

Abstract
Genitopatellar syndrome (GPS) is a skeletal dysplasia with cerebral and genital anomalies for which the molecular basis has not yet been determined. By exome sequencing, we found de novo heterozygous truncating mutations in KAT6B (lysine acetyltransferase 6B, formerly known as MYST4 and MORF) in three subjects; then by Sanger sequencing of KAT6B, we found similar mutations in three additional subjects. The mutant transcripts do not undergo nonsense-mediated decay in cells from subjects with GPS. In addition, human pathological analyses and mouse expression studies point to systemic roles of KAT6B in controlling organismal growth and development. Myst4 (the mouse orthologous gene) is expressed in mouse tissues corresponding to those affected by GPS. Phenotypic differences and similarities between GPS, the Say-Barber-Biesecker variant of Ohdo syndrome (caused by different mutations of KAT6B), and Rubinstein-Taybi syndrome (caused by mutations in other histone acetyltransferases) are discussed. Together, the data support an epigenetic dysregulation of the limb, brain, and genital developmental programs.
Main Text
Genitopatellar syndrome (GPS) [MIM 606170] is a rare skeletal dysplasia combining hypoplastic or absent patellae, genital anomalies, craniofacial defects, and intellectual disability among other features. It has been described in 18 subjects to date.1–11 In one patient, we discovered a microdeletion encompassing LMX1B (explaining the patellar anomalies) and NR5A1 (explaining the genital anomalies) though this was not found to be a recurrent molecular lesion in the other subjects.11 An important phenotypic difference is that the subject with the microdeletion is not microcephalic although all other subjects are.
To gain insights into the molecular cause of GPS, we recruited subjects with this disease (see Figure 1 for photos, Figure S1 [available online] for pedigrees, and Table 1 and references therein for clinical details). Families provided informed consent to our study approved by the institutional review board of the Baylor College of Medicine. Subject 3 died at 8 years of age from bowel malrotation that led to volvulus and intestinal necrosis. An autopsy showed dramatic pancreatic hyperplasia (103 g for an expected weight of 15 g) with hyperplasia of some of the islets of Langerhans. An enlarged pancreas has never been described in other subjects, even though all other subjects in this study had abdominal ultrasounds. Other significant findings included kidney hypoplasia with multiple small subcapsullar cysts, a prominent suprapubic fat pad with underdeveloped clitoris and labia minora, and an anteriorly placed anus. Skeletal features included flat temporal bones, brachydactyly, flexion deformities of the hips and knees, and markedly hypoplastic patellae. Neuropathology showed microcephaly (851 g for an expected weight of 1,273 g); mild to moderate diffuse cortical atrophy; hypoplasia of the anterior portion of the corpus callosum; generalized mild gliosis; and small perivascular psammomatous calcifications in the basal ganglia, thalamus, corpus callosum, choroid plexus, and periventricular regions. See Figures 2A–2D for histology of some relevant tissues. Psammoma bodies are calcifications frequently seen in meningiomas and other malignancies (and only rarely in benign overgrowths) and are thought to result from calcification of dead cells or an active process to inhibit cell growth.12
Figure 1.
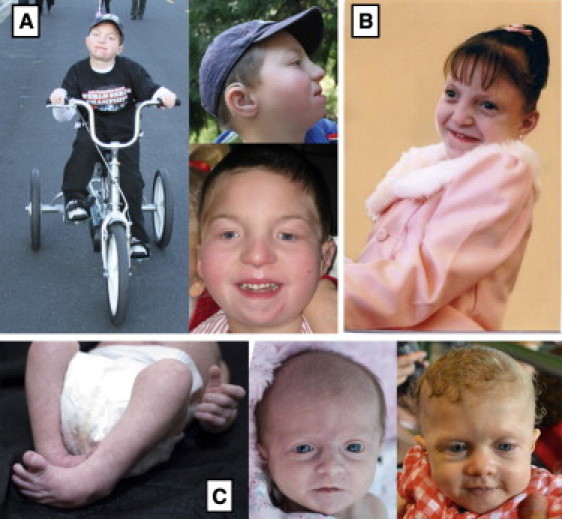
Clinical Presentation of Subjects with GPS
Photographs of (A) subject 2 at 9 years of age, (B) subject 3 at 8 years of age, and (C) subject 5 at birth and at 8 months of age.
