High levels of lactate were detected in his blood. Analysis of respiratory-chain complexes in the homogenate of a muscle biopsy showed reduction of all activities except for that of citrate synthase, which was elevated, whereas activities in fibroblasts and in a liver biopsy were normal. He died at 8 months old as a result of cardiac failure.
The case of individual 62218 was published by De Rosa et al. in 2006.7 At that time, the girl was 7 years old and had clinical features typical of Sengers syndrome; bilateral cataracts were noticed at 10 months of age, and hypertrophic cardiomyopathy was documented by ultrasound examination at 18 months of age and stabilized from 7 years of age on. She is now 12 years old. Although her cardiac conditions are relatively stable under therapy with propranolol and idebenone, her muscle weakness has progressively worsened, and she has required the aid of a wheelchair for distances of more than 100 m. Additional features include reduced body growth (<tenth percentile) and a mild neurogenic electromyography pattern, but she has no sign of CNS involvement (there is no cognitive regression, cerebellar signs, or pyramidal or extrapyramidal abnormalities). A recent brain MRI was normal. One younger brother died at 14 months old as a result of hypertrophic cardiomyopathy with bilateral cataracts. A muscle biopsy showed diffuse complex-IV deficiency with numerous ragged-red fibers. Two younger siblings, a girl and a boy, are alive and well.
All affected individuals carried mutations that are predicted to result in truncated proteins that are missing conserved parts of AGK and that therefore represent loss-of-function alleles (Figure 1 and Figure S1). In two affected individuals from Switzerland, we identified a heterozygous mutation (c.3G>C, p.Met1Ile) altering the AGK initiation codon in combination with two different nonsense mutations. Three individuals from two more families from The Netherlands harbored homozygous mutations that the splice-port algorithm predicted to affect the splice donor site of intron 16 (c.1131+5G>A).12 A second heterozygous splice-site mutation (c.221+1G>A) affecting the donor site of intron 4 was found in an Italian individual (62218) in combination with a heterozygous nonsense mutation (p.Gln405∗). An additional Italian individual, 62217, showed a homozygous deletion of exons 3 and 4. All variants identified were absent from 1,200 European control chromosomes.
Immunoblot experiments confirmed the absence of full-length AGK in muscle tissue from individual 54027 (Figure 1). Measurement of respiratory-chain enzymes in fresh muscle tissue only indicated a slight reduction in the activity of complexes I, II+III, and V (Table 2). However, the analysis of 14C-labeled mitochondrial substrate in 600 g supernatants from fresh muscle tissue13 had already demonstrated a clearly abnormal ratio of CCCP- to ADP-stimulated oxidation rates in the affected individual, indicating a defective ATP synthesis (Table 2 and Figure 2). Defects in ATP synthesis either affect the F1FO ATP synthase, the mitochondrial phosphate carrier, or the adenine nucleotide translocator (ANT).13 Immunodecoration14 with an ANT-specific antibody confirmed the absence of the inner-mitochondrial-membrane ANT in muscle tissue (Figure 2). However, normal ANT levels in undifferentiated myoblast cells generated from the same muscle-biopsy material suggested that a specific posttranslational defect of proper inner-membrane ANT assembly only occurs in differentiated muscle fibers. In agreement with this hypothesis, myoblast cells of affected individual 54027 lost ANT during differentiation (Figure 2 and Figure S2).
Table 2.
Investigations of the Mitochondrial Energy Metabolism in Individual 54027
| Enzymatic Investigations in Muscle Tissue | Individual 54027 | Controls |
|---|---|---|
| Substrate Oxidation Rates [μmol/h/g protein] | ||
| [1-14C]pyruvate + malate + ADP | 34 | 263–900 |
| [1-14C]pyruvate + carnitine + ADP | 52 | 302–856 |
| [1-14C]pyruvate + malate (−ADP) | 15 | 32–102 |
| [1-14C]pyruvate + malate + CCCP | 119 | 304–889 |
| [U-14C]malate + pyruvate + malonate + ADP | 50 | 282–874 |
| [U-14C]malate + acetylcarnitie + malonate + ADP | 30 | 273–678 |
| [U-14C]malate + acetylcarnitine + arsenite + ADP | 32 | 156–378 |
| [1,4-14C]succinate + acetylcarnitine + ADP | 22 | 167–488 |
| Enzyme Activities [unit/g protein] | ||
| Citrate synthase | 161 | 150–338 |
| Complex I | 23 | 28–76 |
| Complex I+III | 92 | 49–218 |
| Complex II | 57 | 39–102 |
| Complex II+III | 32 | 65–180 |
| Complex III | 491 | 304–896 |
| Complex IV (cytochrome c oxidase) | 310 | 181–593 |
| Complex V (oligomycin-sensitive ATPase) | 60 | 86–257 |
Figure 2.
