

Abstract
The human prefrontal cortex (PFC), a mastermind of the brain, is one of the last brain regions to mature. To investigate the role of epigenetics in the development of PFC, we examined DNA methylation in ∼14,500 genes at ∼27,000 CpG loci focused on 5′ promoter regions in 108 subjects range in age from fetal to elderly. DNA methylation in the PFC shows unique temporal patterns across life. The fastest changes occur during the prenatal period, slow down markedly after birth and continue to slow further with aging. At the genome level, the transition from fetal to postnatal life is typified by a reversal of direction, from demethylation prenatally to increased methylation postnatally. DNA methylation is strongly associated with genotypic variants and correlates with expression of a subset of genes, including genes involved in brain development and in de novo DNA methylation. Our results indicate that promoter DNA methylation in the human PFC is a highly dynamic process modified by genetic variance and regulating gene transcription. Additional discovery is made possible with a stand-alone application, BrainCloudMethyl.
Introduction
The human prefrontal cortex (PFC) plays a critical role in complex cognitive behaviors, personality, decision making, and orchestration of thoughts and actions and thus has been referred to as the CEO of the brain. From an evolutionary perspective, it has emerged relatively recently and shows the greatest expansion at the gross anatomy level. The PFC also shows an especially prolonged period of postnatal maturation.1,2 Recent global transcriptome analyses of the developing human PFC have shed light on some of the key processes and genes that contribute to the uniqueness of the human PFC and have implicated involvement of epigenetic mechanisms.3–5 DNA methylation at CpG dinucleotides has long been considered a key mechanism of transcriptional regulation and a critical factor in embryonic development and in cancer.6,7 However, little is known about the normal developmental changes in DNA methylation of the human PFC, particularly during early life. These early epigenetic changes could be particularly relevant for understanding the mechanisms of neurodevelopmental brain disorders, such as autism (MIM 209850) and schizophrenia (MIM 181500).8,9 Tissue-, sex- and age-specific effects on DNA methylation in human samples have been reported,10–17 but there are no reports of genome-wide DNA methylation in the human brain across the lifespan. In this study, we investigated the genome-wide temporal dynamics of DNA methylation in a large cohort of well-characterized human PFC specimens from the second trimester of gestation until old age, identified genetic determinants of DNA methylation in a genome-wide SNP association analysis and examined relationships with mRNA expression. We focus in this report on CpG dinucleotides located primarily in the putative 5′ promoter regions of known genes. Although recent work has emphasized the importance of DNA methylation in gene regions outside 5′ domains,13 understanding how DNA methylation varies in promoter regions in normal brains across the lifespan and how it is associated with genetic variance is an important element in the interpretation of pathological changes in brain disorders and in understanding gene expression and gene function.
Subjects and Methods
Human Postmortem Brain Tissue Collection
Postmortem human brains from nonpsychiatric controls (108) were collected at the Clinical Brain Disorders Branch (National Institute of Mental Health) under protocol 90-M-0142 with informed consent from the next-of-kin and at the Brain and Tissue Bank for Developmental Disorders of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) under protocols NO1-HD-4-3368 and NO1-HD-4-3383. Clinical characterization, neuropathological screening, toxicological analyses, and dissections of the dorsolateral prefrontal cortex were performed as previously described.18 Demographic data for these samples are summarized in Table S1, available online.
Genotyping Methods
SNP genotyping with HumanHap650Y_V3 or Human 1M-Duo_V3 BeadChips (Illumina, San Diego, CA) was carried out according to the manufacturer's instructions with DNA extracted from cerebellar tissue. Genotype data were analyzed with the genotyping analysis module within the BeadStudio software (Illumina). For data analysis, 605,371 SNPs with missing calls < 2%, Hardy-Weinberg equilibrium p values ≥ 0.001, and minor allele frequencies ≥ 0.03 were used, from among a total of 654,333 shared SNPs between the two types of chips.
Methylation Methods
Genomic DNA was extracted from 100 mg of pulverized dorsolateral prefrontal cortex (DLPFC) tissue with the phenol-chloroform method. Bisulfite conversion of 600 ng genomic DNA was performed with the EZ DNA methylation kit (Zymo Research). Methylation of DNA extracted from DLPFC was assessed according to the manufacturer's instructions with Infinium HumanMethylation27 BeadChips (Illumina). Quantitative measurements of DNA methylation were determined for 27,578 CpG dinucleotides spanning 14,495 genes. CpG sites were selected in CpG islands (CpGIs) of the gene proximal promoter regions within 1 kb upstream and 500 bases downstream from the transcription start sites whenever possible, that is for 14,475 consensus coding sequencing (CCDS) in the National Center for Biotechnology Information database (genome build 36). Approximately 40% of genes have no CpGIs in their promoter regions. On average, two assays were selected per CCDS gene and from 3 to 20 CpG sites for more than 200 cancer-related and imprinted genes. Among 27,578 total CpG sites, 20,007 are in CpGIs, 7,571 are in non-CpGIs, and 1,086 are on the X chromosome. A CpGI is defined as a nucleotide sequence of (1) 200 bp or greater in length; (2) 50% or greater in G-C percent and (3) 0.60 or greater in the ratio of observed CpG sites over expected CpG sites. Methylation data were analyzed with the methylation analysis module within the BeadStudio software (Illumina). Methylation status of the interrogated CpG sites was calculated as the ratio of signal from a methylated probe relative to the sum of both methylated and unmethylated probes. This value, known as β, ranges continuously from 0 (completely unmethylated) to 1 (fully methylated). The technical schemes of this array have been described in detail in a previously published paper.19 To ensure data reproducibility, ten samples were analyzed in duplicates starting from the bisulfite conversion step, and high reproducibility was observed (r2 ranged from 0.9973 to 0.9921). For validation, we used 92 samples from the current study and measured methylation status at 34 CpG sites by using an Illumina custom GoldenGate platform. The CpG site positions for the probes were exactly the same as in the Infinium arrays. The correlation between the data from the two platforms was very high (r2 = 0.79; Figure S1).
In this study, DNA was derived from tissue homogenates. We cannot distinguish differential DNA methylation within a population of cells that are stable in their cell type from a change in the abundance of cell types. It is most likely that both phenomena contribute to signals measured in this study.
Statistical Methods
We used surrogate variable analysis to account for known and unknown factors, including batch effects.20 A step-wise analysis was used for model selection for each CpG site. The influences of age and sex on DNA methylation were examined by multiple linear regression with age, sex, race, developmental life stage, and age-by-stage interaction as the primary variables. Age was a continuous predictor, whereas sex (male or female), race (individuals of European descent or African Americans) and developmental stage (fetal period, childhood [0–10 years], and postchildhood [ages older than 10 years]) were categorical predictors. The use of age, stage, and age-by-stage interaction allowed the effects of age on DNA methylation to be independent in each age stage (i.e., to have different slopes between the stages) and thus enabled fitting nonlinear trajectories over the entire lifespan into linear models within the three life stages. In the age analysis, a false-discovery-rate (FDR) correction was applied at the 0.05 level for multiple testing.21 In the sex analysis, Bonferroni correction was applied at the 0.05 level as in a previously published paper.14 In order to eliminate the effects of age, sex, and race in the methylation quanatitative trait loci (mQTL) analysis, we used the residuals from multiple regression (as described above) to analyze the associations with SNP genotypes by utilizing an additive model (multiple regression with allele dosage) in PLINK.22 We also used multidimensional scaling (MDS) in PLINK across all genotypes to identify population clusters and population outliers. Because two races (individuals of European descent and African Americans) showed two distinct clusters in the identity-by-state (IBS) analysis, we also performed separate mQTL analyses in these separate race groups. The methods were the same as those performed in the combined cohort of individuals of European descent and African Americans. SNPs within 1 Mb of a CpG site were arbitrarily defined as cis-SNPs, and all the other SNPs were defined as trans-SNPs, as in previous studies.11,23 A total number of such cis-SNP associations were ∼11 million, whereas a total number of all possible associations were ∼16 billion. In this analysis, FDR was applied at the 0.05 level for multiple testing corrections. We used the web tool DAVID functional annotation to examine enrichment of gene ontology terms in the sets of genes.24 We used weighted correlation network analysis (WGCNA), an R package for weighted correlation network analysis,25 to describe the correlation patterns among CpG loci across all the samples. For calculating correlations between expression and DNA methylation levels, we used Pearson's r.
