

Abstract
Congenital mirror movements (CMM) are characterized by involuntary movements of one side of the body that mirror intentional movements on the opposite side. CMM reflect dysfunctions and structural abnormalities of the motor network and are mainly inherited in an autosomal-dominant fashion. Recently, heterozygous mutations in DCC, the gene encoding the receptor for netrin 1 and involved in the guidance of developing axons toward the midline, have been identified but CMM are genetically heterogeneous. By combining genome-wide linkage analysis and exome sequencing, we identified heterozygous mutations introducing premature termination codons in RAD51 in two families with CMM. RAD51 mRNA was significantly downregulated in individuals with CMM resulting from the degradation of the mutated mRNA by nonsense-mediated decay. RAD51 was specifically present in the developing mouse cortex and, more particularly, in a subpopulation of corticospinal axons at the pyramidal decussation. The identification of mutations in RAD51, known for its key role in the repair of DNA double-strand breaks through homologous recombination, in individuals with CMM reveals a totally unexpected role of RAD51 in neurodevelopment. These findings open a new field of investigation for researchers attempting to unravel the molecular pathways underlying bimanual motor control in humans.
Main Text
Mirror movements (MM) are involuntary movements of one side of the body that mirror intentional movements on the opposite side. Mild MM are physiological in young children and gradually disappear within the first decade of life probably because of the maturation of the motor network.1 Congenital mirror movements (CMM [MIM 157600]) persisting after age 10 in subjects with no other clinical feature constitute a rare disorder that is mainly inherited in an autosomal-dominant fashion although sporadic cases also exist. MM predominate in the upper limbs, with muscles controlling the fingers and hands being constantly involved, and their intensity increases with the complexity of the voluntary movement. MM impair the ability to perform tasks requiring skilled bimanual coordination and are associated with pain in the upper limbs during sustained manual activities. In this setting, MM reflect multiple dysfunctions and structural abnormalities of the motor network, including altered decussation of the corticospinal tracts.2
Recently, heterozygous mutations in DCC (deleted in colorectal carcinoma [MIM 120470]), the gene encoding the receptor for netrin 1 (NTN1 [MIM 601614]), have been identified in families with autosomal-dominant CMM.3,4 Impairment of DCC/netrin 1 signaling, which promotes attraction and guidance of developing axons toward the midline, results in alterations of axonal fiber crossing and abnormal ipsilateral connections.3,4 MM are genetically heterogeneous, however; no DCC mutations have been identified in several familial and sporadic cases.4,5
We have previously ruled out DCC mutations in a French family (Family A) with autosomal-dominant MM.4 Written informed consent was obtained from all patients before blood sampling. The study received approval from ethical standards committee on human experimentation (INSERM, CHU Pitié-Salpêtrière). Genome-wide linkage analysis with SNP microarrays followed by genotyping with 92 microsatellite markers on uninformative or positive regions in this family identified a single linked region with a maximal multipoint LOD score value (+2.4) in chromosome region 15q14-q21.2 (Figure 1A). A common haplotype, delimited by markers D15S102 and D15S982 and encompassing a 14.4 Mb region, segregated in all eight affected family members and in eight asymptomatic relatives, including three obligate carriers. The multipoint LOD score, recalculated from microsatellite marker genotypes and including all eight affected members, reached +2.7, its maximal expected value in view of the pedigree structure (Figure 1B). The region contained 223 known genes, but only one, SEMA6D (MIM 609295), was potentially involved in neuronal migration. Direct sequencing of its coding sequence detected no mutations in Family A or in a second family (from Germany) with two affected members (Family B). In addition, analysis of the Family A proband by high-resolution CGH array (Nimblegen HD-2 microarrays, Roche) revealed no microdeletion or duplication within the candidate interval.
Figure 1.