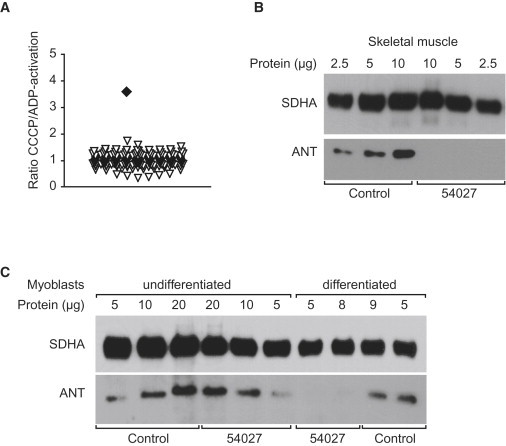
Deficiency in ATP Synthesis and ANT in Sengers Syndrome
The increased ratio of [1-14C]pyruvate + malate + CCCP over [1-14C]pyruvate + malate + ADP oxidation (A) in affected individual 54027 (filled square) versus controls (triangles) indicates a deficiency in ATP synthesis. A decreased amount of ANT (B) was detected with immunoblot analysis in skeletal muscle and in differentiated myoblasts (C) of affected individual 54027. Primary antibodies and their conditions are as follows: ANT antibody (1:1,000; mouse monoclonal; MSA02, Mitosciences) and SDHA (1:30,000; mouse monoclonal; MS204, Mitosciences).
AGK is commonly described as a multisubstrate lipid kinase that catalyzes the phosphorylation of diacylglycerol (DAG) and monoacylglycerol (with lower affinity).15 The resulting products, phosphatidic acid (PA) or lysophosphatidic acid (LPA), respectively, can either act as signaling molecules15,16 or take part in the synthesis of phospholipids. Furthermore, AGK scavenges mitochondrial DAG, which has been reported to induce reactive oxygen species (ROS) signaling via a pathway mediated by protein kinase D1.17 It has been shown that DAG kinase 1 in S. cerevisiae has an essential function in membrane-lipid biosynthesis in the case of growth resumption after a stationary phase, especially when the de novo fatty-acid biosynthesis was inhibited by cerulenin.18 AGK might play a similar function in the mitochondria (Figure 3) of proliferating or differentiating tissues (e.g., muscle and heart tissue) that predominately procure their energy from fat and are devoid of fatty-acid synthesis; in Sengers syndrome, such tissues are affected the most. Indeed, the most striking effect of AGK depletion in PC-3 cells by siRNA has been a reduction of LPA and PA in mitochondria.15 PA and its downstream product, cardiolipin, are both found in crystals of ANT,19 the most abundant mitochondrial protein, which is severely decreased in the muscle tissue of individuals with Sengers syndrome. A disturbed membrane-lipid composition might also be responsible for the zonular nuclear cataract, a region of differentiating cells where the cellular architecture and exact arrangement of the cells are critical for light transmission and lens transparency.
Figure 3.
