

Abstract
We have identified KIF11 mutations in individuals with syndromic autosomal-dominant microcephaly associated with lymphedema and/or chorioretinopathy. Initial whole-exome sequencing revealed heterozygous KIF11 mutations in three individuals with a combination of microcephaly and lymphedema from a microcephaly-lymphedema-chorioretinal-dysplasia cohort. Subsequent Sanger sequencing of KIF11 in a further 15 unrelated microcephalic probands with lymphedema and/or chorioretinopathy identified additional heterozygous mutations in 12 of them. KIF11 encodes EG5, a homotetramer kinesin motor. The variety of mutations we have found (two nonsense, two splice site, four missense, and six indels causing frameshifts) are all predicted to have an impact on protein function. EG5 has previously been shown to play a role in spindle assembly and function, and these findings highlight the critical role of proteins necessary for spindle formation in CNS development. Moreover, identification of KIF11 mutations in patients with chorioretinopathy and lymphedema suggests that EG5 is involved in the development and maintenance of retinal and lymphatic structures.
Main Text
There is substantial phenotypic overlap between MLCRD (microcephaly, primary lymphedema, and chorioretinal dysplasia) syndrome (MIM 152950) and CDMMR (chorioretinal dysplasia, microcephaly, and mental retardation) syndrome (MIM 156590).1 Both have been observed to segregate with autosomal-dominant inheritance and present with an overlapping, yet variable, spectrum of CNS and ocular developmental anomalies.2,3 Microcephaly, ranging from mild to severe, is the critical component of both syndromes and is often associated with mild to moderate developmental delay and a characteristic facial phenotype4 (Figures 1A and 1B). Chorioretinopathy constitutes the most common eye abnormality (Figure 1C). However, retinal folds, microphthalmia, and myopic and hypermetropic astigmatism have also been reported, and some individuals have no overt ocular phenotype.5 The presence of lymphedema has historically been seen as the critical differentiating feature between the two syndromes. The lymphedema in MLCRD is congenital and typically confined to the dorsa of the feet (Figure 1D), and lymphoscintigraphy reveals the absence of radioactive isotope (technetium 99) uptake from the webspaces between the toes (Figure 1E). We have identified causative variants for this complex disease, and the fact that we show that KIF11 mutations can cause both MLCRD and CDMMR suggests that together, these syndromes should be considered as a single entity that has variable clinical features but a unified molecular basis.
Figure 1.
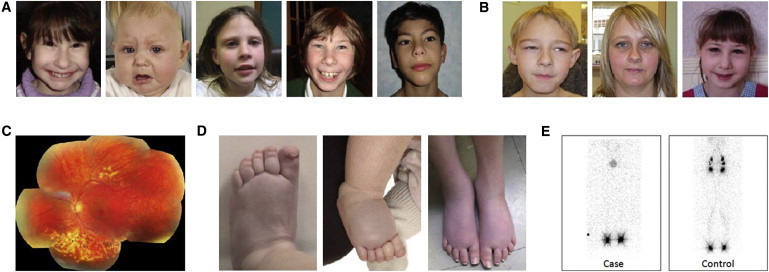
Clinical Features of KIF11 Mutation-Positive Patients with MLCRD and CDMMR
(A) Facial features of individuals diagnosed with MLCRD.
(B) Facial features of individuals diagnosed with CDMMR. The faces in (A) and (B) are characteristic of the syndrome and have upslanting palpebral fissures, a broad nose with a rounded tip, a long philtrum with a thin upper lip, a prominent chin, and prominent ears.
(C) A composite color photograph of the left fundus in patient CDMMR05 II-1 shows focal areas of peripheral chorioretinal atrophy.
(D) Bilateral, congenital lower-limb primary lymphedema in subjects with MLCRD involves the dorsa of the feet, which show pitting edema, deep interphalangeal creases, small dysplastic nails, and wide-caliber veins.