Results
Global Patterns of DNA Methylation
DNA methylation levels across all samples and CpG sites showed a bimodal distribution with 63.3% of loci being relatively hypomethylated (β < 0.2, where β is the ratio of signal from a methylated probe relative to the sum of both methylated and unmethylated probes) and 13% of loci hypermethylated (β > 0.8; Figure S2). Among the 27,578 CpG sites surveyed on the array, 20,007 CpG sites are in CpGIs and 7,571 CpG sites are in non-CpGIs. Consistent with a previous study,19 the average DNA methylation level (β value) across all samples was significantly higher for the sites in non-CpGIs than for the CpGs in CpGIs (0.62 ± 0.26 and 0.14 ± 0.23, respectively, p < 10−15; Figure S3). Moreover, the distance of the methylation loci within CpGIs from transcription start sites (TSS) predicted their level of methylation: the greater the distance, the greater the proportion of hypermethylated sites (Figure S4), consistent with a previous report.26 In contrast, methylation status in non-CpGI sites was not dependent on the distance from TSS. Finally, a relatively higher proportion of hypomethylated sites was observed in larger CpGIs than in small CpGIs (<1,000 bp; Figure S5).
DNA Methylation Is Age Dependent
In order to visualize global patterns of DNA methylation within the entire collection of samples, we used MDS27 and showed that the overall pattern of genome-wide methylation is highly age dependent (Figure 1). For autosomal loci, we observed a tight cluster of fetal samples, a progression of methylation levels in fetuses and children, and another cluster of subjects older than 10 years, indicating similarity of global methylation patterns within these age clusters and relative dissimilarity between the clusters (Figure 1B). Also, the fetal group dramatically stands out as the most dissimilar from all the other samples, reminiscent of our observations of the global prefrontal cortical transcriptome in an expanded cohort of subjects.4 The MDS analysis of methylation patterns for X chromosomal loci showed that, as expected, the samples were distinctly segregated by sex (Figure 1C). Within the sex groups, moreover, we observed further clustering by age; again, fetal samples formed separate clusters distinct from children (0–10 years), whereas children's methylation patterns diverged from the older individuals. These results confirm that both sex and age are important determinants of DNA methylation at CpG sites on the X chromosome.
Figure 1.
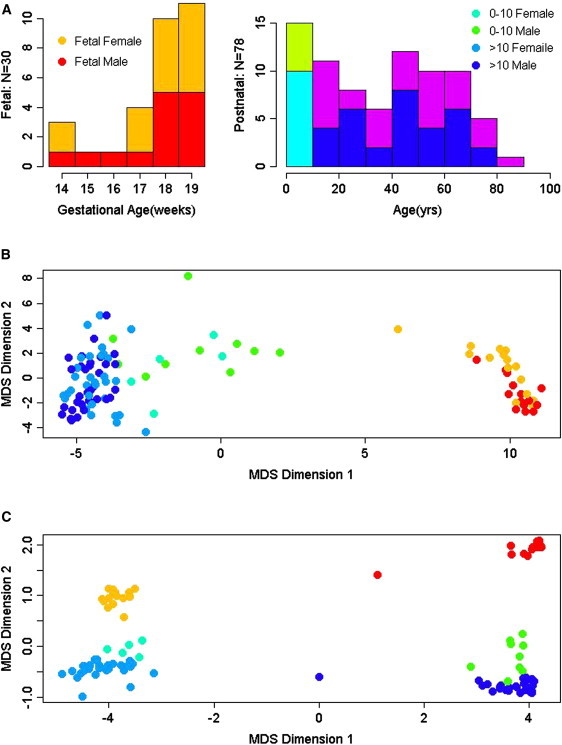
Depiction of Global DNA Methylation Patterns in Human Subjects across the Lifespan
(A) Histograms of subject ages in the brain collection. All fetal samples (left panel) are between 14 and 20 gestational weeks. The following color scale is used: red, male fetal samples (n = 14); orange, female fetal samples (n = 16); light blue, male children (n = 10); green, female children (n = 5); blue, males older than 10 years (n = 32); and purple, females older than 10 years (n = 31). The same color scale is used in (B and C).
(B) Global DNA methylation pattern examined by multidimensional scaling for autosomal CpG sites. Each DNA methylation sample is represented as a single point colored by the age of the subject. There is a tight cluster of the fetal samples, a progression of methylation levels from the fetal period to childhood, and another cluster for postchildhood.
(C) Global DNA methylation pattern of CpG sites on the X chromosome examined by multidimensional scaling. Each DNA methylation sample is represented as a single point colored by the age of the subject and sex. Samples were distinctly segregated by sex. A progression of methylation was also observed from the fetal period to postchildhood within each sex group.
DNA Methylation Changes Rapidly during the Fetal Period
In order for any investigator to conveniently explore DNA methylation patterns as a function of age, sex, and other demographic and genetic factors in our data, we have created a user friendly web interface (BrainCloudMethyl; see Subjects and Methods for details). We found that DNA methylation showed remarkable variation in the DLPFC across human life, as indicated by the rate of changes (Figure 2). By far, the fastest changes in DNA methylation occurred during fetal period with methylation levels dropping or increasing by almost 80% per year (with a 100% change reflecting complete demethylation or full methylation). During childhood and later in life methylation changed at a much slower pace (2−3 orders of magnitude slower, Figure 2B), mimicking again the DLPFC transcriptome data.4
Figure 2.
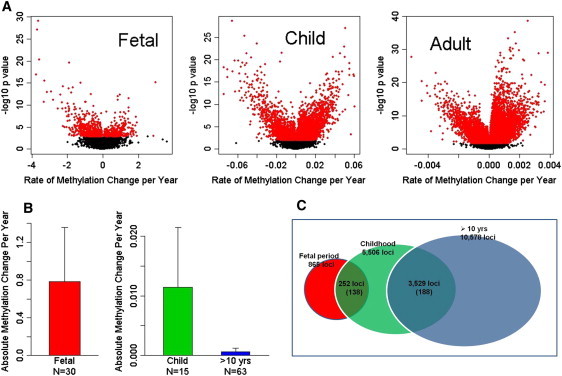
Depiction of Rates of DNA Methylation in Three Life Stages
(A) Rates of methylation change with age during three developmental life stages (the fetal period, childhood, and postchildhood/adult) at each CpG site for 27,578 CpG loci. A rate of change is plotted on the x axis. The −log10 p value is on the y axis, such that increasing values indicate more significant changes in methylation with age within a given stage. Red dots represent significant age-related methylation changes at FDR < 0.05. The fastest rates of methylation changes occurred during the fetal period, involving predominantly decreases in methylation (negative values on the x axis). In contrast, during the childhood and adulthood the changes were much slower and involved mainly increased methylation with aging (positive values on the x axis).