Table 1.
Clinical Features of Enrolled Subjects and 13 Other Subjects from the Literature
| Subjects | 1 | 2 | 3 | 4 | 5 | 6 | Literaturea | Total (Affected/Total) |
|---|---|---|---|---|---|---|---|---|
| Gender | female | male | female | male | female | male | 4 females/9 males | 7 females/12 males |
| Skeletalb | ||||||||
| Absent or hypoplastic patellae | + | + | + | + | + | + | 12 | 18/19 |
| Flexion deformities | + | + | + | + | + | + | 13 | 19/19 |
| Club feet | + | + | + | + | + | + | 12 | 18/19 |
| Thoracolumbar kyphosis or scoliosis | + | + | 3 | 5/19 | ||||
| Pelvic anomalies | + | + | 5 | 7/19 | ||||
| Costal anomalies | + | + | + | 1 | 4/19 | |||
| Neurologicalc | ||||||||
| Microcephaly | + | + | + | + | + | + | 13 | 19/19 |
| Developmental delay or intellectual disability | + | + | + | + | + | + | 10/10d | 16/16 |
| Absent or thin corpus callosum | + | + | + | + | + | + | 9 | 15/19 |
| Colpocephaly or ventriculomegaly | + | + | + | 1 | 4/19 | |||
| Pachygyria | + | 2 | 3/19 | |||||
| Subependymal periventricular nodular heterotopia | + | 1 | 2/19 | |||||
| Optic atrophy or cortical visual impairment | + | + | + | 0 | 3/19 | |||
| Hearing loss | + | + | 1 | 3/19 | ||||
| Anal and Genitale | ||||||||
| Anteriorly positioned anus | + | + | 1 | 3/19 | ||||
| Anal atresia or stenosis | + | + | 0 | 2/19 | ||||
| Hypoplastic labia minora or majora | + | + | + | 3 | 6/6 | |||
| Clitoromegaly | + | + | 3 | 5/6 | ||||
| Scrotal hypoplasia | + | + | + | 9 | 12/12 | |||
| Cryptochidism | + | + | + | 9 | 12/12 | |||
| Renalf | ||||||||
| Hydronephrosis | + | + | + | + | 11 | 16/19 | ||
| Multicystic kidneys | + | + | 4 | 6/19 | ||||
| Cardiacg | ||||||||
| Atrial septal defect | + | + | + | + | 3 | 7/19 | ||
| Ventricular septal defect | + | 3 | 4/19 | |||||
| Facialh | ||||||||
| Facial dysmorphisms | + | + | + | + | + | + | 13 | 19/19 |
| Broad nasal bridge | + | + | 7 | 9/19 | ||||
| Prominent nasal bridge | + | + | 3 | 5/19 | ||||
| Otheri | ||||||||
| Tracheo or laryngomalacia | + | + | 4 | 6/19 | ||||
| Feeding difficulties | + | + | + | + | 2 | 6/19 | ||
| Small bowel malrotation | + | 1 | 2/19 | |||||
| Hypothyroidism | + | 2 | 3/19 | |||||
From Armstrong and Clarke,2 Bergmann et al.,3 Brugha et al.,4 Cormier-Daire et al.,5 Goldblatt et al.,6 Penttinen et al.,9 and Reardon10.
Occasional findings include osteoporosis, radioulnar synostosis, radial head deformity, brachydactyly, short stature, joint laxity, dislocated patellae, undertubulation of long bones, coxa vara, camptodactyly, narrow thorax, and exostoses.
Occasional findings include hypotonia, hypertonia, seizures, and subdural hemorrhage (subject 5).
Ten out of thirteen children survived beyond neonatal period and were included.
Occasional findings include rectal duplication (subject 6) and underdeveloped clitoris (subject 3).
Occasional findings include fused renal ectopia, dysplastic kidneys, and hypoplastic kidneys (subject 3).
Occasional findings include tortuous ascending aorta, dilated aortic arch, patent ductus arteriosus, patent foramen ovale, and stenosis of the pulmonary valve.
Occasional findings include bulbous nose, retrognatia or micrognatia, cleft or high-arched palate, gingival hyperplasia, coarse facies, full cheeks, ear anomalies, dental anomalies, sparse hair, hypertelorism, plagiocephaly, bitemporal flattening, downslanting palpebral fissures, downturned corners of the mouth, short columella, short philtrum, tented upper lip, and midface hypoplasia.