(E) Comparison of lower-limb lymphoscintigraphy (imaging taken 2 hr after injection of radioactive isotope [technetium 99] into the webspaces between the toes) in patient MLCRD01 II-2 and an unaffected control. The patient's lymphoscintigraphy shows no significant main-tract filling and therefore suggests initial lymphatic-vessel dysfunction.
Whole-exome sequencing has proved to be a successful approach to identifying causative mutations in primary lymphoedema.6–8 We therefore sought to establish the molecular genetic basis of MLCRD by whole-exome sequencing the DNA of five unrelated probands (individuals MLCRD01:II-2, MLCRD02:II-2, MLCRD03:III-2, MLCRD04:I-1, and MLCRD05:I-1 in Table 1 and Table S1, available online) with microcephaly and lymphedema. Subjects for this study were recruited from genetic, lymphovascular, and ophthalmic clinics in Europe. Informed consent was obtained from all subjects. All affected individuals and family members underwent a detailed physical examination. Ethical approval for this study was obtained from the South West London Research Ethics Committee (REC Ref: 05/Q0803/257).
Table 1.
Clinical and Genetic Findings in KIF11-Mutation-Positive Patients
| Pedigree | Individual | Gender | Head Circumferenceb | Lymphedema | Eye Abnormalities | Additional Clinical Features | Nucleotide Variant | Exon | Protein Alteration |
|---|---|---|---|---|---|---|---|---|---|
| MLCRD01 | I-1 | male | −3.0 | minimal edema | none | mild LD | c.1159C>T | 10 | p.Arg387∗ |
| II-2a | female | −7.2 | congenital, bilateral, and lower limb | none | mild LD and dysmorphic features | c.1159C>T | 10 | p.Arg387∗ | |
| MLCRD02 | I-1 | female | −1.6 | none | diabetic retinopathy | mild LD | c.3016delA | 21 | p.Ile1006Leufs∗62 |
| II-2a | female | −4.0 | congenital, bilateral, and lower limb | none | mild dysmorphic features | c.3016delA | 21 | p.Ile1006Leufs∗62 | |
| MLCRD03 | II-1 | male | −2.0 | none | none | mild LD | c.1039_1040delCT | 9 | p.Leu347Glufs∗8 |
| III-2a | female | −7.5 | congenital, bilateral, and lower limb plus pleural effusions | hypermetropic astigmatism and chorioretinopathy | mild LD | c.1039_1040delCT | 9 | p.Leu347Glufs∗8 | |
| MLCRD06 | I-1 | male | −4.3 | congenital, bilateral, and lower limb | none | low birth weight, failure to thrive, | c.432T>G | 5 | p.Phe144Leu |
| MLCRD07 | I-1c | male | −2.3 | congenital, bilateral, and lower limb | bilateral chorioretinopathy | mild LD, ASD, and myoclonic epilepsy | c.2830C>T (de novo) | 20 | p.Arg944Cys |
| MLCRD08 | I-1 | male | −3.5 | none | none | none | c.1425_1426delinsAAA | 12 | p.Val476Asnfs∗2 |
| II-1 | male | −5.5 | congenital, mild, bilateral, and lower limb | myopia | moderate LD | c.1425_1426delinsAAA | 12 | p.Val476Asnfs∗2 | |
| MLCRD09 | I-1 | female | −3.0 | none | none | none | c.1592delA | 13 | p.Gln531Argfs∗8 |
| II-1d | male | −4.0 | congenital, bilateral, and lower limb | bilateral chorioretinopathy | ASD and dysmorphic features | c.1592delA | 13 | p.Gln531Argfs∗8 | |
| MLCRD10 | I-1 | male | −5.5 | congenital, mild, bilateral, and mild lower limb (resolved) | bilateral chorioretinopathy | mild LD | c.699-2A>G (de novo) | 6/7 | acceptor splice site |
| MLCRD11 | II-1 | male | low by history | congenital, bilateral, and lower limb plus mild in hands | none | none | c.2547+2T>C | 18/19 | donor splice site |