(B) Average rates of significant methylation changes with age within three developmental stages (red = the fetal period, green = childhood, blue = postchildhood). The absolute rate of the methylation change per year for the significant age-related CpG loci (FDR < 0.05) is on the y axis. The fastest changes in methylation occurred during the prenatal period, followed by childhood and postchildhood. The error bars represent standard deviations.
(C) Diagram showing the numbers of the CpG sites with significant age-related changes during the life stages. Of 27,578 CpG sites, significant age-related changes in DNA methylation (FDR < 0.05) were observed at 865 sites during the fetal period, 5,506 sites during childhood, and 10,578 sites during postchildhood. One hundred and thirty-eight sites out of the 252 sites that overlapped between the fetal period and the childhood and 188 sites out of the 3,529 sites that overlapped between children and postchildhood showed the opposite patterns of methylation (an increase followed by a decrease or vice versa) between these life stages. The transition from the fetal life to childhood is frequently associated with a reversal of direction in DNA methylation profile.
Of the 27,578 CpG sites, significant age-related changes in DNA methylation (FDR < 0.05) were observed at 865 sites during the fetal period (corresponding to 3.1% of all sites; 2.1% decreased and 1% increased), 5,506 sites during childhood (20%; 6.9% decreased and 13.1% increased), and 10,578 sites during postchildhood (38.3%; 12.5% decreased and 25.8% increased; Table S2). It is surprising that the overall methylation changes during the fetal period were much greater but involved fewer loci than in other life stages, though this could reflect in part the limited age span of the fetal samples and/or greater variance in the fetal period.
Consistent with earlier reports,10,28 CpG sites in genes involved in cancer and implicated in processes such as tumor suppression, telomere maintenance, and DNA repair, mostly increased methylation with aging. These genes included MGMT (MIM 156569), ESR1 (MIM 133430), RASSF1 (MIM 605082), RAD50 (MIM 604040), GSTP1/GTS3 (MIM 134660), RARB (MIM 180220), MYOD1 (MIM 159970), LAMB1 (MIM 150240), and the Werner gene WRN (MIM 604611), the latter gene associated with a premature aging syndrome (MIM 277700). Interestingly, changes in age-related methylation of many of these cancer-related genes started to occur during childhood and continued into old age (e.g., PRDM1 [MIM 603423], LOX [MIM 153455], BAP1 [MIM 603089], APC [MIM 611731], and TP53 [MIM 191170], known to be tumor suppressors, as well as some oncogenes; Table S3). Moreover, our data confirmed the ten top CpG sites with increased methylation levels during aging of adult subjects from a recent study of the brain.12
Transition from Fetal to Postnatal Age Is Associated with Reversal of Direction in DNA Methylation
We found that 252 CpG sites showed correlations with age during both the fetal period and childhood. Furthermore, 3,529 CpG sites were significantly correlated with age during both childhood and postchildhood. Interestingly, 138 of 252 sites (54.8%) that overlapped between the fetal period and childhood showed the opposite patterns between these life stages (a shift from a decrease to an increase of methylation with age or vice versa), whereas only 188 sites of 3,529 (5.3%) that overlapped between children and subjects older than 10 years demonstrated this pattern (Figure 2C). This indicates that the transition from fetal life to early childhood is associated with a reversal of direction in DNA methylation change at many loci, most often from demethylation prenatally to increased methylation postnatally. This pattern is again precisely the inverse of that which we have observed at the transcriptional level, where there is a preponderance of genes showing increases in expression with age in utero followed by decreases postnatally.4 DAVID functional annotation clustering analysis of the genes showing these shifts revealed significant enrichment (2- to 4-fold, corrected p < 0.05) for the terms “disulfide bond,” “glycoproteins,” and “signaling peptide,” characteristic of secreted proteins involved in cell-cell signaling and interactions with the extracellular matrix and known to play key roles in morphogenesis, cellular differentiation, angiogenesis, apoptosis, and modulation of the immune responses. Figure 3A shows examples of different patterns of age-related methylation.
Figure 3.
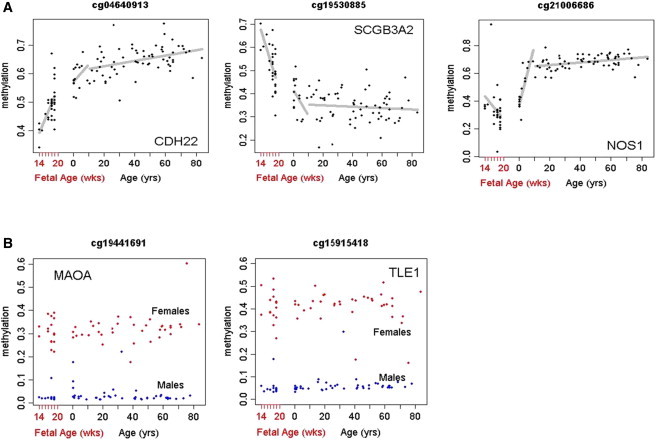
Depiction of Individual Loci's DNA Methylation Signatures across the Human Lifespan
(A) Depiction of individual loci's DNA methylation signatures across the human lifespan, illustrating three patterns of DNA methylation across the fetal, child and adult life periods. Methylation levels are on the y axis, with age on the x axis. The CpG site of CDH22 (cg04640913) showed significant age-related increases during the fetal period, childhood, and postchildhood (p = 2 × 10−6, p = 1 × 10−3, and p = 2.3 × 10−4, respectively). The CpG site of SCGB3A2 (cg19530885) showed significant age-related decreases during the fetal period, childhood, and postchildhood (p = 4.5 × 10−13, p = 9.5 × 10−3, and p = 2.4 × 10−4, respectively).The CpG site of NOS1 (cg21006686) showed a significant age-related decrease during the fetal period (p = 8.6 × 10−4) and increases during childhood and postchildhood (p = 2 × 10−8 and p = 1.1 × 10−2, respectively).
(B) Depiction of individual loci's DNA methylation signatures across the human lifespan, illustrating sex differences. Females are red and males are blue. The CpG sites of MAOA (cg19441691) and TLE1 (cg15915418) showed significant sex differences (p = 1.04 × 10−62 and 1.27 × 10−65, respectively).
Age-Related Changes in the Context of CpGI Location
To examine the effect of CpGI context on age-related changes, we divided significant correlations with age on the basis of location within CpGI and non-CpGI, Figure 4. We found that the effect sizes of age-related DNA methylation changes were larger at CpG sites in non-CpGIs than in CpGIs in all three life stages. This is in agreement with a rat study29 and the human adult brain study of Hernandez et al.,12 which has also found that the effect sizes of age-related effects were larger in non-CpGI sites, although this study reported also more age-related associations in CpGIs. The proportions of age-related changes in non-CpGIs and CpGIs were 5.6% and 2.2% during the fetal period, 28.1% and 16.9% during childhood, 47.6% and 34.9% during postchildhood (Fisher's exact test p = 4.1 × 10−42, p = 3.4 × 10−91, and p = 7.9 × 10−83, respectively). Moreover, those CpG sites in CpGIs that did show age-dependent changes were more likely to become more methylated with age than the sites in non-CpGIs, which were generally demethylated with aging, similar to the report of Christensen et al.10 The ratios between increased and decreased methylation at CpG sites in CpGIs versus non-CpGIs were, respectively, 2.4 and 1.3 during childhood, and 2.7 and 1.2 during postchildhood (two-proportion Z scores = 10 and 11, respectively, p < 10−15).
