

Abstract
Structural variations are among the most frequent interindividual genetic differences in the human genome. The frequency and distribution of de novo somatic structural variants in normal cells is, however, poorly explored. Using age-stratified cohorts of 318 monozygotic (MZ) twins and 296 single-born subjects, we describe age-related accumulation of copy-number variation in the nuclear genomes in vivo and frequency changes for both megabase- and kilobase-range variants. Megabase-range aberrations were found in 3.4% (9 of 264) of subjects ≥60 years old; these subjects included 78 MZ twin pairs and 108 single-born individuals. No such findings were observed in 81 MZ pairs or 180 single-born subjects who were ≤55 years old. Recurrent region- and gene-specific mutations, mostly deletions, were observed. Longitudinal analyses of 43 subjects whose data were collected 7–19 years apart suggest considerable variation in the rate of accumulation of clones carrying structural changes. Furthermore, the longitudinal analysis of individuals with structural aberrations suggests that there is a natural self-removal of aberrant cell clones from peripheral blood. In three healthy subjects, we detected somatic aberrations characteristic of patients with myelodysplastic syndrome. The recurrent rearrangements uncovered here are candidates for common age-related defects in human blood cells. We anticipate that extension of these results will allow determination of the genetic age of different somatic-cell lineages and estimation of possible individual differences between genetic and chronological age. Our work might also help to explain the cause of an age-related reduction in the number of cell clones in the blood; such a reduction is one of the hallmarks of immunosenescence.
Introduction
Structural changes in the human genome have been identified as one of the major types of interindividual genetic variation.1,2 Furthermore, the rate of formation of copy-number variants (CNVs) exceeds the corresponding rate of SNPs by 2–4 orders of magnitude.3–5 In spite of this, little is known about the rate of formation and distribution of de novo somatic CNVs in normal cells and whether these aberrations accumulate with age. There are, however, indications that chromosomal remodeling in the nuclear and mitochondrial genomes increases with age.6–12 Theoretical predictions suggest that somatic mosaicism should be widespread,13,14 and reviews in the field point out that somatic mosaicism, in both healthy and diseased cells, is an understudied aspect of human-genome biology.15–18 A recent estimate of 1.7% for the frequency with which somatic mosaicism causes large-scale structural aberrations in adult human samples is, however, a relatively low number.19 We have shown that adult monozygotic (MZ) twins and differentiated human tissues frequently display somatic CNVs.20,21 We therefore hypothesized that the nuclear genome of blood cells in vivo might accumulate CNVs with age, and we used age-stratified MZ twins as a starting point for testing this hypothesis. Because nuclear genomes of MZ twins are identical at conception, they represent a good model for studying somatic variation. We replicated a MZ-twin-based analysis by using age-stratified cohorts of single-born subjects. Using these resources, we show age-related accumulation of CNVs in the nuclear genomes of blood cells in vivo. Age effects were found for both megabase- and kilobase-range variants.
Material and Methods
Studied Cohorts, DNA Isolation, and Quality Control
Samples were collected with informed consent from all subjects, and the study was approved by the respective local institutional review boards or research ethics committees. The information about studied cohorts of MZ twins and single-born subjects is provided in Tables S1 and S2, available online. We isolated DNA from peripheral blood by using the QIAGEN kit (QIAGEN, Hilden, Germany).
The quality, quantity, and integrity of DNA samples were controlled with NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA), picoGreen fluorescent assay (Invitrogen, Eugene, Oregon, USA), and agarose gels.
Sorting of Subpopulations of Cells from Peripheral Blood and Culturing of Fibroblasts
Peripheral blood mononuclear cells (PBMCs) were isolated from the whole blood with Ficoll-Paque centrifugation (Amersham Biosciences, Uppsala, Sweden), and a mixture of granulocytes was collected from under the PBMC layer. We isolated CD19+ cells from PBMCs by positive selection with CD19 MicroBeads (Miltenyi Biotech, Auburn, CA, USA). First, we negatively selected CD4+ cells by using the CD4+ T cell Isolation Kit II (Miltenyi Biotech, Auburn, CA, USA), and then we positively selected the cells by using CD4 MicroBeads (Miltenyi Biotech, Auburn, CA, USA). The CD19+ and CD4+ cells were incubated for 30 min at 4°C with phycoerythrin- and PerCP-conjugated antibodies (BD Biosciences, San Diego, CA, USA), respectively, for fluorescence-activated cell sorting (FACS) analysis. We measured purities of >90% for CD19+ and >98% for CD4+ cells by flow cytometry (FACS CantoII, BD Biosciences, San Diego, CA,USA). The skin-biopsy-derived fibroblasts were cultured in RPMI medium supplemented with Hams F-10 medium, fetal bovine serum (10%), penicillin, and L-glutamine (all cell culture reagents were from GIBCO, Invitrogen, Paisley, UK) in an incubator at 37°C. After reaching ∼90% confluence, the cells were trypsinized (Trypsin-EDTA, GIBCO, Invitrogen, Paisley, UK), and the fibroblasts were used for DNA isolation. We performed a standard phenol-chloroform extraction to isolate DNA from CD19+ cells, CD4+ cells, fibroblasts, and crude granulocyte fraction.
Genotyping with Illumina SNP Arrays and Calling of Large-Scale CNVs
We performed the SNP genotyping experiments by using several types of Illumina beadchips according to the recommendations of the manufacturer. Such experiments were performed at two facilities: Hudson Alpha Institute for Biotechnology (Huntsville, AL, USA) and the SNP Technology Platform (Uppsala University, Sweden). All Illumina genotyping experiments passed the following quality-control criteria: The SNP call rate for all samples was >98%, and the LogRdev value was <0.2. The results from Illumina SNP arrays consist of two main data tracks: log R ratio (LRR) and B-allele frequency (BAF)22 (see Figure 1). Deviations of consecutive probes from normal states are indicative of structural aberrations. We analyzed Illumina output files by using Nexus Copy Number version 5.1 (BioDiscovery, CA, USA), which applies a “Rank Segmentation” algorithm based on the circular binary segmentation (CBS) approach.23 The applied version, “SNPRank Segmentation,” an extended algorithm in which BAF values are also included in the segmentation process, generated both copy-number and allelic-event calls. We applied the default calling parameters of the program. The array data for large-scale CNVs reported in this paper have been submitted to the Database of Genomic Structural Variation (dbVAR) under the accession number nstd58.
Figure 1.