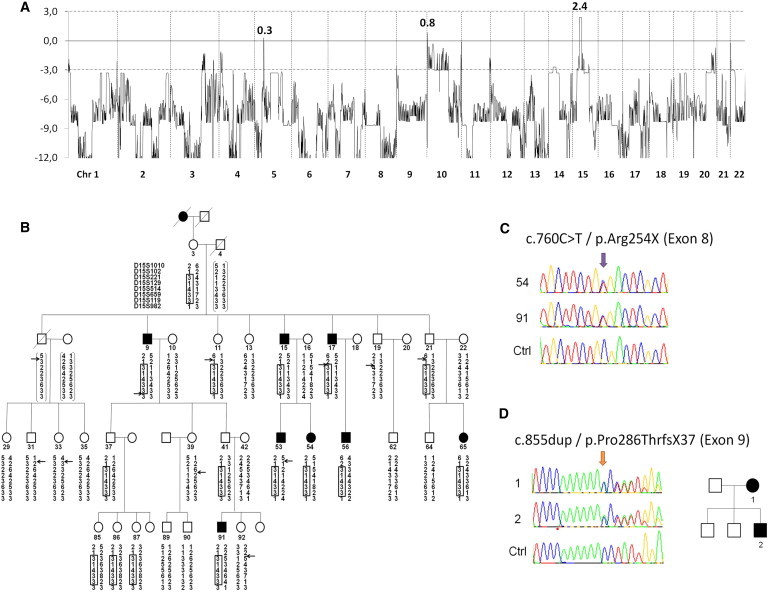
Identification of RAD51 Mutations in Two Independent Families
(A) LOD score plot for genome-wide linkage analysis in Family A, revealing a single locus with a maximal multipoint LOD score value on chromosome arm 15q (Z = +2.4). Twenty-six family members (7 symptomatic individuals, 3 obligate carriers, 12 at-risk asymptomatic relatives, and 4 spouses) were genotyped by linkage-24 microarrays (Illumina). Multipoint LOD scores were calculated with Merlin 1.0 (affected-only analysis, autosomal-dominant trait, disease allele frequency of 0.00001, penetrance of 80%, null phenocopy rate). All regions with LOD scores >−2 other than that on chromosome 15 were further analyzed with microsatellite markers and excluded on the basis of the absence of a common haplotype in all affected family members.
(B) Refinement of the chromosome 15 interval with eight microsatellite markers (D15S1010, D15S102, D15S221, D15S129, D15S514, D15S659, D15S119, D15S982) showing a common haplotype segregating in all affected family members. Multipoint LOD scores were recalculated from the microsatellite markers in the chromosome 15 interval via Allegro 1.0 with the same parameters as those previously used (Zmax = +2.7).
(C) Confirmation of the c.760C>T (p.Arg254∗) mutation in RAD51 by Sanger sequencing in Family A.
(D) The coding region of RAD51 was amplified with 11 primer pairs (sequences available on request) in the index cases of Families B (from Germany) and C (from UK). The c.855dupA (p.Pro286Thrfs∗37) mutation in RAD51 was identified in Family B (pedigree).
We then sequenced the entire exome in two affected members of Family A (individuals 54 and 91). A total of 32,390 and 33,648 variants were identified in each subject (Table S2 available online). Further analysis focused on the eight variants (five SNPs and three indels) that were (1) heterozygous in the two affected subjects, (2) absent from the dbSNP database (version 132), and (3) contained in the linked interval (Table 1). Seven variants were confirmed by Sanger sequencing, four of which were also found in control individuals. The three remaining variants included two intronic substitutions predicted to have no effect on splicing, and a nonsense mutation (c.760C>T [p.Arg254∗], RefSeq accession number NM_002875) in exon 8 of RAD51 (MIM 179617). This nonsense mutation cosegregated with MM in Family A (Figure 1C) and was absent from 644 healthy unrelated European individuals.
Table 1.