Occasional findings include hypogonadotrophic hypogonadism, respiratory distress, apnea, recurrent infections, single palmar crease, and skin laxity.
Figure 2.
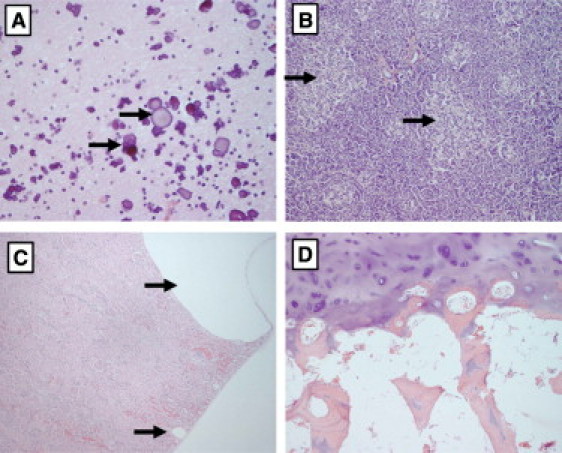
Hematoxylin and Eosin Images of Tissues from the Autopsy of Subject 3
Tissues shown are (A) the brain (400×) where we show a large cluster of perivascular calcifications in the white matter of the cerebral cortex (arrows point to two of them); these are never present in normal tissue; (B) the pancreas (100×) showing numerous islets of Langerhans, some of which are hyperplastic (black arrows), whereas others are of normal size (red arrows); (C) the kidney (100×), where we show one large and one small subcapsular cyst (arrows), and (D) the severely hypoplastic patella (100×), where there is persistence of the cartilaginous core of the trabeculae (black arrow).
We performed whole-exome sequencing on three subjects (subjects 2, 4, and 5). For subjects 2 and 4, exomes were captured on Nimblegen's SeqCap EZ V2.0 library and sequencing was conducted on Illumina HiSeq. Sequences were aligned to the human reference genome (hg18) with Burrows-Wheeler Aligner (BWA) (v 0.5.9)13 and recalibrated with the Genome Analysis Toolkit (GATK). Both samples achieved over 91% targeted bases at 20× coverage. SNPs and insertion-deletion events were called with Samtools Pileup (version 0.1.17).14 Variants were annotated with ANNOVAR15 and protein-impacting variants that were rare (minor allele frequency < 5%), novel, and nonsynonymous were preferentially explored. We narrowed gene candidates by comparing with known functional databases such as dbNSFP16 and SWISS-PROT17 to narrow down the list of plausible causative variants. For subject 5, the exome was captured on the Agilent SureSelect 50 Mb oligonucleotide library. DNA was sheared by sonication to an approximately 200 bp length. Fragment ends were ligated to specific adaptors and capture was performed with the manufacturer's protocol. The captured exome was reamplified by PCR (12 cycles) then applied to a single lane of Illumina HiSeq sequencer. The Illumina reads were aligned to the reference human genome (hg19) with BWA (v. 0.5.9) and Samtools (v. 0.1.12a). Pileup and varFilter commands were used to call variants, and these were filtered to retain SNPs and insertion-deletions with Phred-like quality scores of at least 20 and 50, respectively. ANNOVAR was used to annotate nonsynonymous variants according to the type of mutation, occurrence in dbSNP, SIFT score,18 and 1000 Genomes allele frequency.19
As shown in Table 2, only 13 genes showed rare novel variants that were potentially pathogenic and were shared by the three subjects. Variants were visualized and compared to the exomes of 20 other subjects with unrelated conditions. When keeping only high-quality variants (e.g., removing probable false positive variants in repeat regions or variants seen in unrelated conditions), only KAT6B variants remained. All variants were frameshift insertions-deletions (Figure 3, Figures S2A–2C, available online, and Table 3; RefSeq NM_012330.2 was used for the positions). We confirmed the variants by Sanger sequencing and sequenced the complete coding sequence of the gene in the other individuals recruited in our study (see Table S1 for primers). We have thus identified nonsense mutations in two additional subjects and one of the previously identified frameshift deletions in another (Figure 3 and Figures S2D–2F). Analysis of parental samples from five subjects showed that the mutations were acquired de novo (Figure S2). All mutations lead to a loss of the highly conserved transcription activation domain (Figure 3 and Figure S3). The Exome Variant Server has public information on KAT6B for over 1,100 individuals of European descent and 900 African Americans, with an average coverage of over 85× for the coding sequences of KAT6B. No truncating mutations of KAT6B were identified in this server or in other exomes performed by Baylor College of Medicine's Human Genome Sequencing Center.