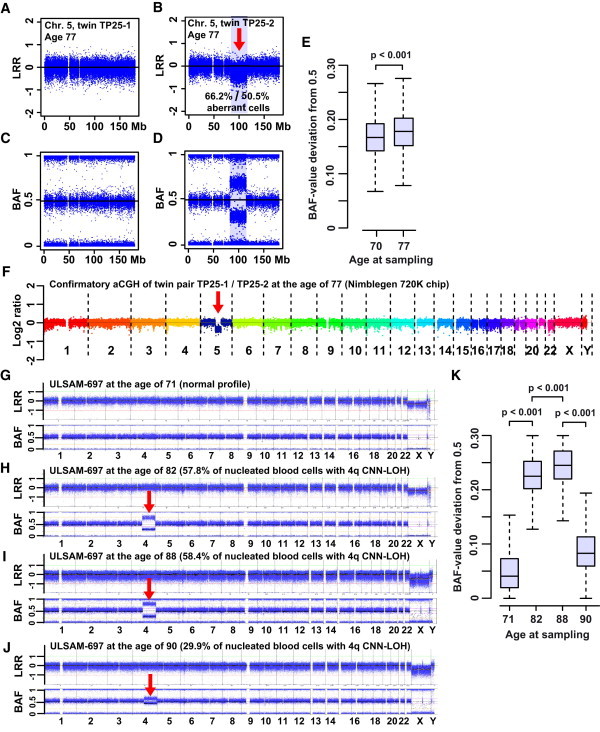
Two Examples of Megabase-Range De Novo Somatic Aberrations
(A) A normal profile of MZ twin TP25-1.
(B) A 32.5 Mb deletion on 5q is shown in nucleated blood cells of co-twin TP25-2. This deletion was uncovered with LRR data from the Illumina SNP array.
(C and D) The BAF profiles of twins TP25-1 (C) and TP25-2 (D). The qPCR experiments showed that 66.2% of nucleated blood cells in TP25-2 had the 5q deletion (i.e., 33.1% fewer copies of the DNA segment, Figure 5). The R-package-MAD (Mosaic Alteration Detection) analysis of the Illumina data suggested that 50.5% of the cells had the 5q deletion when the subjects were 77 years old.
(E) The deviation of BAF values from 0.5 (the allelic fraction of intensity at each heterozygous SNP) was plotted, and the percentage of cells with the 5q deletion was higher when the subjects were 77 years old than when they were 70 years old (t test: p < 0.001). This slow increase in aberrant clones was also supported by the MAD estimate of 48.3% of cells detected when the subjects were 70 years old. The size and position of this deletion is typical of patients with myelodysplastic syndrome (MDS).
(F) A confirmatory array-CGH experiment.
(G–K) Another large somatic event: a terminal CNNLOH encompassing 103 Mb of 4q in ULSAM-697. The LRR and BAF data from Illumina SNP genotyping of samples collected when the subjects were 71, 82, 88, and 90 years old are plotted in (G), (H), (I), and (J), respectively. Percentages of cells with the aberration were calculated with the MAD package and are given for each panel.
(K) The proportion of cells with the 4q aberration changes with time, and the changes are significantly different between all samplings at different ages (ANOVA: F(3,25935) = 39087, p < 0.001; Tukey's test for multiple comparisons). Figure S8 shows other analysis details of the samples collected from ULSAM-697 when he was 90 years old. These analyses include those of fibroblasts and three types of sorted blood cells. The analysis of samples obtained when the subjects were 90 years old was performed in duplicate experiments on Illumina 1M-Duo and Omni-Express arrays.
A Method for Detection of Small-Scale CNVs with Illumina SNP Array Data
We developed and applied an algorithm for testing whether smaller structural variants would also accumulate with age. We used deviations in BAF as the main tool for detecting candidate CNV regions because it can detect mosaicism in as low as 5%–7% of cells24,25 and allows uncovering of deletions and duplications as well as copy-number-neutral loss of heterozygozity (CNNLOH). This method uses an in-house-developed R-script26 to perform scans for deviations in BAF values alone and in BAF values together with LRR values in MZ twins. Figure S1 describes this algorithm, which identifies CNV calls for each MZ pair at user-defined thresholds of either ΔBAF or both ΔBAF and ΔLRR. Our initial tests of the algorithm were based on the entire cohort of 159 MZ pairs. However, a series of “trial and error” tests suggested that the method is sensitive to the quality of input data, given that the results were heavily biased toward detection of putative CNV calls in MZ co-twins with lower quality of genotyping, as measured by the Nexus Quality (NQ) score. The latter is one of the features of Nexus Copy Number software. We therefore defined strict NQ-score-based criteria for inclusion of MZ pairs in the analysis (see Table S3 and Figure S1), which resulted in the selection of 87 pairs that were processed further.
We based the final analysis on 87 twin pairs by identifying candidate CNV loci in which BAF values were different between co-twins when multiple thresholds were used. As expected, the number of putative CNV calls between MZ co-twins was highly dependent on the settings of the ΔBAF filtering (Figures S1–S4). Thus, when the settings were too generous in this step, an age-related signal was hidden in large background variation (Figure S2). By using more strict filtering criteria, we found an age-related correlation (Figures 2A and S4C). We trimmed the list of putative CNVs generated by ΔBAF by using a ΔLRR filter of >0.35 so that only loci with differences in both BAF and LRR remained in the final list (Figures 2B and S4D). Hence, the ΔLRR filter removed all loci with copy-number-neutral variation from the list. In the course of tuning ΔBAF (or both ΔBAF and ΔLRR) filtering parameters, we took advantage of three already-known large-scale aberrations that are described in our dataset (Figures 1A–1F, 3, and S5). These worked as ideal internal controls for the validity of our approach as shown in Figures S2–S4. Hence, by plotting the number of calls both including the probes located within the three known aberrations (Figures S2A–S2B, S3A–S3B, and S4A–S4B) and after excluding the probes located within the known aberrations (Figures S2C–S2D, S3C–S3D, and S4C–S4D), we could compare and evaluate the observed and expected results. For example, in Figure S4B, the twin pair TP25-1/TP25-2 sticks out because the probes positioned within the large de novo aberration of chromosome 5 (Figure 1) are included in the list of calls. When plotting the same data after excluding probes within this region, we found that the twin pair falls into the cluster of variation similar to that of the other MZ twin pairs (Figure S4D). On the basis of such evaluations, we observed that probes within the three large-scale CNVs were detected (or not, depending on the input file used in the analysis) as predicted by our ΔBAF and ΔLRR algorithm. Therefore, these evaluations provided an internal validation of our approach to detecting de novo small-scale CNVs.