List of the Variants Detected by Whole-Exome Analysis that Are Present in Patients 54 and 91 from Family A, Located in the Chromosome 15 Interval, and Absent from the dbSNP Database
| Position on chr15 (in bp) | Gene | RefSeq Accession Number/MIM Number | Exon/Intron | Nucleotide Change | Protein Change | Type | Status in 54 | Status in 91 | Confirmation by Sangera | In Silico Predictionb | Presence in Controlsc | Expression in Brain |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 38543560 | BAHD1 | NM_014952/613880 | intron 4 | c.1975+49A>C | - | intronic | htz | htz | yes | NE | 0/321 | yes |
| 38809110 | RAD51 | NM_002875/179617 | exon 8 | c.760C>T | p.Arg254∗ | nonsense | htz | htz | yes | - | 0/644 | yes |
| 38817215 | FAM82A2 | NM_018145/611873 | exon 10 | c.1127T>G | p.Val376Gly | missense | htz | htz | no | - | - | - |
| 40164933 | PLA2G4D | NM_178034/612864 | intron 5 | c.428+45_ 428+48dup | - | intronic | htz | htz | yes | NE | 21/92 | yes |
| 40167192 | PLA2G4D | NM_178034/612864 | intron 1 | c.46-2del | - | intronic | htz | htz | yes | Decreases the score of the acceptor splice site | 4/356 | yes |
| 40221690 | PLA2G4F | NM_213600/- | exon | c.2334G>C | p.Val778Val | synonymous | htz | htz | yes | NE | 2/93 | yes |
| 40241144 | VPS39 | NM_015289/612188 | intron 24 | c.2552+28del | - | intronic | htz | htz | yes | NE | 21/91 | yes |
| 40481666 | CAPN3 | NM_000070/114240 | intron 12 | c.1536+41C>T | - | intronic | htz | htz | yes | NE | 0/334 | yes |
Genomic positions were based on the the NCBI36/hg18 version of the Human genome. Abbreviations: htz, heterozygous; NE, no major effect on splicing.
The eight variants were confirmed by Sanger sequencing: yes, the variant was present in both individuals; no, the c.1127T>G (p.V376G) variant was absent from both individuals.
In silico predictions were assessed for intronic variants via Alamut 2.0 (Interactive Biosoftware, Rouen, France).
Number of individuals with the variant/total number of controls tested.
As an alternative method to identify the mutated gene in Family A, we searched for deregulated genes in lymphoblasts from affected subjects. Reverse-transcribed RNA from lymphoblasts of four affected subjects and three unaffected noncarrier relatives of Family A were hybridized on Illumina HumanHT-12 beadchips. Two independent statistical analyses restricted to genes contained in the chromosome 15 interval showed that RAD51 mRNA was significantly downregulated in affected versus control individuals (fold difference = 0.7, p = 0.009; Table 2; Figures 2A–2C). By pretreating the lymphoblastic cells of affected members of Family A with emetin, we demonstrated that this downregulation corresponded to the degradation of the mutated mRNA by nonsense-mediated decay (Figure 2D). In addition, no truncated protein could be observed in western blot analysis in untreated lymphoblastic cells of three affected family members (not shown).
Table 2.
Classic Class Comparison Analysis of Transcriptomic Data between Affected Individuals and Controls
| Unique Illumina ID | Gene (MIM Number) | Fold-Change Controls/Patients | Parametric p Value | Controls Gene Expression | Patients Gene Expression | Expression in Brain |
|---|---|---|---|---|---|---|
| ILMN_1794157 | CATSPER2P1 (-) | 1.51 | 0.002 | 172.77 | 114.67 | - |
| ILMN_1697629 | PLA2G4B (606088) | 1.52 | 0.005 | 541.34 | 356.32 | - |
| ILMN_2363027 | RAD51 (179617) | 1.42 | 0.009 | 200.72 | 141.79 | + |
| ILMN_2372379 | MGA (-) | 1.24 | 0.009 | 78.27 | 63.15 | + |
| ILMN_2401906 | CDAN1 (607465) | 1.31 | 0.014 | 11358.92 | 8684.88 | + |
| ILMN_1710329 | MYEF2 (-) | 1.96 | 0.030 | 23.11 | 11.8 | + |
RNA extracted from lymphoblasts of four affected individuals and three spouses were hybridized on lllumina HumanHT12 beadchips. Expression profiles were extracted and normalized with Beadstudio software (Illumina). Normalized expression data were log2 transformed. The 131 genes expressed on bead chips from the 223 candidate genes on chromosome 15 were included for further analysis. Genes differently expressed between affected individuals and controls were selected for a fold difference of at least 1.2 between groups and a univariate p value of 0.05 with BRB array tools software. Six genes were significantly underexpressed in the affected individuals compared to the controls (p < 0.05, fold-change > 1.2), four of which were expressed in the brain (expression data provided by Genecards). No correction for multiple comparisons was used because the number of samples was too small and because this gene list would be intersected with the second approach (Figure 2A).