Figure 4.
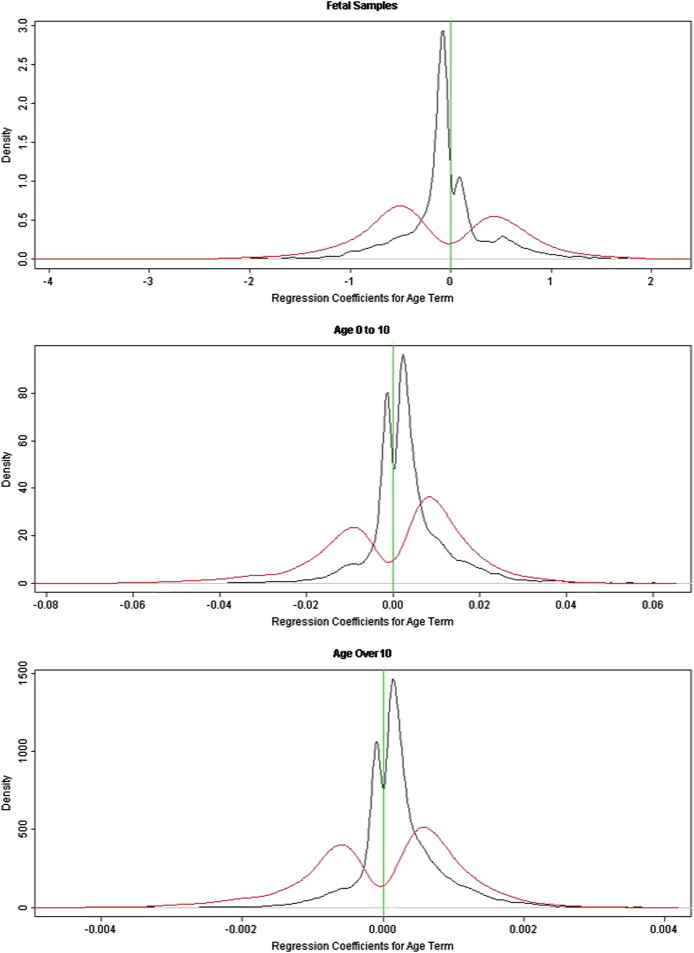
Depiction of Age-Related Changes in DNA Methylation in Three Life Stages as a Function of CpGI Context
Black lines show density distributions of regression coefficients for age term for the CpG loci located in the CpGIs. Red lines show density distribution of regression coefficients for age term for the CpG loci located outside the CpGIs. More loci are demethylated than hypermethylated during the fetal period, whereas increased methylation with age dominates in postnatal life (childhood and postchildhood). The effect sizes are larger for loci outside the CpGIs than those located in the CpGIs.
DNA Methylation Is Sex Biased
A majority of loci (85.8%) on the X chromosome showed a sexually dimorphic pattern of DNA methylation (of 932 CpG sites significantly different between the sexes, 678 showed higher methylation in females and 254 showed higher methylation in males), whereas only 5% of autosomal loci were significantly associated with sex (of 1,333 CpG sites, 986 showed higher methylation in females and 347 showed higher methylation in males, 5% Bonferroni correction, p < 1.8 × 10−6; Table S4). In agreement with a previous report in human blood,30 methylation of X chromosome CpG sites showed markedly larger sex differences than autosomal CpG sites, as indicated by the average absolute values of beta regression coefficients (0.21 and 0.015, respectively). Table 1 lists top autosomal genes that showed sex differences. Examples of sex-biased genes include MAOA (MIM 309850) on X chromosome and TLE1 (MIM 600189) on chromosome 9, a mammalian transcriptional corepressor participating in neuronal differentiation as a negative regulator in the central nervous system31 (Figure 3B). This gene is also associated with hypermethylation and gene silencing in hematologic malignancies.32
Table 1.
Top Ten Autosomal Genes with Most Significant Sex Differences
| CpG Locus | Chromosome | Gene Symbol | p Value (−log) | Difference |
|---|---|---|---|---|
| cg15915418 | 9 | TLE1 | 64.9 | female > male |
| cg07711515 | 9 | BAG1 | 60.8 | female < male |
| cg27063525 | 6 | C6orf68 | 46.7 | female > male |
| cg11673803 | 10 | GLUD1 | 44.3 | female > male |
| cg21243096 | 1 | POUF3F1 | 44.2 | female > male |
| cg04455759 | 11 | SDHD | 28.4 | female < male |
| cg08284151 | 12 | DPPA3 | 20.1 | female > male |
| cg05924191 | 15 | FLI20582 | 17.8 | female > male |
| cg23758485 | 16 | SMPD3 | 17.4 | female > male |
| cg07494248 | 2 | HSPD1 | 16.4 | female > male |
Comethylation Networks
To gain insight into the relationships among all 27,578 loci, we conducted WGCNA.25 The WGCNA is used here to find clusters (modules/groups) of highly correlated CpG sites, to relate modules to each other and to external sample traits (such as age and sex). Modules were created on the basis of high absolute correlations (i.e., they formed an unsigned comethylation network). Using the absolute values of the correlation coefficients between the profiles of CpG loci, we detected five modules, corresponding to the blocks of highly interconnected loci, which contained 10,422; 5,989; 825, 361, and 288 CpG loci in modules 1–5, respectively, Figure 5A. The remaining loci (n = 9,693) did not intercorrelate and thus were not included in the clusters. Module 3 clustered sex-biased CpG loci. Loci clustered in modules 2, 4, and 5 were highly correlated with age and showed module-specific patterns of age-related methylation (Figure S6). This is also clearly seen in the heat map of DNA methylation across all five modules (Figure 5B). To determine whether comethylation modules were biologically meaningful, we used functional enrichment and gene ontology analysis. DAVID functional annotation clustering revealed significant ∼2-fold enrichment of genes related to glycoproteins, glycosylation, disulfide bonds, and signaling in modules 2, 4, and 5 (Benjamini corrected p values = ×10−6 to ×10−30). Interestingly, the most numerous module 1 contained CpG sites, principally located in the CpGIs (92%), which were almost completely unmethylated (average methylation β = 0.033), and by and large showed no age dependence across the life span, suggesting that this module might represent sites involved in permissive transcription.
Figure 5.