Figure 2.
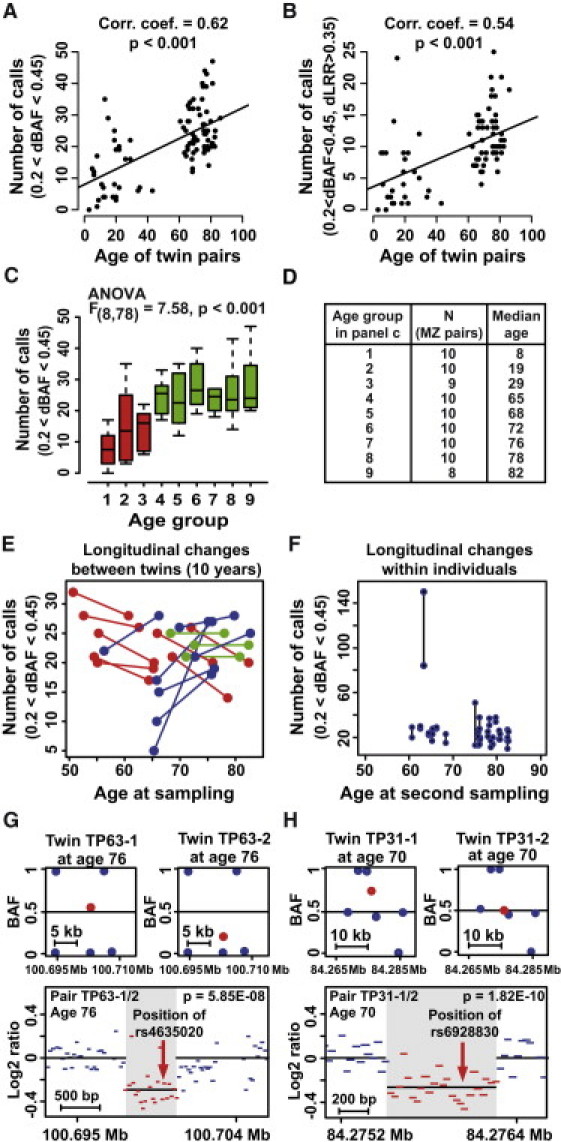
Age-Related Accumulation of Small Somatic Structural Rearrangements in 87 Pairs of MZ Twins
(A and B) Linear regression analyses showing that the number of calls increases with age in MZ twin pairs when ΔBAF values are between 0.2 and 0.45 as well as when ΔBAF values are between 0.2 and 0.45 and when the LRR deviation is >0.35. Each dot represents data from one MZ twin pair. Details regarding the filtering algorithms used are shown in Figure S1.
(C and D) An analysis of statistical significance for nine age groups of MZ twin pairs when ΔBAF values are between 0.2 and 0.45.
(E and F) Longitudinal data analyses comparing the number of ΔBAF reports (between 0.2 and 0.45) of 18 twin pairs that were sampled twice, 10 years apart. Each point in the plot represents the number of differences within one MZ pair (E). Each line (plotted between the two time points for the same MZ pair) thus represents the change over time of the number of differences within a pair (blue line, increase; red line, decrease; green line, no change). The intraindividual changes for each twin over a period of 10 years are shown in (F). The x axis shows individual ages at the later sampling. On the y axis, the number of differences found between the two samples from the same person at the two time points is shown, and vertical lines connect co-twins.
(G and H) Validation of copy-number imbalance between MZ twins in two pairs (chromosomes 10 and 6, respectively), which were detected by the ΔBAF analysis. The small boxes at the top of both (G) and (H) display original data from Illumina arrays for pairs TP63-1/TP63-2 and TP31-1/TP32-2, respectively. The larger boxes at the bottom of (G) and (H) display raw data from Nimblegen tiling-path 135K array for these two twin pairs. Each line is drawn to scale and represents data from one oligonucleotide probe. Statistical significance for the results of the Nimblegen array was calculated with the Mann-Whitney U test; values were analyzed for the region of interest (shaded) and for both areas on either side of the control regions. Twenty additional examples of validation experiments are shown in Figure S6. There was no difference between the rates of validation success for the young (n = 8) and old (n = 26) MZ pairs used in these experiments (t test: t = 0.7062, p value = 0.4819), supporting the results from linear-regression analyses. The detailed description of the Nimblegen array is provided in Figure S6 and Table S4.
Figure 3.
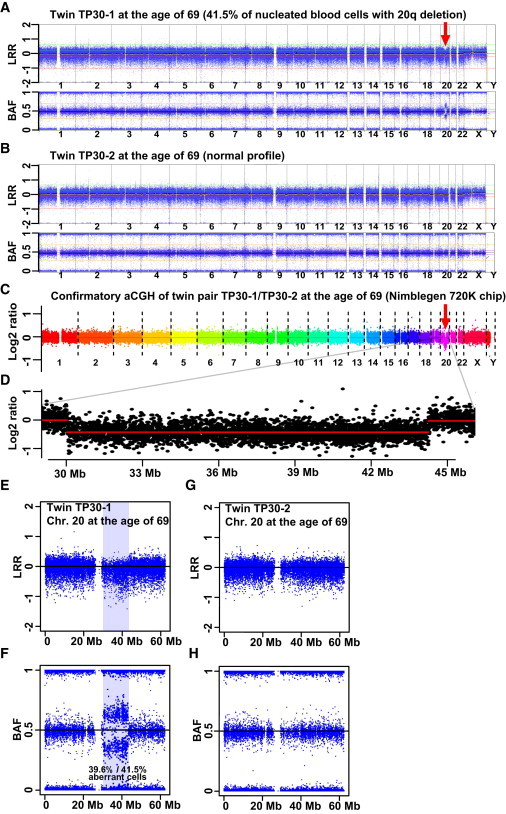
An Example of a Somatic Megabase-Range Aberration
(A, E, and F) A deletion encompassing 12.9 Mb of 20q in MZ twin TP30-1 was sampled when she was 69 years old.
(B, G, and H) The normal profile of co-twin TP30-2, as detected by LRR and BAF after Illumina SNP array genotyping. R-package-MAD analysis of the Illumina data suggested that 41.5% of the blood cells had the 20q deletion. qPCR validation experiments confirmed this result by showing 39.6% aberrant cells (i.e., 19.8% fewer copies of the DNA segment, Figure 5).