| III-2 | female | −4.7 | none | none | mild LD | c.2547+2T>C | 18/19 | donor splice site | |
| IV-1 | male | −4.0 | congenital, bilateral, and lower limb plus mild in hands | none | moderate LD and hypospadias | c.2547+2T>C | 18/19 | donor splice site | |
| IV-3 | male | −3.7 | congenital, mild, bilateral, and lower limb | none | moderate LD | c.2547+2T>C | 18/19 | donor splice site | |
| IV-5 | female | −4.1 | none | none | mild LD | c.2547+2T>C | 18/19 | donor splice site | |
| MLCRD12 | I-1 | male | −6.4 | congenital, mild, bilateral, and lower limb | bilateral chorioretinopathy | moderate LD | c.1963_1964dupAA | 15 | p.His656Serfs∗8 |
| CDMMR01 | II-1 | female | −5.0 | adult onset and post-traumatic mild edema | chorioretinopathy | mild LD | c.1159C>T (de novo) | 10 | p.Arg387∗ |
| III-1 | male | −5.5 | none | hypermetropic astigmatism and chorioretinopathy | moderate LD | c.1159C>T | 10 | p.Arg387∗ | |
| CDMMR02 | I-1 | female | low by history | none | bilateral chorioretinopathy | none | c.704C>G | 7 | p.Ser235Cys |
| II-1 | female | low by history | none | bilateral chorioretinopathy | moderate LD | c.704C>G | 7 | p.Ser235Cys | |
| CDMMR03 | I-1 | male | −3.4 | none | no vision in right eye (retinal detachment) and peripheral retinal atrophy in left eye |
mild LD | c.2304_2305delCA (de novo) | 18 | p.His768Glnfs∗7 |
| CDMMR04 | I-1 | male | −5.1 | none | chorioretinopathy, nystagmus, and exotropia | moderate LD | c.700C>T | 7 | p.Arg234Cys |
| CDMMR05 | II-1 | female | −3.9 | none | chorioretinopathy | mild LD | c.1804C>T | 14 | p.Gln602∗ |
| II-2 | female | −6.1 | none | hypermetropic astigmatism | none | c.1804C>T | 14 | p.Gln602∗ |
Abbreviations are as follows: LD, learning difficulties; and ASD, atrial septal defect. Please see main text for additional abbreviations.
Individuals exome sequenced in primary analysis.
Head circumference measured as occipitofrontal head circumference in cm and corrected for age and sex.
Case description in Vasudevan et al. (2005).4
Case description in Eventon-Friedman et al. (2009).24
Whole-exome capture was performed by in-solution hybridization, followed by massively parallel sequencing with the SureSelect All Exon CCDS (Consensus Coding Sequences) Target Enrichment System (Agilent)9 and sequencing on a Genome AnalyserIIx (Illumina) with 76 bp paired-end reads. Sequence reads were aligned to the reference genome (hg18) with Novoalign (Novocraft Technologies SdnBhd). Duplicate reads, resulting from PCR clonality or optical duplicates, and reads mapping to multiple locations were excluded from downstream analysis. Depth and breadth of sequence coverage were calculated with custom scripts and the BedTools package.10 More than 5.1 Gb of sequence was generated for each subject, such that >75% of the coding bases of the CCDS-defined exome were represented by at least 20 reads (Table S2). Single-nucleotide substitutions and small indel variants were identified and quality filtered within the SamTools software package11 and in-house software tools (Table S3).12 Variants were annotated with respect to genes and transcripts with the Annovar tool.13 We filtered variants for novelty by comparing them to dbSNP132 and 1000 Genomes SNP calls (December 2010) and to variants identified in 250 control exomes (primarily of European origin), which we sequenced and analyzed by the same method described above.