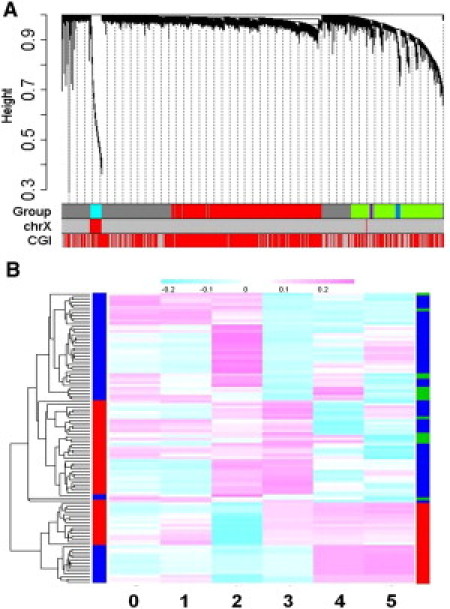
Depiction of Hierarchical Clustering with the Weighted Correlation Network Analysis
(A) Hierarchical clustering dendrogram obtained with the weighted correlation network analysis. The first color row underneath (labeled Group) shows the module assignment determined by the Dynamic Tree Cut. The second color row (labeled chrX) represents autosomal (gray) or X chromosomal (red) location of the loci in the module. The third row (labeled CpGI) shows whether the loci are located in the CpGIs (red) or outside the CpGIs (gray).
(B) Heat map visualization of DNA methylation status of six modules. A hierarchical clustering was conducted across 108 subjects shown in rows (a vertical color bar on the left indicates sex: red = females, blue = males; a vertical color bar on the right of the picture indicates age groups: red = fetal, green = childhood, blue = postchildhood) and six modules in columns (CpG loci were clustered into comethylation modules: groups 0–5, with group 0 containing unclassified loci). Color key on top indicates DNA methylation values: lowest, pink highest, blue lowest.
DNA Methylation Correlates with Expression of a Subset of Genes
Genome-wide transcriptional profiling was performed in the DLPFC in the same subjects with custom-spotted two-color microarrays.4 Expression data are available at the Gene Expression Omnibus (GEO) database. To interrogate relationships between DNA methylation and transcription, we correlated (Pearson's correlation) methylation status at CpG loci in gene promoters with expression of all probes on the array corresponding to the same gene (a total number of such correlations was 45,499 for 12,277 genes). Overall, across all CpG sites, there was poor correlation with expression as indicated by the preponderance of values near zero in the distribution of correlation coefficients across all comparisons, Figure 6A. Moreover, the distribution of correlation coefficients was almost completely symmetrical, indicating that methylation might suppress as well as activate transcription of a subset of genes that showed concordant changes in methylation and expression (3,363 significant correlations after Bonferroni correction, p < ×10−6, of which 1,774 [52.7%] were negative; Table S5). Importantly, CpGI context appeared to play a role in predicting gene expression as there was an over 2-fold higher proportion of significant correlations involving sites outside CpGI than of significant correlations involving those sites in CpGIs (out of all correlations with loci outside CpGIs 12.6% were significant at p < ×10−6, whereas only 5.7% of loci in CpGIs were significantly correlated). Of particular interest is NNAT (neuronatin [MIM 603106]), an imprinted gene on chromosome 20, with the second highest negative correlation between DNA methylation and expression (r = −0.922, p = 1.3 × 10−45). NNAT is expressed only from the paternal allele, is involved in the regulation of ion channels during early brain development, and has been implicated in pituitary cancers by means of loss of expression due to hypermethylation at the promoter CpG sites.33 Our data suggest that promoter DNA methylation of NNAT might also be important in the transcriptional regulation of NNAT during normal PFC development because its expression falls dramatically during the transition from the fetal period to infancy and slowly thereafter, whereas DNA methylation follows exactly the opposite pattern (Figure 6B).
Figure 6.
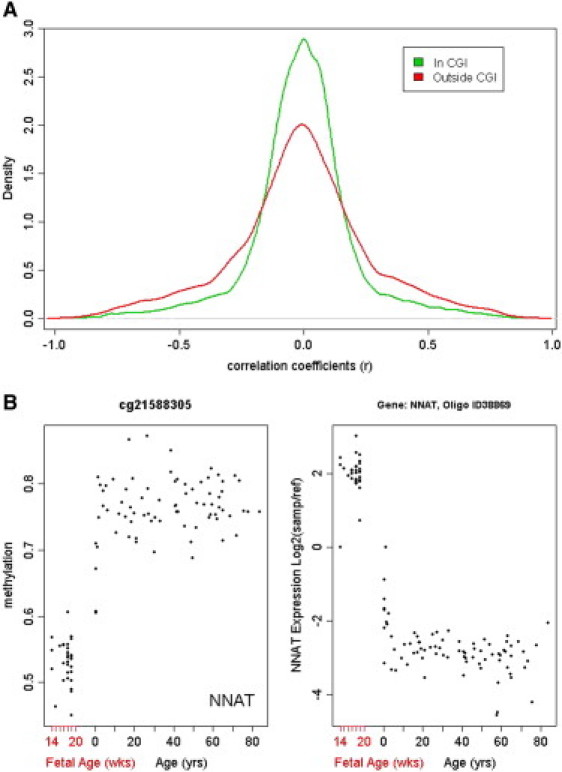
Depiction of Correlations between DNA Methylation and Gene Expression
(A) Histogram of correlation coefficients for Pearson's correlations between DNA methylation and gene expression. Green line shows correlations of expression with methylation at the loci located in CpGIs, red line, with loci outside CpGIs.
(B) An example of opposite patterns of DNA methylation (left) and expression (right) changes with age (x axis) for NNAT (neuronatin).
Remarkably, transcription of a group of methyltransferases of the DNMT3 family, carrying out dramatic reprogramming of DNA methylation during early embryonic development, appears to be, at least in part, also regulated by DNA methylation. Methylation levels at the CpG loci in the promoters of DNMT3A (MIM 602769) and DNMT3B (MIM 602900) as well as DNMT3L (MIM 606588), which lacks the catalytic domain but binds to DNMT3A and DNMT3B variants and facilitates their chromatin targeting, are significantly and inversely correlated with the expression of these genes (r values from −0.66 to −0.46, p values from 5.8 × 10−15 to 3.8 × 10−7). Consistent with their essential role in the establishment of early DNA methylation patterns, expression levels of DNMT3A, DNMT3B, and DNMT3L variants are high during the fetal period and drop by an approximately 2- to 4-fold decrease by birth, whereas methylation follows the opposite trajectory (Figure S7). Our data suggest that genes controlling de novo DNA methylation are themselves subjected to epigenetic regulation and that this process continues well into childhood years.
Methylation Quantitative Trait Loci (mQTL) Analysis
To identify population clusters and potential population outliers, we used IBS analysis across all genotypes and subjects and found that the two races (individuals of European descent and African Americans) formed clearly distinct clusters (Figure S8). We identified one outlier among individuals of European descent, who was deleted from mQTL analysis. We conducted genome-wide association analysis of SNPs with DNA methylation and found a large number of significant mQTLs. Because mQTL analysis was performed with residuals from multiple regression with sex, age, stage, age-by-stage interaction, and race, these effects would not be expected to affect the SNP association results. Quantile-quantile (Q-Q) plots clearly demonstrate the abundance of extreme association effects over that expected by chance in all subjects as well as separately in each race cohort (Figures S9A and S9B). In the cis-analysis (cis defined as within 1 Mb of the CpG site), 2,836 SNP genotype-CpG pairs were significantly correlated (FDR < 0.05; Table S6; for top associations see Table 2). HLA-DQB1 (MIM 604305), with mQTL p value 10−29 (Figure 7A), is a highly polymorphic gene coding a major histocompatibility complex class II protein, which has been implicated in a host of autoimmune diseases, including insulin-dependent diabetes mellitus (MIM 222100), gliomas, multiple sclerosis (MIM 126200), schizophrenia, and other disorders.34,35
Table 2.