(C and D) Array-CGH validation experiments also confirmed the copy-number variation. The genetic change in MZ twin TP30-1 is another example of an MDS-like aberration, which was uncovered in a subject without a clinical diagnosis of MDS.
Design of the Nimblegen 135K Custom-Made Tiling-Path Oligonucleotide Array
This tool was designed according to the instructions from Roche-Nimblegen (Madison, WI, USA) and encompassed 137,545 probes used for validation of the 138 putative CNVs detected by the Illumina SNP array (Figures 2B, S4C, and S4D). In total, the design consisted of 98,894 experimental probes and an additional 38,651 backbone control probes distributed across the genome. The median overlap of probes (i.e., probe spacing) was 30 bp. This array was applied in cohybridizations of 34 MZ twin pairs (Figures 2G, 2H, and S6 and Table S4).
Array-Comparative Genomic Hybridization with Nimblegen 720K and 135K Arrays
We performed DNA labeling for both platforms (3 × 720K and 12 × 135K) by using the random priming with the Nimblegen Dual-Color DNA Labeling kit (Roche-Nimblegen) according to Nimblegen's protocol. In brief, test and reference DNA (500 ng each) samples were labeled with Cy3 and Cy5, respectively. The combined test and reference DNA was cohybridized (for 48 hr at 42°C) onto a human comparative genomic hybridization (CGH) 3 × 720K whole-genome tiling array (100718_HG18_WH_CGH_v3.1_HX3, OID:30853; Roche-Nimblegen) or a 12 × 135K custom-designed array (110131_HG18_LF_CGH_HX18, OID:33469; Roche-Nimblegen). The arrays were washed with the Nimblegen Wash Kit. We performed image acquisition with MS 200 Scanner at 2 μm resolution by using high-sensitivity and autogain settings. We extracted data with NimbleScan v2.6 segMNT, including spatial correction (LOESS) and qspline fit normalization, in order to compensate for differences in signal between the two dyes.27 We generated an experimental metrics report with NimbleScan v2.6 to verify hybridization quality. We performed CNV analysis with Nexus Copy Number software version 5.1 by using default settings (see above). All plots shown in Figures 2G, 2H, and S6 are derived from unaveraged, normalized raw data.
Validation Experiments Involving Quantitative Real-Time Polymerase Chain Reaction
We measured the relative amount of DNA molecules by using quantitative real-time polymerase chain reaction (qPCR) with SYBR green to validate the CNV findings from the arrays. qPCRs were performed in 20 μl reactions containing 5 ng genomic DNA, 0.3 μM of each primer, and 1× Maxima SYBR Green/ROX qPCR Master Mix (Fermentas, Vilnius, Lithuania) (for primer sequences, see Table S5). The reactions were incubated at 95°C for 10 min, after which they underwent 40 cycles of 95°C for 15 s and 60°C for 60 s in a Stratagene Mx3000P (Agilent Technologies) machine. The reactions for evaluation of primer efficiencies were performed in duplicates with control DNA (normal human female genomic DNA, Promega Corporation, Madison, WI, USA), whereas all other reactions with test and reference DNA were performed in triplicates; in both instances, the averages were used in analyses. Each primer pair's efficiency and standard curve are described in Figure S7. Melting-curve analysis was performed in all the experiments, and the results were analyzed with MxPro v4.10 software. We used ultra-conserved elements on human chromosomes 3 and 6 (UCE3 and UCE6) as control loci as previously described.28,29 We used the average cycle threshold (Ct) value of UCE6 to normalize the average Ct values of UCE3 and test loci. We used these normalized Ct values to calculate copy-number ratios of test regions. Using the estimated copy-number ratios from UCE3 and the test loci from multiple replicate experiments, we performed t tests for statistical testing.
Statistical Methods
The statistical analyses were performed with the R 2.12–2.13 software.26 We used methods such as linear regression, t tests, and one-way analyses of variance (ANOVAs) when suitable, as further specified in the text. Prior to testing, we controlled the data so that no test assumptions were violated. For multiple comparisons (i.e., Figures 1K and S8G), we used the Tukey honest-significant-difference method by implementing the TukeyHSD function in R. When appropriate, we performed the nonparametric Fisher's exact test and Mann-Whitney U test, as described in the text.
Boxplots of Longitudinal-Analysis Data
Heterozygous SNPs have a theoretical expected BAF value of 0.5, and deviations from this normal state can be indicative of structural aberrations.24 We can therefore use changes in the magnitude of these deviations in the subjects' longitudinal samples to measure intraindividual changes over time and to estimate the proportion of cells affected by large-scale aberrations. We produced the boxplots in Figures 1E, 1K, 4J, S9D, S9G, and S8G to visualize such changes in BAF variation. In these figures, we plotted the absolute deviation of BAF values from 0.5 for all heterozygous SNPs in the region of interest (i.e., ABS (0.5−BAF)) on the y axes. We only included heterozygous SNPs (i.e., those with a BAF value between 0.2 and 0.8) in these calculations to increase quality and accuracy of the plots. A larger BAF value deviation from 0.5 corresponds to a larger degree of mosaicism, i.e., a higher proportion of cells with a specific aberration. We used t tests (in cases with two factor levels) or one-way ANOVAs (in cases with >2 factor levels) to test for significance of such differences. For the model illustrated in Figures 1K and S8G, we used the Tukey post-hoc test for multiple comparisons to compute differences between factor-level means after adjusting p values for the multiple testing.
Figure 4.