Table 2.
Number of Variants Identified
| Subject 2 | Subject 4 | Subject 5 | Shared (2 of 3) | Shared (3 of 3) | |
|---|---|---|---|---|---|
| Total variants | 2654026 | 2760221 | 490885 | ||
| Total variants after base quality filtering | 1090174 | 1115186 | 256344 | ||
| Novel variants (dbSNP129/1000G) | 872368 | 886839 | 237419 | ||
| Genes with rare nonsynonymous variants, splice site variants, insertions or deletions variants in coding regions. | 440 | 367 | 273 | 76 | 13 |
Figure 3.
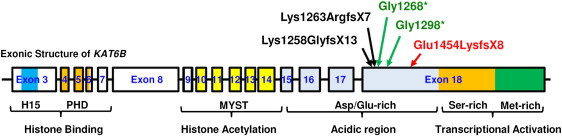
Location of Mutations Identified in KAT6B
The five different mutations form a cluster within the C-terminal acidic domain of KAT6B. The 16 coding exons and the corresponding introns of KAT6B are depicted with boxes and solid lines, respectively, with introns not shown to scale. The encoding domains are indicated below the exon-intron organization, along with the corresponding functions. The longest isoform is shown (isoform 1 in uniprot, a.k.a. MORF-beta, CCDS7345, 2073 aa). The following abbreviations are used: H15, histone H1- or H5-like domain; PHD, tandem plant homeodomain-linked zinc fingers. The amino acid changes resulting from five independent mutations present in 6 different subjects are indicated above the schematic exon-intronic structure. The mutations result in C-terminal truncation and remove the transcriptional activation domain, with the resulting mutant impaired in transcriptional activation. Deletions are shown in black arrows, nonsense mutations in green, and the complex insertion-deletion mutation in red. Sequence details of the mutations are shown in Table 3.
Table 3.
Mutations Identified in the Subjects
| Subject | Mutation (DNA) | Mutation (Protein) | Parents Tested | Reference |
|---|---|---|---|---|
| 1 | c.3892G>T | p.Gly1298∗ | not available | subject 1 in Abdul-Rahman et al.1 See also Schlaubitz et al.11 |
| 2 | c.4360_4368delinsAAAAACCAAAA | p.Glu1454LysfsX8 | de novo | subject 2 in in Abdul-Rahman et al.1 See also Schlaubitz et al.11 |
| 3 | c.3802G>T | p.Gly1268∗ | de novo | Lammer and Abrams7 Schlaubitz et al.11 |
| 4 | c.3769_3772delTCTA | p.Lys1258GlyfsX13 | de novo | Lifchez et al.8 |
| 5 | c.3788_3789delAA | p.Lys1263ArgfsX7 | de novo | this report |
| 6 | c.3769_3772delTCTA | p.Lys1258GlyfsX13 | de novo | this report |
Lymphoblastoid cells were established by Epstein-Barr virus infection for subjects 1 through 4. One million cells were collected, and RNA was extracted with Trizol, treated with DNase I, then phenol-chloroform extracted. The first strand of cDNA was synthesized with oligo dT primers via Invitrogen's SuperScript III First-Strand synthesis kit. For RT-PCR, a 5′ nuclease assay from Integrated DNA Technologies (Coralville, IA) was designed with probes having a 5′ fluorescein amidite fluorophore, a 3′ IBFQ quencher, and an internal ZEN quencher (see Table S1). Quantitative real-time PCR was performed in an ABI 7900 HT machine with ABI's TaqMan Universal PCR Master Mix according to the manufacturer's instructions (Figure 4A). We also amplified cDNA by using primers encompassing the last exon-exon junction and the most 5′ mutations and sequenced the products (see Table S1 for primers). These experiments demonstrate that the mutant mRNAs do not undergo nonsense-mediated decay (Figures 4A and 4B), which is consistent with localization of the premature stop codons in the last exon. To assess the expression pattern of Myst4 in organs known to be affected by GPS, we performed immunohistochemistry on mice of various developmental ages. The primary antibody used is Sigma AV38985 (1:200 dilution), the secondary is Invitrogen A-11012 (1:600 dilution). The specificity of the antibody for the mouse protein was confirmed by showing a staining pattern in the brain compatible with published RNA in situ experiments20 (see Figure 5A). Myst4 is strongly expressed in the telencephalic vesicles, trigeminal ganglion, spinal cord, dorsal root ganglia, digestive tract, pancreas liver and ribs of developing embryos (Figures 5A and 5B). After birth, it is strongly expressed in the diaphysis of the long bones, the kidney, and the patella, among other organs (Figures 5C–5K).