Figure 2.
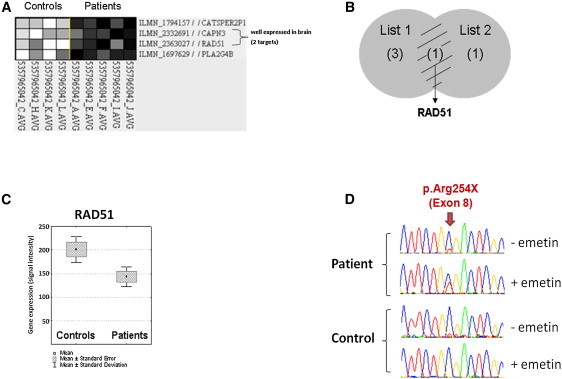
Downregulation of RAD51 mRNA in Lymphoblasts from Affected Individuals and Degradation of the Mutated mRNA by Nonsense-Mediated Decay
In parallel with the whole-exome analysis, we used transcriptomic analysis to identify the gene responsible for MM in Family A, postulating that the mutation might decrease the mRNA expression in lymphoblasts from affected individuals compared to healthy spouses from the same family. Total RNA extracted from lymphoblasts of four affected individuals and three spouses were hybridized on lllumina HumanHT12 beadchips. Expression profiles were extracted and normalized with Bead studio software (Illumina). Normalized expression data were log2 transformed. The 131 genes expressed on bead chips from the 223 candidate genes on chromosome 15 were included for further analysis. Two complementary statistical analyses by two independent investigators were performed to identify genes differentially expressed between the groups. Results of classic class comparison analysis are presented in Table 2.
(A) Gene clustering approach with the pattern discovery tool of GeneATWork software (IBM Research). The filtering criteria for a gene's inclusion in a pattern are a maximum deviation of 0.05 and a p value of 0.001. The best patterns classifying the “phenotype” and “control” groups were retained. This approach distinguished the affected individuals from the controls on the basis of four genes underexpressed in the affected subjects. Among these four genes (CATSPER2P1, CAPN3, RAD51, and PLA2G4B), only two (RAD51 and CAPN3) were expressed in the brain.
(B) RAD51 was the only gene lying at the intersection of the gene lists obtained with the two statistical analyses.
(C) Expression data obtained for RAD51 on HumanHT12 beadchips.
(D) Lymphoblastic cells from three affected individuals and three asymptomatic spouses from Family A were treated overnight with 10 μg/ml emetin to inhibit nonsense-mediated decay (NMD). Total RNA was extracted with the QIAGEN RNeasy Mini kit (Invitrogen) and reverse-transcribed with the SuperScript III First-Strand Kit (Invitrogen). RAD51 cDNA was amplified and sequenced with specific primers located in exons 7 (Forward) and 10 (Reverse). Chromatograms for one affected individual and one spouse, showing lower levels of mutated mRNA compared to WT mRNA in untreated cells and a comparable expression levels of both mRNA in cells pretreated with emetin, are shown.
To confirm that mutations in RAD51 can cause MM, we screened the RAD51 coding sequence by Sanger sequencing in the index cases of Family B and of a third family from the UK in which point mutations in DCC had been ruled out (Family C). Duplication of an adenine (c.855dupA), introducing a premature termination codon (p.Pro286Thrfs∗37), was identified in exon 9 of RAD51 in Family B (Figure 1D) and was absent from the 644 control subjects. No mutation was identified in Family C. Altogether, these results show that heterozygous mutations introducing premature termination codons in RAD51 cause congenital mirror movements in two unrelated families. Because RAD51 mRNA was significantly downregulated in individuals with CMM of Family A resulting from the degradation of the mutated mRNA by nonsense-mediated decay, haploinsufficiency is the main consequence of the mutations and the disease probably occurs once the amount of functional RAD51 falls below a critical level.