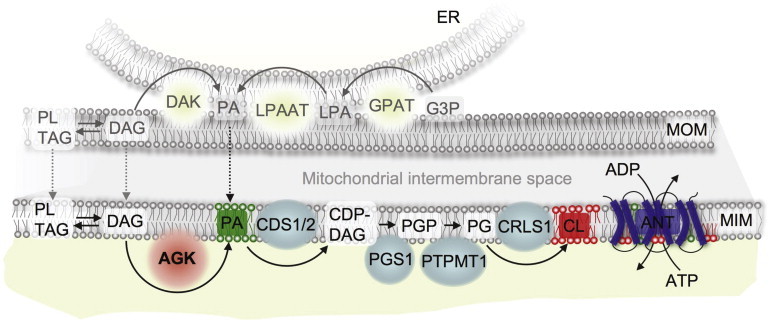
Potential Role of AGK in the Mitochondrial Lipid Metabolism
Abbreviations are as follows: AGK, acylglycerol kinase; CDS, CDP-diacylglycerol synthase; PGS, phosphatidylglycerophosphate synthase; PTPMT, phosphatidylglycerol-phosphate phosphatase; CRLS, cardiolipin synthase; ANT, adenine nucleotide translocator; DAK, diacylglycerol kinase; GPAT, glycerol-3-phosphate acyltransferase; LPAAT, 1-acylglycerol-3-phosphate O-acyltransferase; G3P, glycerol 3-phosphate; LPA, lyso-phosphatidic acid; PA, phosphatidic acid; DAG, diacylglycerol; TAG, triacylglycerol; PL, phospholipid; CDP-DAG, cytidine diphosphate diacylglycerol; PGP, phosphatidylglycerol-phosphate; PG, phosphatidylglyceroll; CL, cardiolipin; ER, endoplasmic reticulum; OMM, outer mitochondrial membrane; and IMM, inner mitochondrial membrane.
The clinical manifestation of Sengers syndrome resembles defects of mitochondrial ATP synthesis, either of F1FO ATP synthase (e.g., TMEM70 [MIM 612418]20 and ATP5E [MIM 606153]21) or of the mitochondrial phosphate carrier, SLC25A3 (MIM 600370)13. Symptoms of Sengers syndrome also resemble the known cardiolipin-metabolism defect (such as in X-linked Barth syndrome [MIM 302060],22 which is caused by TAZ [MIM 300394]23 mutations) in terms of neonatal onset of the disease, cardiomyopathy, and myopathy. Lactic acidosis is found in Sengers syndrome and in the other defects of ATP synthesis but usually not in Barth syndrome. Elevated excretion of 3-methylglutaconic acid (3-MGA) in urine is found in F1FO-ATP-synthase deficiency20,21 and in Barth syndrome.24 However, it is usually not found in Sengers syndrome with ANT deficiency or in the SLC25A3 defect,13 although a mild elevation of 3-MGA in urine was reported in individual 62218.7 Therefore, Sengers syndrome includes aspects of both ATP-synthesis and cardiolipin-metabolism defects.
At the moment, it is purely speculative whether the tissue expression of the four different ANT isoforms correlates with the pathomechanism in Sengers syndrome. ANT1 encoded by SLC25A4 is the major isoform in muscle and heart tissue, whereas ANT3 (SLC25A6 [MIM 300151]) is ubiquitously expressed.25 ANT2 (SLC25A5 [MIM 300150]) is mainly associated with smooth muscle cells,25 and ANT4 (SLC25A31 [MIM 610796]) is mainly expressed in the liver, testes, and brain.26
In summary, the biochemical data are consistent with a defined role of AGK in driving the assembly of ANT and, in some circumstances, respiratory-chain complexes as a result of its effects on phospholipid metabolism in mitochondria. Given the strong pre-existing evidence of mitochondrial dysfunction in Sengers syndrome, exome sequencing of a single affected individual coupled with the appropriate filtering of candidates for mitochondrial functions allowed us to identify AGK variants as the cause of Sengers syndrome in a significant proportion of individuals and to reveal an unexpected link between lipid metabolism and ATP synthesis.