Figure 4.
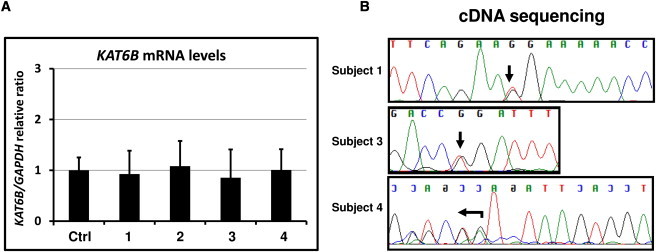
Mutant Transcripts Do Not Undergo Nonsense-Mediated Decay
(A) Messenger RNA levels of KAT6B, normalized to GAPDH, in lymphoblastoid cell lines derived from subjects 1–4, and from control cell lines (n = 3) (data are represented as mean ± standard error of the mean).
(B) Sequencing of cDNA from those cells to demonstrate definitively that the mutant transcripts do not undergo nonsense-mediated decay.
Figure 5.
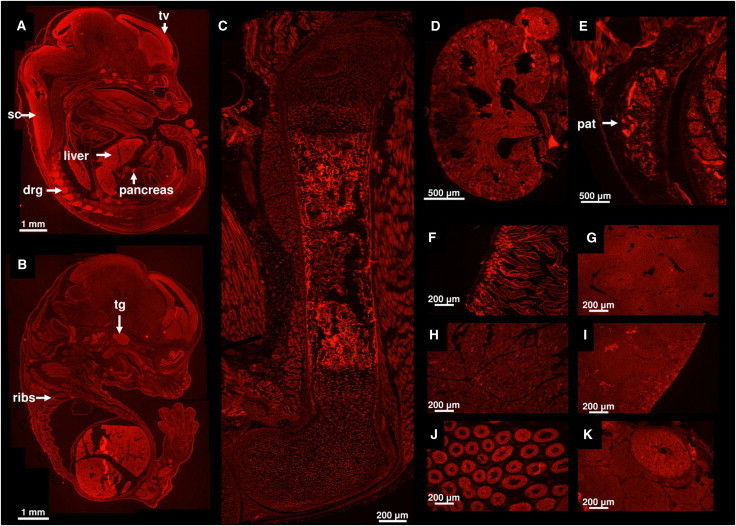
Myst4 Expression in Wild-Type C57BL/6 Mice Detected by Immunohistochemistry
Tissues shown are (A) and (B) whole embryos at embryonic day 15.5, (C) femur at postnatal day 1, and (D) kidney at postnatal day 1. The other tissues are at 8 weeks of age: (E) patella, (F) duodenum (which required 2.5 s of exposure time instead of 5), (G) liver, (H) pancreas, (I) spleen, (J) testis, and (K) ovary. Arrows point to the telencephalic vesicles (tv), spinal cord (sc), liver, pancreas, dorsal root ganglia (drg), trigeminal ganglion (tg), ribs, and patella (pat).