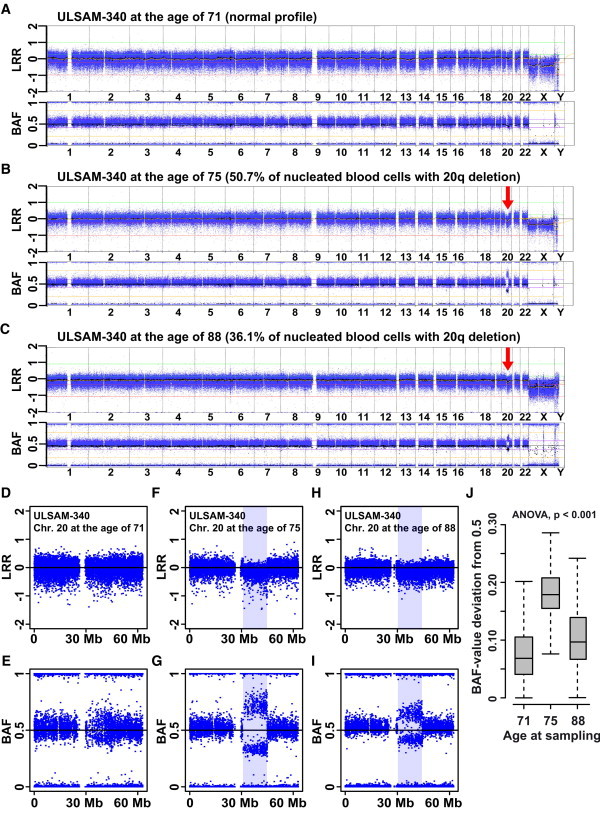
Longitudinal Analysis of ULSAM-340, a Single-Born Subject Containing a 13.8 Mb Deletion on 20q, as Detected by LRR and BAF with the Illumina SNP Array
The size and position of this deletion is typical of MDS patients. This subject, however, has not been diagnosed with MDS. When the patient was 71 years old, the deletion was only carried by a small proportion of blood cells and was barely detectable, and neither Nexus Copy Number software nor R-package MAD reported this aberration at this age (A, D, and E). R-package MAD suggested that 50.7% of the nucleated cells had the deletion when ULSAM-340 was 75 years old (B, F, and G) and that when he was 88 years old, the corresponding proportion of cells was 36.1% (C, H, and I). qPCR validation experiments showed that the sample taken when the patient was 88 years old contained 14.5% fewer copies of DNA in the segment as compared to the sample taken when he was 75 years old (Figure 5). The deviations from 0.5 of the BAF values within the deleted region in the three different sampling stages are illustrated in (J).
Quantification of the Number of Cells Affected by Megabase-Range Aberrations
We calculated the approximate percentage of cells affected by aberrations in the megabase range by using data from qPCR experiments (the data are described in Figure 5). The qPCR measurements provided the approximate number of DNA molecules that are affected by an aberration. Assuming that an aberration affects only one chromosome (i.e., an aberration that is a heterozygous event) in a diploid genome, we used this number and converted it to the approximate number of affected cells. Our assumption is reasonable, given that we are studying normal cells and that the size of these large-scale aberrations renders them unlikely to affect both chromosomes (i.e., they are unlikely to be homozygous [biallelic] events). For example, the relative number of DNA copies in nucleated blood cells of twin TP25-2 at the age of 77 years confirmed the array data. To determine these numbers, we used two primer pairs (41.1 and 42.1) designed within the deleted region and took five independent measurements for both primer pairs. These experiments suggested that, at the age of 77, twin TP25-2 had 30.8% (when primer pair 41.1 was used) and 35.4% (when primer pair 42.1 was used)—an average of 33.1%—fewer DNA copies with a 32.5 Mb 5q deletion than did her co-twin at the same age (Figure 5). If one assumes that this deletion is affecting one chromosome in a diploid cell, our calculations suggest that 66.2% of cells contain this deletion.
Figure 5.
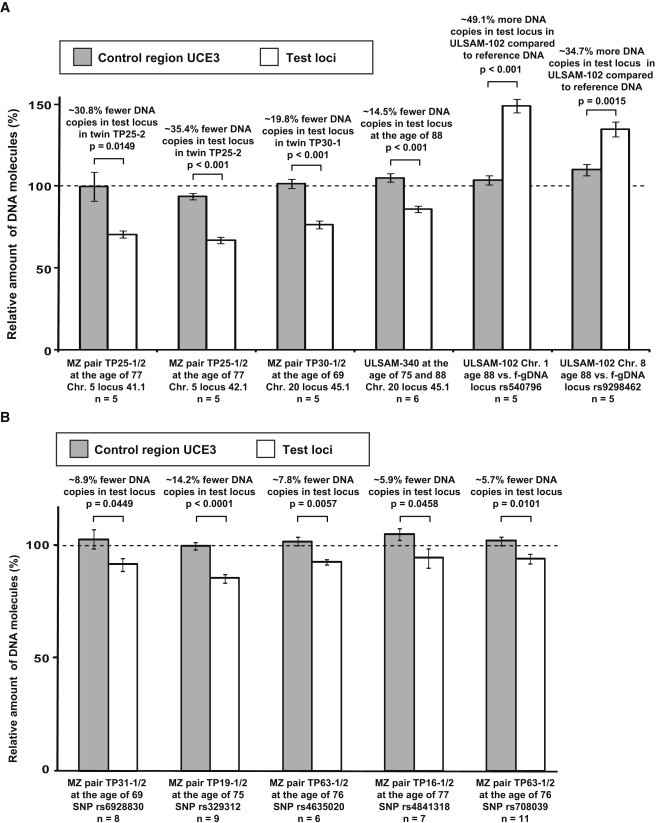
Validation of de novo CNVs by qPCR with SYBR Green
Eleven independent qPCR experiments, each composed of multiple (5–11) independent measurements, are shown. The relative number of DNA copies in both test loci (white bars) and the control region UCE3 (gray bars) were plotted. Before we plotted and performed statistical analyses with t tests, we normalized all Ct values by using the control region UCE6. Figure S7 shows the determination of primer efficiency for each of the primer pairs.
(A and B) Validations for five large-scale (A) and five small-scale (B) aberrations. The dotted line drawn at 100% represents the copy-number state in control DNA (i.e., that from the normal MZ co-twin, or human female control DNA, or DNA from the same subject sampled at another age), and error bars indicate standard error of means.
(A) The 5q deletion in twin TP25-2 (Figure 1) was validated with two primer pairs (41.1 and 42.1) designed within the deleted region. In total, ten independent qPCR experiments showed that ∼66.2% of all nucleated blood cells in TP25-2 had the 5q deletion (i.e., an average of 33.1% [30.8% with primer pair 41.1 and 35.4% with primer pair 42.1] fewer copies of the DNA segment). Similarly, the 20q deletion in twin TP30-1 (Figure 3) was validated with primer pair 45.1 in five experiments. The 19.8% fewer DNA copies found in the test locus indicates that 39.6% of the nucleated blood cells had the deletion. For ULSAM-340, the array data indicated a longitudinal somatic change in the number of cells carrying the 20q deletion. Six independent qPCR experiments comparing DNA sampled when ULSAM-340 was 75 and 88 years old showed that the subject had 14.5% fewer copies of the DNA segment when he was 88 years old. In ULSAM-102, the Illumina array identified a duplication event on both chromosomes 1 and 8 (Figure S9). Given that the proportion of cells with a gained segment in this subject was relatively stable over time, we used human female genomic DNA as control DNA in these experiments. The qPCR experiments validated both somatic CNVs.