Acknowledgments
We are indebted to the subjects and their families involved in the study. We gratefully acknowledge the support of J. Schum, R. Hellinger, and A. Löschner in genotyping and cell-culture work. T.M. and H.P. were supported by the Impulse and Networking Fund of the Helmholtz Association in the framework of the Helmholtz Alliance for Mental Health in an Ageing Society (HA-215) and the German Federal Ministry of Education and Research (BMBF)-funded Systems Biology of Metabotypes grant (SysMBo 0315494A) and German Network for Mitochondrial Disorders (mitoNET 01GM0867). T.M. is supported by the BMBF-funded German Center for Heart Research. T.M. and T.M.S. were supported by the European Commission 7th Framework Program (N. 261123), the Genetic European Variation in Disease Consortium, and the German Ministry for Education and Research (01GR0804-4). J.A.M. was supported by the Wissenschaftspreis 2008 of the Austrian Paediatric Society (ÖGKJ), W.S. was supported by the Jubiläumsfonds of Oesterreichische Nationalbank (12568), and J.A.M., F.Z., and W.S. were supported by the Vereinigung zur Förderung pädiatrischer Forschung und Fortbildung Salzburg. Telethon-Italy is gratefully acknowledged for grants GPP10005 and GGP1011 to M.Z.; M.Z. is also supported by Fondazione Cariplo (grant 2011-0526) and the Italian Association of Mitochondrial Patients and Families (Mitocon). E.L. is supported by Fondazione Pierfranco e Luisa Mariani–Organizzazione Non Lucrativa di Utilità Sociale.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
MitoP2, http://www.mitop.de
References
- 1.Sengers R.C., Trijbels J.M., Willems J.L., Daniels O., Stadhouders A.M. Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise. J. Pediatr. 1975;86:873–880. doi: 10.1016/s0022-3476(75)80217-4. [DOI] [PubMed] [Google Scholar]
- 2.Cruysberg J.R., Sengers R.C., Pinckers A., Kubat K., van Haelst U.J. Features of a syndrome with congenital cataract and hypertrophic cardiomyopathy. Am. J. Ophthalmol. 1986;102:740–749. doi: 10.1016/0002-9394(86)90402-2. [DOI] [PubMed] [Google Scholar]
- 3.Lalive d'Epinay S., Rampini S., Arbenz U., Steinmann B., Gitzelmann R. Infantile cataract, hypertrophic cardiomyopathy and lactic acidosis following minor muscular exertion—a little known metabolic disease. Klin. Monbl. Augenheilkd. 1986;189:482–485. [PubMed] [Google Scholar]
- 4.van Ekeren G.J., Stadhouders A.M., Smeitink J.A., Sengers R.C. A retrospective study of patients with the hereditary syndrome of congenital cataract, mitochondrial myopathy of heart and skeletal muscle and lactic acidosis. Eur. J. Pediatr. 1993;152:255–259. doi: 10.1007/BF01956157. [DOI] [PubMed] [Google Scholar]
- 5.Smeitink J.A., Sengers R.C., Trijbels J.M., Ruitenbeek W., Daniëls O., Stadhouders A.M., Kock-Jansen M.J. Fatal neonatal cardiomyopathy associated with cataract and mitochondrial myopathy. Eur. J. Pediatr. 1989;148:656–659. doi: 10.1007/BF00441527. [DOI] [PubMed] [Google Scholar]
- 6.Morava E., Sengers R., Ter Laak H., Van Den Heuvel L., Janssen A., Trijbels F., Cruysberg H., Boelen C., Smeitink J. Congenital hypertrophic cardiomyopathy, cataract, mitochondrial myopathy and defective oxidative phosphorylation in two siblings with Sengers-like syndrome. Eur. J. Pediatr. 2004;163:467–471. doi: 10.1007/s00431-004-1465-2. [DOI] [PubMed] [Google Scholar]
- 7.Di Rosa G., Deodato F., Loupatty F.J., Rizzo C., Carrozzo R., Santorelli F.M., Boenzi S., D'Amico A., Tozzi G., Bertini E. Hypertrophic cardiomyopathy, cataract, developmental delay, lactic acidosis: A novel subtype of 3-methylglutaconic aciduria. J. Inherit. Metab. Dis. 2006;29:546–550. doi: 10.1007/s10545-006-0279-y. [DOI] [PubMed] [Google Scholar]
- 8.Valsson J., Laxdal T., Jonsson A., Jansson K.K., Helgason H. Congenital cardiomyopathy and cataracts with lactic acidosis. Am. J. Cardiol. 1988;61:193–194. doi: 10.1016/0002-9149(88)91331-8. [DOI] [PubMed] [Google Scholar]
- 9.Jordens E.Z., Palmieri L., Huizing M., van den Heuvel L.P., Sengers R.C., Dörner A., Ruitenbeek W., Trijbels F.J., Valsson J., Sigfusson G. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann. Neurol. 2002;52:95–99. doi: 10.1002/ana.10214. [DOI] [PubMed] [Google Scholar]