KAT6B has a highly conserved acetyltransferase domain21 and has been shown to fuse with p300 and CBP following chromosomal translocations in acute myeloid leukemia and myelodysplastic syndrome.22 KAT6B has been reported to interact with the RUNX family of transcription factors22 and form a tetrameric complex with BRPFs, ING5, and EAF6.23,24 KAT6B was pulled down in a PPAR-alpha interacting cofactor complex,25 and a yeast two-hybrid screen identified Atrophin-1 as a binding partner of KAT6B.26 Independently to its cloning in humans, the mouse ortholog of KAT6B was identified by screening a gene-trap library for genes highly expressed in the telencephalon.20 Mice carrying a hypomorphic mutation of Myst4 have short stature, an absence of fusion of the tibia and fibula, microcephaly with neurogenesis defects, early demise, and infertility.27 A subject with a Noonan-like phenotype has recently been identified to harbor a chromosomal translocation disrupting KAT6B after exon 3, and the mRNA levels in lymphoblastoid cells were half of normal.28 Given the phenotypic difference between the subject with a Noonan-like phenotype and subjects with GPS (Table 4), the persistent expression of a truncated protein in GPS, containing intact N-terminal domain and HAT domains but lacking the C-terminal transcriptional activation domain, most likely leads to dominant-negative or gain-of-function effects on cellular signaling. Of relevance, leukemia-associated translocations also generate similar KAT6B fragments fused to p300 and CBP.22 Rubinstein-Taybi syndrome [MIM 180849] is caused by de novo mutations inactivating the histone acetyltransferase activity of the latter two enzymes, and both haploinsufficiency and dominant-negative models have been postulated for this condition.29,30 Although the presentation is distinct from GPS, the two conditions share some facial features, as well as intellectual disability, malformations of the heart and kidneys, and undescended testes. Recently, similar de novo truncating in KAT6B mutations were identified in subjects with the Say-Barber-Biesecker variant of Ohdo syndrome [MIM 249620], which overlaps with Genitopatellar syndrome in terms of facial features and congenital heart defects.31,32 The mutations are however usually located more distally in the C terminus, specifically in the transcriptional activation domain, and there are several clinical differences: structural brain defects, skeletal defects, anal anomalies, genital anomalies and renal defects are more severe or frequent in GPS, whereas ocular, dental, palatal, and thyroid defects are more severe or frequent in the Say-Barber-Biesecker variant of Ohdo syndrome. A comparison of the phenotypes of GPS, the Say-Barber-Biesecker variant of Ohdo syndrome, the child with a translocation involving KAT6B, and Rubinstein-Taybi syndrome is shown in Table 4.
Table 4.
Phenotypic Differences between GPS, the Say-Barber-Biesecker Variant of Ohdo Syndrome, a Subject with a Translocation Truncating KAT6B after Exon 3, and Rubinstein-Taybi Syndrome
| Genitopatellar Syndrome | Say-Barber-Biesecker Variant of Ohdo Syndrome | Subject with Noonan-Like Phenotype and N-Terminal Truncation of KAT6B | Rubinstein Taybi Syndrome | |
|---|---|---|---|---|
| Neurological anomalies | DD/ID,a microcephaly in all, agenesis of the corpus callosum, colpocephaly | DD/ID, microcephaly in minority, hypotonia, no structural defects | microcephaly, ADHD,b IQ 75-80, no structural defects | DD/ID, seizures, no structural defects |
| Facial anomalies | broad or prominent nasal bridge, bulbous nose in minority, full cheeks in minority | blepharophimosis, ptosis, broad and flat nasal bridge, bulbous nose, full cheeks, abnormal ears, small mouth, expressionless or mask-like facies | blepharophimosis, ptosis, arched eyebrows, abnormal ears, smooth philtrum, retrognatia, high-arched palate | arched eyebrows, downslanting palpebral fissures, beaked nose with the columella extending below the nares, high-arched palate, mild micrognathia, grimacing smile |
| Musculo-skeletal anomalies | absent or hypoplastic patellae in majority, flexion contractures, club feet, costo-vertebral anomalies, pelvic anomalies | long thumbs and toes, patellar anomalies in minority | short stature, retarded bone age, ligamentous laxity | short stature, joint hypermobility, broad thumbs and broad big toes |
| Anal and genital anomalies | anal anomalies, hypoplastic labia, clitoromegaly, scrotal hypoplasia, cryptorchidism | cryptorchydism and hypospadias | ||
| Heart anomalies | congenital heart defects | congenital heart defects | congenital heart defects | |
| Structural eye defects | none | frequent | frequent | |
| Dental anomalies | rare | frequent | ||
| Cleft palate | rare | frequent | ||
| Hearing impairment | rare | frequent | ||
| Thyroid abnormalities | rare | frequent | ||
| Renal anomalies | hydronephrosis or cysts in a majority | vesicoureteric reflux in one individual | ||
| Other | feeding difficulties, tracheomalacia, respiratory difficulties, small bowel malrotation | feeding difficulties | feeding difficulties, respiratory difficulties, skin anomalies (hirsutism, naevus flammeus on the forehead, and keloid formation), malignancies |
DD/ID is used as an abbreviation for developmental delay or intellectual disability.