Analysis of the exome-variant profiles was performed under a model of a rare autosomal-dominant disorder; this model required at least one previously unobserved, heterozygous nonsynonymous or splice-site substitution or an insertion or deletion in the same gene in all five individuals. This analysis failed to identify a single gene matching these criteria (Table S4). Further evaluation of the data with an expectation of genetic heterogeneity among these five cases revealed KIF11 to be the only gene that harbored previously unobserved variants in three of the five individuals (Table S4). All three allelic mutations (a nonsense variant [p.Arg387∗], a single-nucleotide deletion [p.Ile1006Leufs∗62], and a dinucleotide deletion [p.Leu347Glufs∗8]) are predicted to cause changes to the KIF11 protein product (Table 1). All were confirmed by Sanger sequencing (primer sequences are listed in Table S5). Sequencing in the two subjects, in whom exome sequencing did not reveal novel KIF11 variants, confirmed the wild-type coding sequence. Our findings indicate that heterozygous mutations of KIF11 underlie a significant proportion of MLCRD, and we therefore assessed KIF11 for mutations in other MLCRD cases by Sanger sequencing in probands from nine additional families, including a large multigenerational pedigree (MLCRD11, Figure S1). In these probands, sequencing revealed seven more independent heterozygous KIF11 variants: three frameshift insertions and deletions, two missense substitutions, an acceptor splice-site substitution, and a donor splice-site change that was observed in the proband from the multigenerational pedigree (Table 1). Each of the ten identified KIF11 alleles was assessed in all available relatives; two were shown to have arisen de novo, and eight demonstrated cosegregation with microcephaly, variable in its severity, and with a spectrum of eye and lymphatic abnormalities (Table 1). In total, we identified KIF11 mutations in 10 of the 14 MLCRD-affected families examined.
Within the extended MLCRD-affected families, we observed individuals who had heterozygous mutations in KIF11 and in whom there was no evidence of lymphatic involvement (Table 1). The considerable and recognizable overlap between the two conditions led us to address the hypothesis that MLCRD and CDMMR are allelic disorders. Sequencing of KIF11 in a cohort of six independent families in which microcephaly and eye abnormalities had been observed in the absence of lymphedema revealed five independent variants: Two missense substitutions and three variants, including one nonsense mutation (p.Arg387∗) that we had previously identified in pedigree MLCRD01, were predicted to lead to premature termination of the protein product (Table 1). In total, we identified 27 carriers of 14 mutant KIF11 alleles in 15 independent families. None of the 14 variants is present in dbSNP, has been identified by the 1000 Genomes Project, or was observed in a cohort of 250 control samples.
We failed to identify coding variation in KIF11 in five families from which four probands had previously been diagnosed with MLCRD and one with CDMMR (Table S1). These cases might represent KIF11 alleles comprising noncoding or undetected coding variants. They might represent phenocopies of MLCRD and CDMMR or be explained by locus heterogeneity. We were unable to investigate the latter because none of the KIF11-mutation-negative families were of sufficient size to allow exclusion of the locus by linkage analysis.
KIF11 encodes EG5, a bipolar, homotetrameric, slow-processive, plus-end-directed spindle motor protein of the kinesin-5 family.14 Each monomer contains a conserved N-terminal motor, a central coiled-coil domain, and a C-terminal tail that contains a bimC box, a conserved sequence of positively charged amino acid residues (Figure 2).15,16 The homotetramer consists of four monomers that are arranged in an antiparallel fashion so that the resulting molecule possesses two motor domains and two nonmotor tails at each end of a central stalk. The contact of EG5 with microtubules is established through the C-terminal tails, whereas the subsequent sliding of the antiparallel microtubules is driven by the motor domains.17 Seven of the identified allelic mutations, two nonsense and five frameshift, including insertions and deletions, are predicted to lead to premature termination of the protein. A sixth frameshift variant, a single-base deletion in the second-to-last exon, is predicted to result in substitution of the terminal 50 residues of the 1,056 amino acid wild-type protein and extension of the reading frame by a further 12 residues. The two splice-site mutations are both predicted in HSF (Human Splicing Finder v.2.4.1)18 to have significant impact on the splicing of the 5 kb transcript (Table S6). The four missense mutations all alter evolutionarily conserved amino acid residues and are predicted to have a damaging effect on protein function according to SIFT and PolyPhen; three (p.Phe144Leu, p.Arg234Cys, and p.Ser235Cys) are located within the motor domain,15 and the fourth (p.Arg944Cys) is located within the bimC box in the C-terminal tail of the molecule (Figure S2).16
Figure 2.