RAD51 is essential for maintaining genomic integrity through its involvement in the repair of DNA double-strand breaks by homologous recombination (HR).6–8 The RAD51 protein interacts with BRCA1 (BRCA1 [MIM 113705]) and BRCA2 (BRCA2 [MIM 600185])9–11 and defective HR is predicted to contribute to genomic instability and tumor development. Therefore, mutations in RAD51 have long been predicted to increase the risk of developing cancers12 or to modulate the tumor response or resistance to chemotherapy.13,14 However, a single constitutional missense variant was reported in two siblings with breast cancer, indicating that RAD51 is not a major cancer predisposition gene.15 Our findings are consistent with this observation and reveal an unexpected role of this gene in mammalian neurodevelopment.
To gain further insight into this function and to identify a possible relationship between RAD51 and DCC, we compared the expression of the two genes in the developing mouse cortex. DCC expression increased from embryonic day 12 (E12) to embryonic day 15 (E15), whereas RAD51 expression was strongest at E12; expression of both genes declined thereafter (Figure 3A). The spatial distribution of RAD51 was different from that of DCC and evolved during brain development: at E12, RAD51 was mostly detected in the cortical ventricular zone (proliferative zone; Figures 3B and 3C), whereas DCC was present in the preplate (postmitotic zone; Figures 3F and 3G), confirming previous observations.16,17 In the cortex of newborn mice (P0), RAD51 was mainly present in the subplate (SP) and, in lesser amounts, in layer V (Figures 4A and 4B), whereas DCC was selectively located in axons innervating the cortex (Figures 4C, 4D, 4G, and 4H). Strikingly, RAD51 was detected in a subpopulation of corticospinal axons at the pyramidal decussation in 2-day-old (P2) mice (Figures 4I and 4J), suggesting that RAD51 deficiency could specifically alter the decussation process. RAD51 is therefore specifically present in the developing mouse cortex, in brain tissues and at stages that are critical for the establishment of the corticospinal tract.
Figure 3.
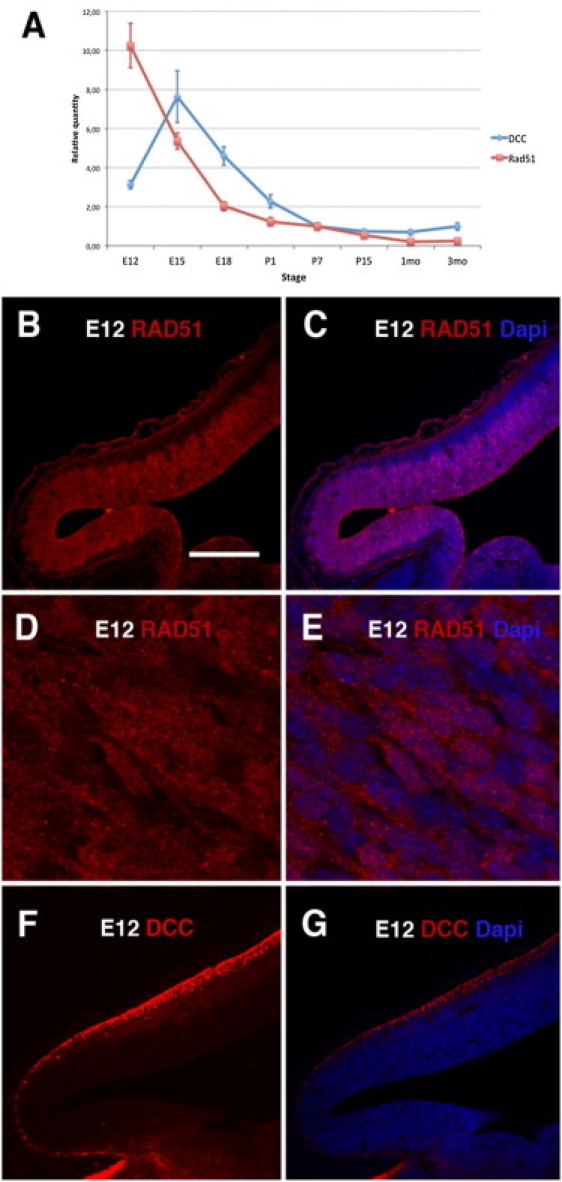
Comparative Expression and Localization of RAD51 and DCC in Developing Mouse Cortex
(A) Quantification of RAD51 and DCC expression in mouse cerebral cortex sampled in quadruplicate at several stages of development (E12, E15, E18, P1, P7, P15, 1 month, and 3 months) by real-time PCR. Quantification of each sample was carried out with the QIAGEN QuantiTect primer assays for DCC and RAD51. PPIA and PGK1 were used as control genes. Each sample was run in triplicate on a Lightcycler-1536 apparatus (Roche). Forty-five two-step cycles (15 s at 95°C and 30 s at 60°C) were performed. Analysis was performed with qbase Plus software (Biogazelle).