(B) qPCR validation of five loci with small-scale de novo CNVs within MZ twins. These loci were identified by Illumina array genotyping and were confirmed on the Nimblegen 135K array (see also Figures 2G, 2H, and S6). The layout of this panel is similar to that of (A), described above. For example, the first locus (rs6928830) illustrates de novo CNVs in twin TP31-1 (Figure 2H).
In order to quantify the level of mosaicism, we also applied an alternative, published method19,30 based on calculations of the deviation of BAF values from the expected value of 0.5 for the heterozygous SNPs in a normal state. This method has been tailored for data derived from the Illumina SNP platform. The R-package MAD (Mosaic Alteration Detection) version 0.5–930 identifies the aberrant regions, such as deletions, gains, and CNNLOHs, and calculates the B deviation (Bdev, deviation from the expected BAF value of 0.5 for heterozygous SNPs) value, which is then used for calculation of the number of cells affected by the aberration. We used the following modified version of the published19 formula for deletions, gains, and CNNLOHs:
Results
Age-Related Accumulation of Megabase-Range Structural Variants
Our analysis of 159 MZ pairs involved genotyping with Illumina 600K SNP arrays, confirmation of monozygozity (>99.9% genotype concordance), CNV calling with Nexus Copy Number software (BioDiscovery, CA, USA), followed by inspection of genomic profiles. Validation was performed with a different Illumina array, Nimblegen array, and qPCR. Comparison of MZ twin pairs, including 19 previously reported pairs,21 identified five large de novo aberrations of >1 Mb among 81 young or middle-aged (≤55 years) and 78 elderly (≥60 years) pairs studied (Figures 1, 3, 5, and S5). All five large rearrangements occurred in the older twins, suggesting a relationship between age and the presence of changes. Tables S1 and S2 show a description of subjects, cohorts, and statistical support for the use of Illumina data for the detection of variants. We expanded on the results from twins by using two age-stratified groups of single-born subjects. First, we genotyped DNA from 108 men, all 88 years old, from the ULSAM (Uppsala Longitudinal Study of Adult Men) cohort by using the Illumina-1M-Duo array. We found that four subjects had large-scale rearrangements at the age of 88 years, and the somatic nature of such rearrangements was established by examination of samples taken from the same individuals at other time points (Figures 1, 4, 5, and S8–S10 and Table S1). Second, for the young or middle-aged single-born control cohort (33–55 years), we used existing Illumina 550K data from 180 controls from the ADVANCE (Atherosclerotic Disease, Vascular Function, and Genetic Epidemiology) study.31,32 Analogous analysis of ADVANCE subjects did not reveal any cases of large-scale aberrations. The genotyping quality of 550K experiments is at least as good as the quality of 1M-Duo arrays, and the resolution of the 550K array is sufficient for detection of ∼1Mb aberrations that have been uncovered in the ULSAM cohort (Figures S11 and S12 and Table S6). In fact, we described a 1.6 Mb deletion by using the 300K array in twin D8,21 and literature comparing arrays suggests that the 250K level is sufficient for uncovering submegabase-range changes.28,33 By studying the twins and the single-born individuals and by analyzing the two groups together, we obtained firm statistical support for age-related accumulation of large structural variants (with Fisher's exact test; p value = 0.00052) (Table S2). Overall, 3.4% of the studied population ≥60 years old carries cells containing megabase-range somatic aberrations that are readily detectable by array-based scanning, whereas none of the younger controls carried aberrations in this size range. The sensitivity of our analysis to detect aberrant clones is about 5% of nucleated blood cells.24,25 A previous estimate of 1.7% for somatic mosaicism was performed in an analysis that was not stratified by age.19
Five subjects harboring large CNVs (twin TP25-2 and ULSAM-102, -298, -340, and -697) were followed in repeated samplings collected up to 19 years apart. They all showed accumulation of aberrant cells with a variation in the rate of this process. Twin TP25-2 is an example of slow accumulation of a 5q-deletion clone (Figure 1); when this twin was 77 years old, two independent methods (q-PCR and MAD-program-based) suggested that 66.2% and 50.5% of cells, respectively, contained a deletion on one copy of chromosome 5. The change in deviation of BAF within the deleted region when twin TP25-2 was 70 and 77 years old translates into a 2.2% increase in cells with the 5q deletion. The latter estimation was based on analysis with the MAD program. It is noteworthy that the size and position of this 5q deletion are typical of myelodysplastic syndrome (MDS).34–38 However, twin TP25-2 has not been diagnosed with this disease. ULSAM-102 is another example of slow accumulation and contains gains on 1p and 8q (Figure S9). The 1p gain is stable, whereas the 8q gain shows a statistically significant (ANOVA: p value <0.05) increase over a period of 10 years. Consequently, ULSAM-102 probably carries two coexisting clones with different aberrations. In ULSAM-340 and -697, the rate of accumulation was faster and there was a decrease in the proportion of cells with aberrations at later samplings. ULSAM-340 contains a 20q deletion, which was barely detectable at the age of 71 (Figure 4). The number of cells containing the 20q deletion was estimated by analysis with the MAD program to be 50.7% when ULSAM-340 was 75 years old and to be 36.1% when he was 88 years old. ULSAM-340 is another example of an aberration typical of MDS in a subject without this diagnosis. However, his clinical history includes thrombocytopenia, which is normally a part of MDS clinical features. We therefore speculate that this symptom might be due to clonal expansion of cells with a 20q deletion and suppression of normal thrombocyte production. Finally, ULSAM-697 was analyzed four times and shows the most pronounced increase and decrease in the number of cells with CNNLOH of 4q (Figures 1 and S8). This aberration was not detectable at the age of 71, reached 58.4% at the age of 88, and decreased radically to 29.9% of cells at the age of 90. When ULSAM-697 was 90 years old, we profiled sorted CD4+ cells, CD19+ cells, granulocytes, and fibroblasts, in addition to whole-blood DNA. CD4+ cells, granulocytes, and whole blood showed similar levels of aberrant cells, whereas CD19+ cells and fibroblasts appeared normal. We performed all experiments on samples taken when ULSAM-697 was 90 years old in duplicate with different types of arrays. Thus, in ULSAM-697, both lymphoid and myeloid cells were affected, except for, quite surprisingly, CD19+ B cells. Overall, the analyses performed on ULSAM-340 and ULSAM-697 suggest that the cells with aberrations have a higher proliferative potential than do other cells in the immune system, but they are not immortalized because they apparently disappear from circulation.