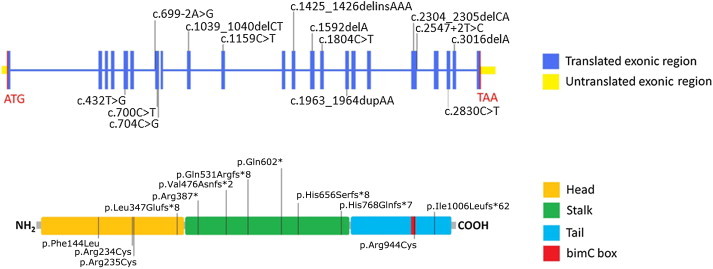
Location of the Identified Mutations in KIF11
Upper panel: Mutations are indicated with respect to the genomic organization of KIF11.
Lower panel: KIF11 domain structure excludes the two identified splice-site mutations.
Kif11 has been shown to be widely expressed during murine embryonic development and is elevated in proliferating tissues.19,20 During zebrafish development, kif11 is dynamically expressed in tissues that are associated with rapid proliferation (Figure S3). Interestingly, homozygous disruption of Kif11 leads to early embryonic lethality, and signs of a proliferation defect are shown at embryonic day 2.5.19,20 It has been previously demonstrated that EG5 localizes to spindle microtubules during mitosis and also contributes to the assembly of the bipolar spindle,21 as well as the regulation of axonal outgrowth22 and CNS development.23 Several genes (CENPJ, MCPH1, ASPM, CDK5RAP2, STIL, CEP152, and WDR62 [MIM 608393, 251200, 608716, 604804, 612703, 604321, and 604317, respectively]) mutated in recessively inherited microcephaly have products with roles in centrosome formation and spindle development. Our identification of heterozygous KIF11 mutations in dominant forms of microcephaly provides further evidence of the critical role of molecules involved in mitotic spindle function in CNS development. The observation of lymphedema and chorioretinopathy provides evidence of a role of EG5 in the development and maintenance of the lymphatic and retinal structures. It is currently unclear whether MLCRD and CDMMR result from disruption of the mitotic function of EG5 or from other roles of EG5 in the cell. The recently defined function of EG5 as a brake on microtubule activity as part of axonal turning22 provides the basis for speculating that the dominant mutations observed in this study might disrupt the control of lymphatic development in a similar manner.
Our findings demonstrate a pleiotropic phenotypic expression of mutant KIF11 alleles and show that MLCRD and CDMMR are allelic disorders. Beyond the observed variability of lymphatic and eye involvement, there is also a range in the severity of microcephaly. Microcephaly was primary (congenital) in all subjects, and there was some correlation between the degree of microcephaly and the severity of the learning disorder. Although all probands were chosen because they had microcephaly, there was marked intrafamilial variation (Table 1); one parent had a normal head circumference, mild learning difficulties, no lymphedema, and only a retinopathy associated with her diabetes (MLCRD02 I-1).
Chorioretinopathy was a highly specific finding in patients with KIF11 mutations, but none of the patients had retinal folds or microphthalmia to suggest that the condition originally described by Jarmas et al.5 might be nonallelic. A number of patients with additional abnormalities (e.g., thrombocytopenia and craniosynostosis) were not found to have KIF11 mutations. It could be relevant for clinical differentiation that chorioretinopathy was not observed in any of the KIF11-mutation-negative subjects (Table S1), although numbers are too small for this to be a definitive observation.
The lymphedema in the mutation-positive subjects was present at birth and was restricted to both lower limbs and rarely extended above the knees. The dorsa of the feet were particularly affected and had small, dysplastic nails and deep interphalangeal creases. There were often large-caliber veins in the lower limbs. Clinically, the findings resemble those seen in Milroy disease (Figure 1D). The lymphedema was usually persistent but responded well to compression garments. Interestingly, there appeared to be some intrafamilial consistency among the clinical features regardless of the type of mutation, but such consistency is difficult to verify with such small numbers.