- 10.Graham B.H., Waymire K.G., Cottrell B., Trounce I.A., MacGregor G.R., Wallace D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 11.Elstner M., Andreoli C., Klopstock T., Meitinger T., Prokisch H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009;457:3–20. doi: 10.1016/S0076-6879(09)05001-0. [DOI] [PubMed] [Google Scholar]
- 12.Dogan R.I., Getoor L., Wilbur W.J., Mount S.M. SplicePort–an interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285-291. doi: 10.1093/nar/gkm407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayr J.A., Merkel O., Kohlwein S.D., Gebhardt B.R., Böhles H., Fötschl U., Koch J., Jaksch M., Lochmüller H., Horváth R. Mitochondrial phosphate-carrier deficiency: A novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 2007;80:478–484. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feichtinger R.G., Zimmermann F., Mayr J.A., Neureiter D., Hauser-Kronberger C., Schilling F.H., Jones N., Sperl W., Kofler B. Low aerobic mitochondrial energy metabolism in poorly- or undifferentiated neuroblastoma. BMC Cancer. 2010;10:149. doi: 10.1186/1471-2407-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bektas M., Payne S.G., Liu H., Goparaju S., Milstien S., Spiegel S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J. Cell Biol. 2005;169:801–811. doi: 10.1083/jcb.200407123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y., Natarajan V. Lysophosphatidic acid signaling in airway epithelium: Role in airway inflammation and remodeling. Cell. Signal. 2009;21:367–377. doi: 10.1016/j.cellsig.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowell C.F., Döppler H., Yan I.K., Hausser A., Umezawa Y., Storz P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J. Cell Sci. 2009;122:919–928. doi: 10.1242/jcs.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fakas S., Konstantinou C., Carman G.M. DGK1-encoded diacylglycerol kinase activity is required for phospholipid synthesis during growth resumption from stationary phase in Saccharomyces cerevisiae. J. Biol. Chem. 2011;286:1464–1474. doi: 10.1074/jbc.M110.194308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epand R.M., Epand R.F., Berno B., Pelosi L., Brandolin G. Association of phosphatidic acid with the bovine mitochondrial ADP/ATP carrier. Biochemistry. 2009;48:12358–12364. doi: 10.1021/bi901769r. [DOI] [PubMed] [Google Scholar]
- 20.Cízková A., Stránecký V., Mayr J.A., Tesarová M., Havlícková V., Paul J., Ivánek R., Kuss A.W., Hansíková H., Kaplanová V. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008;40:1288–1290. doi: 10.1038/ng.246. [DOI] [PubMed] [Google Scholar]
- 21.Mayr J.A., Havlícková V., Zimmermann F., Magler I., Kaplanová V., Jesina P., Pecinová A., Nusková H., Koch J., Sperl W., Houstek J. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum. Mol. Genet. 2010;19:3430–3439. doi: 10.1093/hmg/ddq254. [DOI] [PubMed] [Google Scholar]
- 22.Barth P.G., Scholte H.R., Berden J.A., Van der Klei-Van Moorsel J.M., Luyt-Houwen I.E., Van 't Veer-Korthof E.T., Van der Harten J.J., Sobotka-Plojhar M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983;62:327–355. doi: 10.1016/0022-510x(83)90209-5. [DOI] [PubMed] [Google Scholar]
- 23.Bione S., D'Adamo P., Maestrini E., Gedeon A.K., Bolhuis P.A., Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996;12:385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 24.Kelley R.I., Cheatham J.P., Clark B.J., Nigro M.A., Powell B.R., Sherwood G.W., Sladky J.T., Swisher W.P. X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J. Pediatr. 1991;119:738–747. doi: 10.1016/s0022-3476(05)80289-6. [DOI] [PubMed] [Google Scholar]
- 25.Stepien G., Torroni A., Chung A.B., Hodge J.A., Wallace D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]
- 26.Dolce V., Scarcia P., Iacopetta D., Palmieri F. A fourth ADP/ATP carrier isoform in man: Identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005;579:633–637. doi: 10.1016/j.febslet.2004.12.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.