(B–G) Sagittal sections of the neocortex of E12 mouse embryos, fixed overnight in paraformaldehyde 4%, and immunolabeled with anti-RAD51 (1/50, sc-6862, Santa Cruz Biotechnology, Santa Cruz, CA) (B–E) or anti-DCC (1/100, sc-6535, Santa Cruz) (F and G) and counterstained with DAPI (C, E, G). Confocal plane (D, E). Scale bar represents 220 μm (B, C, F, G) or 15 μm (D, E).
Figure 4.
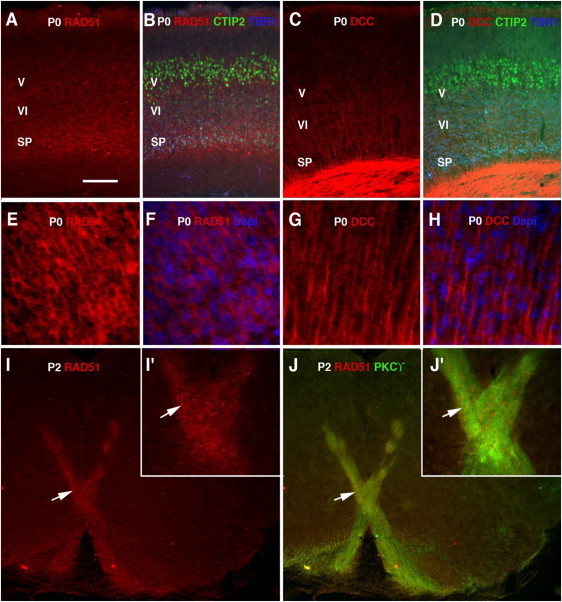
Comparative Localization of RAD51 and DCC in Mouse Brain at Postnatal Stages
(A–H) Cortical coronal section of a newborn (P0) mouse triple immunostained with anti-TBR1 (1/500, Millipore, Molsheim, France) to label the subplate and layer VI, anti-CTIP2 (1/500, Abcam, Cambridge, UK) to label layer V, and either anti-RAD51 (1/50, sc-6862, Santa Cruz) (A, B) or anti-DCC (1/100, sc-6535, Santa Cruz) (C, D), or immunolabeled only with anti-RAD51 (E, F) or anti-DCC (G, H) and counterstained with DAPI (F, H).
(I and J) Coronal section of a P2 mouse at the pyramidal decussation, immunostained with anti-RAD51 (I, J) and anti-PKCγ (1/100, sc211, Santa-Cruz) to label the corticospinal tract (J).
(I′ and J′) Enlargements of (I) and (J); arrows point to the same area.
Scale bar represents 120 μm (A–D), 30 μm (E–H), 250 μm (I, J), or 100 μm (I′, J′).
Interestingly, the subcellular location of RAD51 also changed with the stage of development: RAD51 was mostly detected in the nucleus of progenitor cells at E12 (Figures 3D and 3E) whereas it was mainly localized in the cell soma in the subplate at P0 (Figures 4E and 4F) and had a punctiform distribution at the pyramidal decussation in P2 mice (Figures 4I′ and 4J′). These results suggest that RAD51 could have several functions related to different cellular localizations.