In conclusion, we have identified KIF11 mutations that cause autosomal-dominant forms of microcephaly that are variably associated with congenital lymphedema and/or chorioretinopathy, demonstrating that MLCRD and CDMMR are allelic disorders. The extreme variability of the phenotype, even between individuals with the same KIF11 mutation, suggests that there might be additional genetic or environmental factors that contribute to the extent of disruption. In addition, our findings also provide a substantial foundation for the existence of a link between EG5 and the development and maintenance of retinal and lymphatic structures. It remains to be elucidated how far the established mitotic function of EG5 can account for the different phenotypical aspects of MLCRD and CDMMR and whether at least some defects are consequences of a different role of EG5 during development.
Acknowledgments
This study was possible by the participation of the families concerned, and we are indebted to them. We would like to thank the clinicians involved: Denise Williams (Birmingham Women's National Health Services [NHS] Foundation Trust, Birmingham) and Sixto Garcia Minaur (Hospital Universitario La Paz, Madrid). We thank Carolyn Moores (Crystallography, Birkbeck College, London) for advice on EG5 function and Anukampa Barth (University College, London) for help with documentation and analysis of the in situ hybridizations. This work was supported by British Heart Foundation grants RG/08/006/25302, PG/10/58/28477, and FS/06/063/21445. We also acknowledge support from the Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy's and St. Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust, the specialist Biomedical Research Centre award for Ophthalmology to Moorfields Eye Hospital and the University College London Institute of Ophthalmology, the Sheffield Childrens' Hospital charity, Marie Curie Fellowship, and Cancer Research UK. These studies were also partially supported by the Interuniversity Attraction Poles initiated by the Belgian Federal Science Policy, network 6/05; Concerted Research Actions–Convention No 07/12-005 of the Belgian French Community Ministry; the Fonds de la Recherche Scientifique-Fonds National de la Recherche Scientifique; and la Communauté Française de Wallonie-Bruxelles and la Lotterie Nationale, Belgium. This research utilized the resources of the Biomics Centre, St. George's University of London, and the Advanced Sequencing Facility of the Cancer Research UK London Research Institute. Special thanks go to Nik Matthews.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
Consensus Coding Sequence Project (CCDS) project, http://www.ncbi.nlm.nih.gov/projects/CCDS/
Human Splicing Finder, http://www.umd.be/HSF/
Online Mendelian Inheritance in man (OMIM), http://www.omim.org/
PolyPhen, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
Accession Numbers
The accession number for the KIF11 sequence reported in this paper is NM_004523.
References
- 1.Fryns J.P., Smeets E., Van den Berghe H. On the nosology of the “primary true microcephaly, chorioretinal dysplasia, lymphoedema” association. Clin. Genet. 1995;48:131–133. doi: 10.1111/j.1399-0004.1995.tb04072.x. [DOI] [PubMed] [Google Scholar]
- 2.Leung A.K.C. Dominantly inherited syndrome of microcephaly and congenital lymphedema. Clin. Genet. 1985;27:611–612. doi: 10.1111/j.1399-0004.1985.tb02047.x. [DOI] [PubMed] [Google Scholar]
- 3.Warburg M., Heuer H.E. Chorioretinal dysplasia-microcephaly-mental retardation syndrome. Am. J. Med. Genet. 1994;52:117. doi: 10.1002/ajmg.1320520124. [DOI] [PubMed] [Google Scholar]
- 4.Vasudevan P.C., Garcia-Minaur S., Botella M.P., Perez-Aytes A., Shannon N.L., Quarrell O.W.J. Microcephaly-lymphoedema-chorioretinal dysplasia: Three cases to delineate the facial phenotype and review of the literature. Clin. Dysmorphol. 2005;14:109–116. [PubMed] [Google Scholar]