Small-Scale Structural Aberrations Also Display Positive Correlation with Age
Given the above results, we tested whether smaller structural variants would also accumulate with age, and we used deviations in BAF as the main detection tool because they can detect mosaicism in as low as 5%–7% of cells24,25 and allow detection of deletions and duplications as well as CNNLOH. We performed scans for deviations in BAF values alone and BAF together with LRR in twins by using a new R-script (Figure S1) that identifies CNV calls for each MZ pair at various thresholds of ΔBAF and ΔLRR. Early analyses showed that the algorithm was sensitive to the quality of genotyping because calls were preferentially observed in co-twins with lower data quality. We therefore applied strict inclusion criteria by using the NQ score, which is based on genome-wide noise measurements. This resulted in the selection of 87 out of 159 MZ pairs (Table S3). We found that small putative CNVs increased with age (Figure 2A, linear regression F(1,85) = 54.00, p < 0.001, Figures S2–S4). We further narrowed the number of calls by combining the ΔBAF and ΔLRR values >0.35 from both twins in each MZ twin pair, and this process also indicated that these CNVs accumulate with age (Figure 2B; F(1,85) = 34.60, p < 0.001). We also tested whether genotyping quality (ΔNQ value is the absolute value of the difference in quality score within pairs) might explain the observed pattern. Importantly, there was no effect of ΔNQ on age (F(1,85) = 1.85, p > 0.05), suggesting that the positive correlation with age reflects true aberrations. Figure 2B displays a total of 827 CNV calls at 378 loci in 87 pairs with an age span of 3–86 years. Plotting of the 378 calls against the genome shows the nonrandom distribution and recurrent nature of these CNV calls (Figure S13). On the basis of frequency and/or location in the vicinity of known genes, we selected 138 loci for validation by using a tiling-path array (Nimblegen 135K) in 34 twin pairs. With this platform, 15% of putative CNVs were validated in the same twin pairs in which they were first detected by ΔBAF and ΔLRR analysis. There was no bias in the success rate of validation between younger and older groups (t test: t = 0.7062, p value = 0.4819). In total, 52 of the 138 loci (38%) included on the 135K array showed CNVs within 32 of the 34 MZ pairs tested (Figures 2G, 2H, and S6), and the majority of CNVs encompassed <1 kb. The reason for the discrepancy (i.e., 15% versus 38%) in the validation success rates mentioned above is probably due, at least in part, to the high stringency of the ΔBAF and ΔLRR analysis that only reported a subset of preferentially strong calls representing structural variants and the recurrent nature of loci that are affected by the small-scale variation. Hence, some true structural variants were validated in (often multiple) MZ pairs on the 135K array, even though the initial ΔBAF and ΔLRR analysis did not pick them up because the filtering parameters were too stringent. We selected 5 of these 52 loci for further validation with qPCR, and all five were confirmed by this alternative approach (Figure 5). We also performed breakpoint-PCR validation in 17 out of the above 52 loci by using PCR across the deleted region in instances that were presumed to represent the shortest deletions based on the Illumina and Nimblegen 135K array data. However, these attempts were not successful. We obtained correctly sized PCR bands representing wild-type alleles for tested loci. However, we could not detect any shorter, mutated alleles that were mapped to the correct genomic regions. These validation experiments included gel purification of PCR fragments, PCR-fragment analysis, subcloning in plasmids, and Sanger sequencing (details not shown). These results suggest that the vast majority of the uncovered small structural variants are due to more complex rearrangements involving deletions or gains embedded together with other structural changes. These results are in agreement with a recent sequencing-based validation analysis of CNV loci; the analysis showed that as few as 5% of CNVs suspected to represent gains or deletions are in fact “pure blunt-end breakpoints.”39 Details for the 52 validated loci are shown in Table S4, which includes information about genes affected by the variation. The results presented in Table S4 and Figure S13 emphasize the recurrent nature of the 52 validated loci. For example, out of the 52 loci, 13 only occurred once in any of the 34 tested twin pairs, whereas the remaining 39 were recurrent and occurred 2–16 times in the same set of MZ twin pairs. The number of CNVs per pair validated with the 135K Nimblegen array ranged from 1 to 32 (median 6) (Table S7). In summary, the deviation between MZ co-twins ranged from 0 to 51,040 bp (median 4,995 bp), and the latter corresponds to ∼0.0000016% genome-wide divergence.
By using the small-scale CNV pipeline, we analyzed 18 pairs of MZ twins that were sampled twice, 10 years apart (Figures 2E, 2F, and S1 and Table S8). Analyses were performed in two ways: as an interindividual comparison of one twin to its co-twin at the first and second sampling and as an intraindividual comparison of the two samplings of a single twin. Both types of comparisons suggest variation in the dynamics of changes between co-twins and show both increases and decreases over a period of 10 years in the number of calls in different twin pairs. Interestingly, this evidence for the dynamics of small-scale CNVs over time (Figure 2E) is consistent with the results from longitudinal analyses of large-scale aberrations in ULSAM-697 and ULSAM-340 (Figures 1 and 4), suggesting both increases and decreases over time in the number of cells containing different variants.
Discussion
The phenotypic consequences of accumulating aberrations are an interesting aspect of our results. In two subjects diagnosed with chronic lymphocytic leukemia (CLL), we detected multiple changes consistent with the disease (Figure S10). These findings are not unexpected: Our population-based cohort was not preselected against any diagnoses, and CLL is the most prevalent leukemia among the elderly.40 However, it is surprising that apparently healthy subjects have aberrations characteristic of MDS. A typical 5q deletion (observed in one subject) and a 20q deletion (observed in two subjects) are among the most common aberrations in patients diagnosed with MDS.34–38 Trisomy 8 is also a recurrent aberration in MDS, and ULSAM-102 displays a restricted 8q gain; it remains unclear whether this gain is related to MDS. None of the above-mentioned individuals were diagnosed with MDS, and their cases might represent an indolent, subclinical form of MDS. In two individuals followed in longitudinal sampling (i.e., ULSAM-340 and -697), we observed not only an increase but also a clear subsequent decrease in the proportion of nucleated blood cells with aberrations (Figures 1, 4, and S8). These results suggest an “autocorrection” of the immune system, given that the aberrant clones are apparently disappearing from circulation. Similar expansions of preleukemic clones containing gene fusions specific to acute leukemia have been described in newborns;41 the gene fusions TEL-AML1 and AML1-ETO were present in cord blood at a frequency 100× greater than the frequency that is associated with the risk of developing the corresponding leukemia.