The precise mechanisms linking RAD51 deficiency to MM are unclear, and the possible involvement of the DNA repair function in MM pathogenesis remains to be demonstrated. Insufficient RAD51-related DNA repair during early corticogenesis might lead to excessive apoptosis and altered central nervous system development, as observed in mice lacking XRCC2, another gene of the RAD51 family also involved in HR-mediated DNA repair.18,19 The location of RAD51 in the cytoplasm of cortical cells during mouse brain development, as previously described in other cell types, suggests, however, a role of RAD51 different from its function in HR occurring within the nucleus.20 It might have a direct or indirect role in axonal guidance, as shown for DCC. In keeping with this hypothesis, high RAD51 levels are associated with increased expression of genes involved in actin remodeling in nonneuronal cells.21 Nevertheless, the different cellular locations of DCC and RAD51 during cortical development suggest that these proteins do not interact directly.
Interestingly, homozygous Rad51−/− rodent zygotes show altered cell proliferation and abnormal cell morphology and are unable to undergo embryonic development after embryonic day 6.22,23 Heterozygous Rad51+/− mice are viable, fertile, and appear normal in outer appearance, but neither the morphological organization of their central nervous system nor their motor phenotype has yet been investigated.22,23 Therefore, it is currently unclear whether the Rad51+/− mice reproduce, at least in part, the human phenotype. Further characterization of mouse models is necessary to address this issue and to unravel the mechanisms by which RAD51 mutation leads to mirror movements in humans.
A striking feature is the reduced penetrance associated with RAD51 mutations: in Family A, 8 out of the 16 individuals with the p.Arg254∗ mutation were asymptomatic at examination, for a penetrance of 50%. The absence of mirror movements in these individuals could be, for example, due to a higher expression of RAD51 from the remaining normal allele or to other genetic or epigenetic modifiers. Interestingly, embryonic development of Rad51−/− mice progressed further in a p53-null background, supporting the hypothesis that the genetic background modulates the phenotype induced by the RAD51 mutation.23 So far, DCC and RAD51 seem to account for most MM families: over the four families studied in our laboratory, mutations in DCC were identified in one family4 and mutations in RAD51 were identified in two other families. One family (Family C) was negative for both genes but the existence of intragenic microrearrangements missed by sequencing was not tested and therefore it is uncertain whether a third gene responsible for MM exists.
Study of MM subjects or models provide a unique opportunity to investigate the network and mechanisms underlying the bimanual motor control, which are, so far largely unknown.2 Our findings therefore open a field of investigation for researchers attempting to unravel the molecular pathways underlying bimanual motor control in humans.
Acknowledgments
We thank the families for their participation, the DNA and cell bank for DNA extraction and cell culture, Khadija Tahiri for RNA preparation, and Agnès Rastetter for technical support in western blot analysis. This work was supported by INSERM, by Djilalli Mehri, and by an unrestricted research grant from IPSEN. C.K. was supported by the Hermann and Lilly Schilling Foundation. M.C.'s unit was supported by Ente Cassa di Risparmio di Firenze.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BRB array tools software, http://linus.nci.nih.gov/BRB-ArrayTools.html
dbSNP database, http://www.ncbi.nlm.nih.gov/projects/SNP/
Genecards, http://www.genecards.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Bonnet C., Roubertie A., Doummar D., Bahi-Buisson N., Cochen de Cock V., Roze E. Developmental and benign movement disorders in childhood. Mov. Disord. 2010;25:1317–1334. doi: 10.1002/mds.22944. [DOI] [PubMed] [Google Scholar]
- 2.Galléa C., Popa T., Billot S., Méneret A., Depienne C., Roze E. Congenital mirror movements: a clue to understanding bimanual motor control. J. Neurol. 2011;258:1911–1919. doi: 10.1007/s00415-011-6107-9. [DOI] [PubMed] [Google Scholar]