ADHD is used as an abbreviation for Attention deficit hyperactivity disorder.
We have thus identified mutations in the epigenetic regulator KAT6B in several subjects with GPS. Because this acetyltransferase is a ubiquitous transcriptional coactivator, searching for more binding partners and studying its role in skeletogenesis and development in general might lead to important insights into epigenetic dysregulation in GPS and related diseases.
Acknowledgments
We thank the families for participating in this study. We thank Alyssa Tran, Stephanie Dugan, and Andrea Kwan for help enrolling subjects; Kyu Sang Joeng, Yangjin Bae, Jianning Tao, and Terry Bertin for help and advice with the experiments; and the University Center for Fetal Medicine of the University of Mississippi for help collecting clinical information. Philippe Campeau is funded in part by the Clinician-Scientist Training award of the Canadian Institutes of Health Research.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Exome Variant Server, http://evs.gs.washington.edu/EVS/
GATK, http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Polyphen-2, http://genetics.bwh.harvard.edu/pph2/
Samtools Pileup, http://samtools.sourceforge.net/
Swiss-Prot, http://web.expasy.org/docs/swiss-prot_guideline.html
SIFT, http://sift.jcvi.org/
UCSC Genome Browser (hg18 and hg19), http://genome.ucsc.edu/
References
- 1.Abdul-Rahman O.A., La T.H., Kwan A., Schlaubitz S., Barsh G.S., Enns G.M., Hudgins L. Genitopatellar syndrome: Expanding the phenotype and excluding mutations in LMX1B and TBX4. Am. J. Med. Genet. A. 2006;140:1567–1572. doi: 10.1002/ajmg.a.31258. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong L., Clarke J.T.R. Report of a new case of “genitopatellar” syndrome which challenges the importance of absent patellae as a defining feature. J. Med. Genet. 2002;39:933–934. doi: 10.1136/jmg.39.12.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergmann C., Spranger S., Javaher P., Ptok M. Genitopatellar syndrome, sensorineural hearing loss, and cleft palate. Oral Maxillofac Surg. 2011;15:103–106. doi: 10.1007/s10006-009-0202-4. [DOI] [PubMed] [Google Scholar]
- 4.Brugha R., Kinali M., Aminu K., Bridges N., Holder S.E. Genitopatellar syndrome: A further case. Clin. Dysmorphol. 2011;20:163–165. doi: 10.1097/MCD.0b013e328345a1dd. [DOI] [PubMed] [Google Scholar]
- 5.Cormier-Daire V., Chauvet M.L., Lyonnet S., Briard M.L., Munnich A., Le Merrer M. Genitopatellar syndrome: A new condition comprising absent patellae, scrotal hypoplasia, renal anomalies, facial dysmorphism, and mental retardation. J. Med. Genet. 2000;37:520–524. doi: 10.1136/jmg.37.7.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldblatt J., Wallis C., Zieff S. A syndrome of hypoplastic patellae, mental retardation, skeletal and genitourinary anomalies with normal chromosomes. Dysmorphol. Clin. Genet. 1988;2:91–93. [Google Scholar]
- 7.Lammer E.J., Abrams L. Genitopatellar syndrome: Delineating the anomalies of female genitalia. Am. J. Med. Genet. 2002;111:316–318. doi: 10.1002/ajmg.10582. [DOI] [PubMed] [Google Scholar]
- 8.Lifchez C.A., Rhead W.J., Leuthner S.R., Lubinsky M.S. Genitopatellar syndrome: Expanding the phenotype. Am. J. Med. Genet. A. 2003;122A:80–83. doi: 10.1002/ajmg.a.20268. [DOI] [PubMed] [Google Scholar]
- 9.Penttinen M., Koillinen H., Niinikoski H., Mäkitie O., Hietala M. Genitopatellar syndrome in an adolescent female with severe osteoporosis and endocrine abnormalities. Am. J. Med. Genet. A. 2009;149A:451–455. doi: 10.1002/ajmg.a.32644. [DOI] [PubMed] [Google Scholar]
- 10.Reardon W. Genitopatellar syndrome: A recognizable phenotype. Am. J. Med. Genet. 2002;111:313–315. doi: 10.1002/ajmg.10590. [DOI] [PubMed] [Google Scholar]