Top Ten Most Significant cis mQTLs
| CpG Locus | CpG Gene Symbol | SNP Symbol | SNP Gene Symbol | Chromosome | p Value (−log) |
|---|---|---|---|---|---|
| cg17749961 | LCLAT1 | rs1662955 | LCLAT1 | 2 | 39.9 |
| cg17749961 | LCLAT1 | rs1723167 | LCLAT1 | 2 | 36.4 |
| cg19766460 | C21orf128 | rs1571737 | UMODL1 | 21 | 36.1 |
| cg17749961 | LCLAT1 | rs1612616 | LCLAT1 | 2 | 35.8 |
| cg01889448 | HLA-DQB1 | rs1063355 | HLA-DQB1 | 6 | 29.4 |
| cg12339802 | C1orf109 | rs11264091 | EPHA10 | 1 | 28.7 |
| cg06665322 | GPA33 | rs2281962 | GPA33 | 1 | 28.3 |
| cg14129786 | MGMT | rs7076950 | MGMT | 10 | 27.0 |
| cg06509940 | CD80 | rs4330287 | ADPRH | 3 | 26.9 |
| cg08679985 | KLF17 | rs663818 | KLF17 | 1 | 26.8 |
Figure 7.
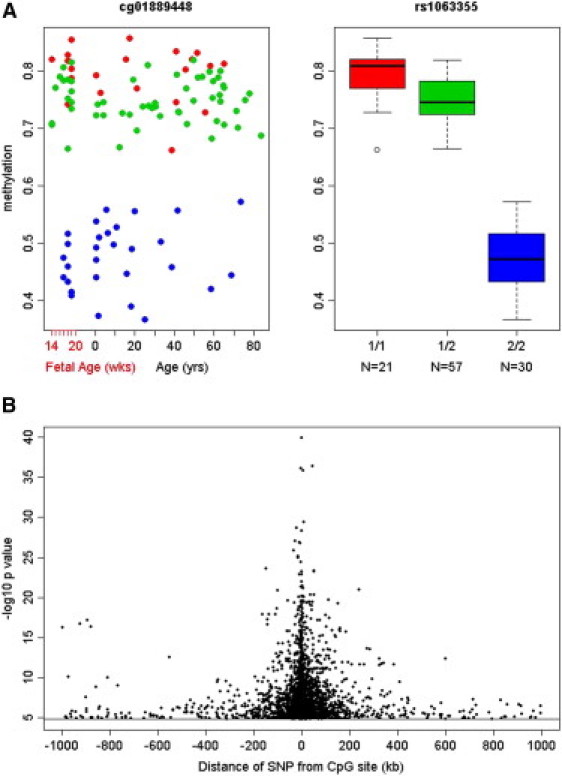
Depiction of Associations between the Genotype and DNA Methylation
(A) Depiction of the mQTL for the CpG site cg01889448 and rs1063355 in HLA-DQB1 (p = 10−29). Three genotypes of rs1063355 are 1/1 in red (n = 21), 1/2 in green (n = 57), and 2/2 in blue (n = 30). The lines in the bar graph indicate the median, and the bars (whiskers) represent the minimum and maximum of the data after the removal of the outliers.
(B) The relationship between the strength of association and the distance of the SNP from the CpG site. Only significant cis-SNPs associations are plotted (FDR < 0.05). The distance of SNPs from the CpG sites in base pairs is on the x axis and the −log10 p values of SNP association with DNA methylation. Plotting p values against the distance of SNPs from each CpG site showed that the highest associations were with the SNPs located close to CpG site.
Plotting p values against the distance of SNPs from the CpG sites showed that the strongest associations were with the SNPs located relatively close to CpG sites, but the association signal extended at least up to 300 kb and in some cases as far as 1 Mb from the gene (Figure 7B). The average distance between significantly associated cis-SNPs and CpG sites was 86.2 kb. One hundred and sixty-five SNPs were associated with two or more different CpG sites. We found 401 trans-mQTLs (FDR < 0.05; Table S7). Table 3 lists the top ten trans-SNP-CpG association pairs with the most significant p values. To address the possibility that the highly significant association data were driven by population stratification, the association analysis was also performed in samples from African American and individuals of European descent separately. The top 200 African American cis-associations and 91.5% of the top 200 individuals of European descent cis-associations were also found to be significant in the analysis of all subjects (FDR < 0.05; Tables S8A and S8B). Finally, it should be noted again that the reported significant associations are, by design, not age (or stage) dependent because age and developmental stage were used to generate residuals for mQTLs. This is further illustrated in the examples of the genotypic groups at highly associated SNPs, showing consistently different methylation across the lifespan (Figure S10). Overall, our results revealed that DNA methylation is associated with genetic variance in numerous CpG sites, and cis-SNP associations are more frequent and more pronounced than trans-SNP associations. Further exploration of methylation profiles for individual genes across the lifespan and the genetic associations of those genes is made possible by an easily accessible stand-alone application, BrainCloudMethyl. This will enable additional discovery that might have profound consequences for our understanding of molecular mechanisms by which risk-associated SNPs exert their pathological effects in brain disorders.
Table 3.
Top Ten Most Significant trans mQTLs
| CpG Locus | CpG Gene Symbol | SNP Symbol | SNP Gene Symbol | p Value (−log) |
|---|---|---|---|---|
| cg18984499 | RPL26 | rs11847580 | C14orf72 | 24.1 |
| cg17704839 | UBL5 | rs733675 | RHOT1 | 23.5 |
| cg18634211 | LIN28 | rs2288322 | FKBP7 | 21.5 |
| cg18634211 | LIN28 | rs10207436 | PRKRA | 21.4 |
| cg25299176 | YWHAE | rs4281963 | LOC647002 | 19.7 |
| cg03923277 | TDG | rs326387 | TMEM132B | 19.5 |
| cg25299176 | YWHAE | rs6716175 | LOC440917 | 18.8 |
| cg18984499 | RPL26 | rs4906142 | PPP2R5C | 18.5 |
| cg13514129 | MACF1 | rs12130070 | SMYD3 | 18.4 |
| cg13514129 | MACF1 | rs2878079 | SMYD3 | 18.4 |
Discussion
We report a genome-wide study of age-related DNA methylation, focused on 5′ promoter regions of genes, in the developing human DLPFC, including the prenatal period. The fastest changes in methylation occurred during the prenatal period, and the transition from fetal life to early childhood was associated with a reversal of direction of DNA methylation, typically from demethylation to increased methylation. As demonstrated before, variations in methylation during aging exist even between monozygotic twins, that is independently of the genetic sequence,36,37 and are associated with phenotypic discordances between the twins, including disease traits.38–40 Although the causes of age-related methylation changes remain to be determined, increases in methylation with age are thought to reflect the accumulation of stochastic methylation events over time (epigenetic drift), whereas decreases might be related to altered fidelity of DNA methyltransferases.41 This suggests that epigenetic factors might play an important role in the pathogenesis of diseases and implies that examining epigenetic mechanisms associated with aging and contributing to the accumulation of methylation errors might be essential to understand pathogenesis of age-related brain disorders.