- 5.Jarmas A.L., Weaver D.D., Ellis F.D., Davis A. Microcephaly, microphthalmia, falciform retinal folds, and blindness. A new syndrome. Am. J. Dis. Child. 1981;135:930–933. doi: 10.1001/archpedi.1981.02130340036013. [DOI] [PubMed] [Google Scholar]
- 6.Ostergaard P., Simpson M.A., Brice G., Mansour S., Connell F.C., Onoufriadis A., Child A.H., Hwang J., Kalidas K., Mortimer P.S. Rapid identification of mutations in GJC2 in primary lymphoedema using whole exome sequencing combined with linkage analysis with delineation of the phenotype. J. Med. Genet. 2011;48:251–255. doi: 10.1136/jmg.2010.085563. [DOI] [PubMed] [Google Scholar]
- 7.Ostergaard P., Simpson M.A., Jeffery S. Massively parallel sequencing and identification of genes for primary lymphoedema: A perfect fit. Clin. Genet. 2011;80:110–116. doi: 10.1111/j.1399-0004.2011.01706.x. [DOI] [PubMed] [Google Scholar]
- 8.Ostergaard P., Simpson M.A., Connell F.C., Steward C.G., Brice G., Woollard W.J., Dafou D., Kilo T., Smithson S., Lunt P. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat. Genet. 2011;43:929–931. doi: 10.1038/ng.923. [DOI] [PubMed] [Google Scholar]
- 9.Coffey A.J., Kokocinski F., Calafato M.S., Scott C.E., Palta P., Drury E., Joyce C.J., Leproust E.M., Harrow J., Hunt S. The GENCODE exome: Sequencing the complete human exome. Eur. J. Hum. Genet. 2011;19:827–831. doi: 10.1038/ejhg.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 13.Li K., Stockwell T.B. VariantClassifier: A hierarchical variant classifier for annotated genomes. BMC Res Notes. 2010;3:191. doi: 10.1186/1756-0500-3-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valentine M.T., Fordyce P.M., Krzysiak T.C., Gilbert S.P., Block S.M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nat. Cell Biol. 2006;8:470–476. doi: 10.1038/ncb1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawin K.E., Mitchison T.J. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc. Natl. Acad. Sci. USA. 1995;92:4289–4293. doi: 10.1073/pnas.92.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rapley J., Nicolàs M., Groen A., Regué L., Bertran M.T., Caelles C., Avruch J., Roig J. The NIMA-family kinase Nek6 phosphorylates the kinesin Eg5 at a novel site necessary for mitotic spindle formation. J. Cell Sci. 2008;121:3912–3921. doi: 10.1242/jcs.035360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinger J.S., Qiu M., Yang G., Kapoor T.M. A nonmotor microtubule binding site in kinesin-5 is required for filament crosslinking and sliding. Curr. Biol. 2011;21:154–160. doi: 10.1016/j.cub.2010.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castillo A., Justice M.J. The kinesin related motor protein, Eg5, is essential for maintenance of pre-implantation embryogenesis. Biochem. Biophys. Res. Commun. 2007;357:694–699. doi: 10.1016/j.bbrc.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chauvière M., Kress C., Kress M. Disruption of the mitotic kinesin Eg5 gene (Knsl1) results in early embryonic lethality. Biochem. Biophys. Res. Commun. 2008;372:513–519. doi: 10.1016/j.bbrc.2008.04.177. [DOI] [PubMed] [Google Scholar]
- 21.Sawin K.E., LeGuellec K., Philippe M., Mitchison T.J. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature. 1992;359:540–543. doi: 10.1038/359540a0. [DOI] [PubMed] [Google Scholar]
- 22.Myers K.A., Baas P.W. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J. Cell Biol. 2007;178:1081–1091. doi: 10.1083/jcb.200702074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferhat L., Cook C., Chauviere M., Harper M., Kress M., Lyons G.E., Baas P.W. Expression of the mitotic motor protein Eg5 in postmitotic neurons: Implications for neuronal development. J. Neurosci. 1998;18:7822–7835. doi: 10.1523/JNEUROSCI.18-19-07822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eventov-Friedman S., Singer A., Shinwell E.S. Microcephaly, lymphedema, chorioretinopathy and atrial septal defect: A case report and review of the literature. Acta Paediatr. 2009;98:758–759. doi: 10.1111/j.1651-2227.2008.01161.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.