- 11.Schlaubitz S., Yatsenko S.A., Smith L.D., Keller K.L., Vissers L.E., Scott D.A., Cai W.W., Reardon W., Abdul-Rahman O.A., Lammer E.J. Ovotestes and XY sex reversal in a female with an interstitial 9q33.3-q34.1 deletion encompassing NR5A1 and LMX1B causing features of Genitopatellar syndrome. Am. J. Med. Genet. A. 2007;143A:1071–1081. doi: 10.1002/ajmg.a.31685. [DOI] [PubMed] [Google Scholar]
- 12.Das D.K. Psammoma body: A product of dystrophic calcification or of a biologically active process that aims at limiting the growth and spread of tumor? Diagn. Cytopathol. 2009;37:534–541. doi: 10.1002/dc.21081. [DOI] [PubMed] [Google Scholar]
- 13.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X., Jian X., Boerwinkle E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.UniProt Consortium The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38(Database issue):D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., the 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas T., Voss A.K., Chowdhury K., Gruss P. Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development. 2000;127:2537–2548. doi: 10.1242/dev.127.12.2537. [DOI] [PubMed] [Google Scholar]
- 21.Champagne N., Bertos N.R., Pelletier N., Wang A.H., Vezmar M., Yang Y., Heng H.H., Yang X.J. Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J. Biol. Chem. 1999;274:28528–28536. doi: 10.1074/jbc.274.40.28528. [DOI] [PubMed] [Google Scholar]
- 22.Yang X.J., Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007;26:5408–5419. doi: 10.1038/sj.onc.1210609. [DOI] [PubMed] [Google Scholar]
- 23.Doyon Y., Cayrou C., Ullah M., Landry A.-J., Côté V., Selleck W., Lane W.S., Tan S., Yang X.-J., Côté J. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Ullah M., Pelletier N., Xiao L., Zhao S.P., Wang K., Degerny C., Tahmasebi S., Cayrou C., Doyon Y., Goh S.-L. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell. Biol. 2008;28:6828–6843. doi: 10.1128/MCB.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surapureddi S., Yu S., Bu H., Hashimoto T., Yeldandi A.V., Kashireddy P., Cherkaoui-Malki M., Qi C., Zhu Y.-J., Rao M.S., Reddy J.K. Identification of a transcriptionally active peroxisome proliferator-activated receptor alpha -interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc. Natl. Acad. Sci. USA. 2002;99:11836–11841. doi: 10.1073/pnas.182426699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim J., Hao T., Shaw C., Patel A.J., Szabó G., Rual J.-F., Fisk C.J., Li N., Smolyar A., Hill D.E. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 27.Voss A.K., Thomas T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays. 2009;31:1050–1061. doi: 10.1002/bies.200900051. [DOI] [PubMed] [Google Scholar]
- 28.Kraft M., Cirstea I.C., Voss A.K., Thomas T., Goehring I., Sheikh B.N., Gordon L., Scott H., Smyth G.K., Ahmadian M.R. Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J. Clin. Invest. 2011;121:3479–3491. doi: 10.1172/JCI43428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petrij F., Giles R.H., Dauwerse H.G., Saris J.J., Hennekam R.C., Masuno M., Tommerup N., van Ommen G.J., Goodman R.H., Peters D.J. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 30.Oike Y., Hata A., Mamiya T., Kaname T., Noda Y., Suzuki M., Yasue H., Nabeshima T., Araki K., Yamamura K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: Implications for a dominant-negative mechanism. Hum. Mol. Genet. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- 31.Clayton-Smith J., O'Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., Voss A.K., Thomas T., Biesecker L.G., Smith P. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day R., Beckett B., Donnai D., Fryer A., Heidenblad M., Howard P., Kerr B., Mansour S., Maye U., McKee S. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin. Genet. 2008;74:434–444. doi: 10.1111/j.1399-0004.2008.01087.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.