Consistent with the findings in tissue differentiation and cancer,13 we found that age-related DNA methylation changes were more likely to occur outside the so-called CpGIs than in CpGIs. This trend could be explained by specific protection mechanisms of CpGIs from de novo methylation.42,43 Moreover, those CpG sites in CpGIs that showed age-dependent changes were more likely to gain rather than loose DNA methylation with aging in accordance with Christensen et al.10 and Hernandez et al.12 who interrogated cancer-related autosomal loci in human tissues and in adult human brains, respectively. Our results of differential age-related patterns between CpGIs and non-CpGIs across more than 27,000 loci primarily in 5′ promoter regions suggest that global methylation changes within these regions can be affected by promoter CpG density during development. A recent paper reporting that most tissue-specific DNA methylation occurs not in CpGIs but in CpGI shores, which are located within 2 kb of islands,13 might support this notion.
Neurodevelopmental behavioral disorders, such as autism and schizophrenia, are thought to have their origins in utero or around birth, although the main symptoms emerge later in life, typically around adolescence/young adulthood in the case of schizophrenia.44 Our results showing remarkable DNA methylation changes, especially in early life stages, suggest that altered methylation during these periods might be critical for the pathogenesis of developmental brain disorders. Consistently, we found dynamic age-related methylation changes across the life span, and particularly in the transition from fetal to postnatal life stage, for genes, such as DLG4 (MIM 602887), DRD2 (MIM 126450), NOS1 (MIM 163731), NRXN1 (MIM 600565), and SOX10 (MIM 602229), that have been implicated in schizophrenia and autism (Figure S11). DNMT1 (MIM 126375), a maintenance DNA methyltransferase, is overexpressed in the cerebral cortex of patients with schizophrenia and bipolar disorder (MIM 125480).45 It is possible that aberrant DNA methylation in schizophrenia might be a consequence of altered normal developmental trajectories triggered either by dysregulation of methyltransferase activity and/or the involvement of environmental genetic factors affecting DNA methylation status. DNA methyltransferases participating in de novo methylation also might be involved (e.g., DNMT3A, DNMT3B, and DNMT3L). DNMT3A, shown to form complexes and work in concert with DNMT3B and DNMT3L, can specifically regulate expression of neurogenic genes and affect synaptic plasticity, learning, and memory in forebrain excitatory neurons.46–48 Consistent with this notion, the subset of our transcriptome data, which correlate inversely with DNA methylation levels, show dramatic age-related changes in the DLPFC across the lifespan, particularly during the transition from the fetal to the postnatal period, suggesting dynamic regulation of expression of these genes by DNA methylation during development.
This genome-wide study also identifies sex-biased autosomal genes in DNA methylation in normal human DLPFC and thus might be useful to evaluate sex differences in various brain disorders. Interestingly, five of the ten most significant associations with sex among autosomal genes (C6orf68/NUS1 [MIM 610463], DPPA3 [MIM 608408], FLJ20852, TLE1, and GLUD1 [MIM 138130], which encodes an enzyme central to glutamate metabolism and implicated in schizophrenia and cognition49,50) were also identified in a previous study that used saliva,14 suggesting that at least some epigenetic differences between sexes are not tissue specific.
Our data also revealed, consistent with previous reports11,23 that DNA methylation at numerous CpG sites is associated with genetic variance. Not surprisingly, we found that cis-SNP associations are more frequent and more pronounced than trans-SNP associations. Six of our ten most significant cis-SNP-CpG association pairs and eight of our ten most significant trans-SNP-CpG association pairs were reported by Gibbs et al.11 Our findings of SNP associations with DNA methylation are consistent with the possibility that some SNPs associated with increased risk for neuropsychiatric diseases might affect gene expression through DNA methylation, in addition to their direct effects on mRNA expression.51 Finally, despite similarities in the global patterns of transcription and DNA methylation in the DLPFC over the human lifespan, we found a relatively limited number of intercorrelated CpG site-expression probe pairs. This confirms earlier evidence that transcription is only partially regulated by promoter DNA methylation at the CpG sites examined in the study and/or that the limited coverage of transcripts (especially of alternative splice variants that might be differentially regulated by methylation) and of CpG sites (in particular those outside the CpGIs) precludes fully capturing these relationships. Another limitation is our inability to distinguish between methylation and 5-hydroxymethylation of cytosine (5hmC) that appears to be abundant in brain and has recently been discovered to play a critical role in dynamic regulation of genes silenced by methylation.52 Finally, it is also possible that recent advances in the knowledge about gene structure will require re-evaluation of the putative promoter regions. Further studies will be needed to reveal how genetic and epigenetic variation, together and independently of each other, are involved in the pathophysiology of neuropsychiatric diseases.
Acknowledgments
We thank Liqin Wang and Vesna Imamovic for their technical expertise; Mary Herman for her contribution to the Clinical Brain Disorders Branch/National Institute of Mental Health brain collection; Abdel Elkahloun at National Human Genome Research Institute Microarray Core Facility for scanning data; and Ronald Zielke, Robert Johnson, and Robert Vigorito at the NICHD Brain and Tissue Bank for Developmental Disorders, University of Maryland School of Medicine, for their collection of brains. We also thank the families of the deceased for the donations of brain tissue and their time and effort devoted to the consent process and interviews. This research was supported by the Intramural Research Program of the National Institute of Mental Health at the National Institutes of Health.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BrainCloudMethyl, http://BrainCloud.jhmi.edu
GEO database, http://www.ncbi.nlm.nih.gov/geo
References
- 1.Bystron I., Blakemore C., Rakic P. Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 2008;9:110–122. doi: 10.1038/nrn2252. [DOI] [PubMed] [Google Scholar]
- 2.Hill R.S., Walsh C.A. Molecular insights into human brain evolution. Nature. 2005;437:64–67. doi: 10.1038/nature04103. [DOI] [PubMed] [Google Scholar]
- 3.Johnson M.B., Kawasawa Y.I., Mason C.E., Krsnik Z., Coppola G., Bogdanović D., Geschwind D.H., Mane S.M., State M.W., Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colantuoni C., Lipska B.K., Ye T., Hyde T.M., Tao R., Leek J.T., Colantuoni E.A., Elkahloun A.G., Herman M.M., Daniel R., Weinberger, Kleinman J.E. Temporal dynamics and genetic control of the human neocortical transcriptome across the lifespan. Nature. 2011;478:519–524. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geiman T.M., Muegge K. DNA methylation in early development. Mol. Reprod. Dev. 2010;77:105–113. doi: 10.1002/mrd.21118. [DOI] [PubMed] [Google Scholar]
- 7.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 8.Connor C.M., Akbarian S. DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics. 2008;3:55–58. doi: 10.4161/epi.3.2.5938. [DOI] [PubMed] [Google Scholar]