- 3.Srour M., Rivière J.B., Pham J.M., Dubé M.P., Girard S., Morin S., Dion P.A., Asselin G., Rochefort D., Hince P. Mutations in DCC cause congenital mirror movements. Science. 2010;328:592. doi: 10.1126/science.1186463. [DOI] [PubMed] [Google Scholar]
- 4.Depienne C., Cincotta M., Billot S., Bouteiller D., Groppa S., Brochard V., Flamand C., Hubsch C., Meunier S., Giovannelli F. A novel DCC mutation and genetic heterogeneity in congenital mirror movements. Neurology. 2011;76:260–264. doi: 10.1212/WNL.0b013e318207b1e0. [DOI] [PubMed] [Google Scholar]
- 5.Djarmati-Westenberger A., Brüggemann N., Espay A.J., Bhatia K.P., Klein C. A novel DCC mutation and genetic heterogeneity in congenital mirror movements. Neurology. 2011;77:1580. doi: 10.1212/WNL.0b013e318230b140. [DOI] [PubMed] [Google Scholar]
- 6.Park J.Y., Yoo H.W., Kim B.R., Park R., Choi S.Y., Kim Y. Identification of a novel human Rad51 variant that promotes DNA strand exchange. Nucleic Acids Res. 2008;36:3226–3234. doi: 10.1093/nar/gkn171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X., Heyer W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 9.Jensen R.B., Carreira A., Kowalczykowski S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carreira A., Hilario J., Amitani I., Baskin R.J., Shivji M.K., Venkitaraman A.R., Kowalczykowski S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–1043. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pellegrini L., Yu D.S., Lo T., Anand S., Lee M., Blundell T.L., Venkitaraman A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 12.Thacker J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005;219:125–135. doi: 10.1016/j.canlet.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Klein H.L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst.) 2008;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupianek A., Schmutte C., Tombline G., Nieborowska-Skorska M., Hoser G., Nowicki M.O., Pierce A.J., Fishel R., Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 15.Kato M., Yano K., Matsuo F., Saito H., Katagiri T., Kurumizaka H., Yoshimoto M., Kasumi F., Akiyama F., Sakamoto G. Identification of Rad51 alteration in patients with bilateral breast cancer. J. Hum. Genet. 2000;45:133–137. doi: 10.1007/s100380050199. [DOI] [PubMed] [Google Scholar]
- 16.Ajioka I., Maeda T., Nakajima K. Identification of ventricular-side-enriched molecules regulated in a stage-dependent manner during cerebral cortical development. Eur. J. Neurosci. 2006;23:296–308. doi: 10.1111/j.1460-9568.2005.04544.x. [DOI] [PubMed] [Google Scholar]
- 17.Shu T., Valentino K.M., Seaman C., Cooper H.M., Richards L.J. Expression of the netrin-1 receptor, deleted in colorectal cancer (DCC), is largely confined to projecting neurons in the developing forebrain. J. Comp. Neurol. 2000;416:201–212. doi: 10.1002/(sici)1096-9861(20000110)416:2<201::aid-cne6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 18.Deans B., Griffin C.S., Maconochie M., Thacker J. Xrcc2 is required for genetic stability, embryonic neurogenesis and viability in mice. EMBO J. 2000;19:6675–6685. doi: 10.1093/emboj/19.24.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francis F., Meyer G., Fallet-Bianco C., Moreno S., Kappeler C., Socorro A.C., Tuy F.P., Beldjord C., Chelly J. Human disorders of cortical development: from past to present. Eur. J. Neurosci. 2006;23:877–893. doi: 10.1111/j.1460-9568.2006.04649.x. [DOI] [PubMed] [Google Scholar]
- 20.Sage J.M., Gildemeister O.S., Knight K.L. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J. Biol. Chem. 2010;285:18984–18990. doi: 10.1074/jbc.M109.099846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orre L.M., Fält S., Szeles A., Lewensohn R., Wennborg A., Flygare J. Rad51-related changes in global gene expression. Biochem. Biophys. Res. Commun. 2006;341:334–342. doi: 10.1016/j.bbrc.2005.12.185. [DOI] [PubMed] [Google Scholar]
- 22.Tsuzuki T., Fujii Y., Sakumi K., Tominaga Y., Nakao K., Sekiguchi M., Matsushiro A., Yoshimura Y., Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim D.S., Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.