- 9.Mill J., Tang T., Kaminsky Z., Khare T., Yazdanpanah S., Bouchard L., Jia P., Assadzadeh A., Flanagan J., Schumacher A. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen B.C., Houseman E.A., Marsit C.J., Zheng S., Wrensch M.R., Wiemels J.L., Nelson H.H., Karagas M.R., Padbury J.F., Bueno R. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibbs J.R., van der Brug M.P., Hernandez D.G., Traynor B.J., Nalls M.A., Lai S.L., Arepalli S., Dillman A., Rafferty I.P., Troncoso J. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez D.G., Nalls M.A., Gibbs J.R., Arepalli S., van der Brug M., Chong S., Moore M., Longo D.L., Cookson M.R., Traynor B.J., Singleton A.B. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irizarry R.A., Ladd-Acosta C., Wen B., Wu Z., Montano C., Onyango P., Cui H., Gabo K., Rongione M., Webster M. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J., Morgan M., Hutchison K., Calhoun V.D. A study of the influence of sex on genome wide methylation. PLoS ONE. 2010;5:e10028. doi: 10.1371/journal.pone.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rakyan V.K., Down T.A., Maslau S., Andrew T., Yang T.P., Beyan H., Whittaker P., McCann O.T., Finer S., Valdes A.M. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teschendorff A.E., Menon U., Gentry-Maharaj A., Ramus S.J., Weisenberger D.J., Shen H., Campan M., Noushmehr H., Bell C.G., Maxwell A.P. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xin Y., Chanrion B., Liu M.M., Galfalvy H., Costa R., Ilievski B., Rosoklija G., Arango V., Dwork A.J., Mann J.J. Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS ONE. 2010;5:e11357. doi: 10.1371/journal.pone.0011357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipska B.K., Deep-Soboslay A., Shannon Weickert C., Hyde T.M., Martin C.E., Herman M.M., Kleinman J.M. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol. Psychiatry. 2006;60:650–658. doi: 10.1016/j.biopsych.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 19.Bibikova M., Le J., Barnes B., Saedinia-Melnyk S., Zhou L., Shen R., Gunderson K.L. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 20.Leek J.T., Storey J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc., B. 1995;57:289–300. [Google Scholar]
- 22.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang D., Cheng L., Badner J.A., Chen C., Chen Q., Luo W., Craig D.W., Redman M., Gershon E.S., Liu C. Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 2010;86:411–419. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 25.Langfelder P., Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straussman R., Nejman D., Roberts D., Steinfeld I., Blum B., Benvenisty N., Simon I., Yakhini Z., Cedar H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009;16:564–571. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 27.Cox T.F., Cox M.A.A. Chapman and Hall; London: 1994. Multidimensional scaling. 213. [Google Scholar]
- 28.Kwabi-Addo B., Chung W., Shen L., Ittmann M., Wheeler T., Jelinek J., Issa J.P. Age-related DNA methylation changes in normal human prostate tissues. Clin. Cancer Res. 2007;13:3796–3802. doi: 10.1158/1078-0432.CCR-07-0085. [DOI] [PubMed] [Google Scholar]
- 29.Thompson R.F., Atzmon G., Gheorghe C., Liang H.Q., Lowes C., Greally J.M., Barzilai N. Tissue-specific dysregulation of DNA methylation in aging. Aging Cell. 2010;9:506–518. doi: 10.1111/j.1474-9726.2010.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cotton A.M., Avila L., Penaherrera M.S., Affleck J.G., Robinson W.P., Brown C.J. Inactive X chromosome-specific reduction in placental DNA methylation. Hum. Mol. Genet. 2009;18:3544–3552. doi: 10.1093/hmg/ddp299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buscarlet M., Perin A., Laing A., Brickman J.M., Stifani S. Inhibition of cortical neuron differentiation by Groucho/TLE1 requires interaction with WRPW, but not Eh1, repressor peptides. J. Biol. Chem. 2008;283:24881–24888. doi: 10.1074/jbc.M800722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fraga M.F., Berdasco M., Ballestar E., Ropero S., Lopez-Nieva P., Lopez-Serra L., Martín-Subero J.I., Calasanz M.J., Lopez de Silanes I., Setien F. Epigenetic inactivation of the Groucho homologue gene TLE1 in hematologic malignancies. Cancer Res. 2008;68:4116–4122. doi: 10.1158/0008-5472.CAN-08-0085. [DOI] [PubMed] [Google Scholar]
- 33.Revill K., Dudley K.J., Clayton R.N., McNicol A.M., Farrell W.E. Loss of neuronatin expression is associated with promoter hypermethylation in pituitary adenoma. Endocr. Relat. Cancer. 2009;16:537–548. doi: 10.1677/ERC-09-0008. [DOI] [PubMed] [Google Scholar]
- 34.Dyment D.A., Ebers G.C., Sadovnick A.D. Genetics of multiple sclerosis. Lancet Neurol. 2004;3:104–110. doi: 10.1016/s1474-4422(03)00663-x. [DOI] [PubMed] [Google Scholar]
- 35.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P., International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fraga M.F., Ballestar E., Paz M.F., Ropero S., Setien F., Ballestar M.L., Heine-Suñer D., Cigudosa J.C., Urioste M., Benitez J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaminsky Z.A., Tang T., Wang S.C., Ptak C., Oh G.H., Wong A.H., Feldcamp L.A., Virtanen C., Halfvarson J., Tysk C. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009;41:240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 38.Javierre B.M., Fernandez A.F., Richter J., Al-Shahrour F., Martin-Subero J.I., Rodriguez-Ubreva J., Berdasco M., Fraga M.F., O'Hanlon T.P., Rider L.G. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuratomi G., Iwamoto K., Bundo M., Kusumi I., Kato N., Iwata N., Ozaki N., Kato T. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol. Psychiatry. 2008;13:429–441. doi: 10.1038/sj.mp.4002001. [DOI] [PubMed] [Google Scholar]
- 40.Mastroeni D., McKee A., Grover A., Rogers J., Coleman P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease. PLoS ONE. 2009;4:e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu S.C., Zhang Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brandeis M., Frank D., Keshet I., Siegfried Z., Mendelsohn M., Nemes A., Temper V., Razin A., Cedar H. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371:435–438. doi: 10.1038/371435a0. [DOI] [PubMed] [Google Scholar]
- 43.Ushijima T., Watanabe N., Okochi E., Kaneda A., Sugimura T., Miyamoto K. Fidelity of the methylation pattern and its variation in the genome. Genome Res. 2003;13:868–874. doi: 10.1101/gr.969603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinberger D.R., Levitt P. Third Edition. Wiley; Oxford: 2010. The neurodevelopmental origins of schizophrenia. Schizophrenia. [Google Scholar]
- 45.Veldic M., Guidotti A., Maloku E., Davis J.M., Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc. Natl. Acad. Sci. USA. 2005;102:2152–2157. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu H., Coskun V., Tao J., Xie W., Ge W., Yoshikawa K., Li E., Zhang Y., Sun Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng J., Zhou Y., Campbell S.L., Le T., Li E., Sweatt J.D., Silva A.J., Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LaPlant Q., Vialou V., Covington H.E., 3rd, Dumitriu D., Feng J., Warren B.L., Maze I., Dietz D.M., Watts E.L., Iñiguez S.D. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia P., Wang L., Meltzer H.Y., Zhao Z. Common variants conferring risk of schizophrenia: A pathway analysis of GWAS data. Schizophr. Res. 2010;122:38–42. doi: 10.1016/j.schres.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Need A.C., Keefe R.S., Ge D., Grossman I., Dickson S., McEvoy J.P., Goldstein D.B. Pharmacogenetics of antipsychotic response in the CATIE trial: A candidate gene analysis. Eur. J. Hum. Genet. 2009;17:946–957. doi: 10.1038/ejhg.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinman J.E., Law A.J., Lipska B.K., Hyde T.M., Ellis J.K., Harrison P.J., Weinberger D.R. Genetic neuropathology of schizophrenia: New approaches to an old question and new uses for postmortem human brains. Biol. Psychiatry. 2011;69:140–145. doi: 10.1016/j.biopsych.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhutani N., Burns D.M., Blau H.M. DNA methylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.