The presented data are probably only part of all the somatic changes that actually occurred in the studied cohorts because balanced inversions and translocations escape our detection and because we interrogated a fraction of all the nucleotides in the genome. Furthermore, we only detected high-frequency aberrations, presumably because these aberrations provided the affected cells with a proliferative advantage, which lead to clonal expansion above the detection limit of ∼5% of cells. It follows from this reasoning that deleterious aberrations leading to proliferative disadvantage or aberrations that are neutral from the point of view of the proliferative potential go undetected. Nevertheless, the chromosomal regions (e.g., those that contain the 20q deletion) and loci affected in a recurrent fashion (Figure S13 and Table S4) are candidates for containing common and redundant age-related defects in human blood cells. These mutations are presumed to provide the affected cells with a mild proliferative advantage without transforming the affected cells into immortalized cancer clones. However, the proliferative advantage for a limited number of cells will most likely affect the overall complexity of cell clones present in blood and should therefore be discussed in the context of immunosenescence, which, in fact, involves loss of complexity of cell clones in both B and T cell lineages.42,43 Our results might therefore help to explain the cause of age-related reduction in the number of cell clones in the blood. This reduction could lead to a less diverse immune system caused by the accumulation of genetic changes that induce the expansion of a limited number of clones. We also anticipate that extension of our work will allow determination of the genetic age of different somatic cell lineages and estimation of possible individual differences between genetic and chronological age.
Acknowledgments
We thank Lars Feuk, Brigitte Schlegelberger, Jacek Witkowski, Greg Cooper, Richard Rosenquist Brandell, Eva Hellström-Lindberg, Chris Gunther, and Eva Tiensuu Janson for critical review of the manuscript and Larry Mansouri and Juan R. Gonzalez for methodological advice. This study was sponsored by grants from the Ellison Medical Foundation (J.P.D. and D.A.) and from the Swedish Cancer Society, the Swedish Research Council, and the Science for Life Laboratory-Uppsala (J.P.D.). A.P. acknowledges FOCUS 4/2008 and FOCUS 4/08/2009 grants from the Foundation for Polish Science. Genotyping was performed in part by the SNP&SEQ Technology Platform, which is supported by Uppsala University, Uppsala University Hospital, the Science for Life Laboratory–Uppsala, and the Swedish Research Council (contracts 80576801 and 70374401).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenePipe PrimerZ, http://genepipe.ngc.sinica.edu.tw/primerz/
Illumina Beadchip information, http://www.illumina.com/documents/products/appnotes/appnote_cytogenetics.pdf
R 2.12–2.13 software, http://www.r-project.org/
Roche-Nimblegen array CGH Protocols, http://www.nimblegen.com/
R-package MAD version 0.5–9, http://www.creal.cat/jrgonzalez/software.htm
Surveillance Epidemiology and End Results (SEER) Program Fast Stats, http://seer.cancer.gov/faststats/
The Gene Ontology, http://www.geneontology.org/
The Genetic Association Database, http://geneticassociationdb.nih.gov/
The HUGO Gene Nomenclature Committee, http://www.genenames.org/
University of California Santa Cruz Human Genome Browser, http://genome.cse.ucsc.edu/cgi-bin/hgGateway
Accession Numbers
The array data for large-scale CNVs reported in this paper have been submitted to the Database of Genomic Structural Variation (dbVAR) under the accession number nstd58.
References
- 1.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Ommen G.J. Frequency of new copy number variation in humans. Nat. Genet. 2005;37:333–334. doi: 10.1038/ng0405-333. [DOI] [PubMed] [Google Scholar]
- 4.Lupski J.R. Genomic rearrangements and sporadic disease. Nat. Genet. 2007;39(7 Suppl):S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 5.Itsara A., Wu H., Smith J.D., Nickerson D.A., Romieu I., London S.J., Eichler E.E. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harley C.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri H., Schächter F., Uchida I., Wei L., Zhu X., Effros R., Cohen D., Harley C.B. Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am. J. Hum. Genet. 1993;52:661–667. [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H.C., Pang C.Y., Hsu H.S., Wei Y.H. Differential accumulations of 4,977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta. 1994;1226:37–43. doi: 10.1016/0925-4439(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 9.Fraga M.F., Ballestar E., Paz M.F., Ropero S., Setien F., Ballestar M.L., Heine-Suñer D., Cigudosa J.C., Urioste M., Benitez J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohamed S.A., Hanke T., Erasmi A.W., Bechtel M.J., Scharfschwerdt M., Meissner C., Sievers H.H., Gosslau A. Mitochondrial DNA deletions and the aging heart. Exp. Gerontol. 2006;41:508–517. doi: 10.1016/j.exger.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Flores M., Morales L., Gonzaga-Jauregui C., Domínguez-Vidaña R., Zepeda C., Yañez O., Gutiérrez M., Lemus T., Valle D., Avila M.C. Recurrent DNA inversion rearrangements in the human genome. Proc. Natl. Acad. Sci. USA. 2007;104:6099–6106. doi: 10.1073/pnas.0701631104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sloter E.D., Marchetti F., Eskenazi B., Weldon R.H., Nath J., Cabreros D., Wyrobek A.J. Frequency of human sperm carrying structural aberrations of chromosome 1 increases with advancing age. Fertil. Steril. 2007;87:1077–1086. doi: 10.1016/j.fertnstert.2006.08.112. [DOI] [PubMed] [Google Scholar]
- 13.Frank S.A. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: Mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 1):1725–1730. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345–352. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Youssoufian H., Pyeritz R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 16.Erickson R.P. Somatic gene mutation and human disease other than cancer: An update. Mutat. Res. 2010;705:96–106. doi: 10.1016/j.mrrev.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 17.De S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011;27:217–223. doi: 10.1016/j.tig.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Dumanski J.P., Piotrowski A. Structural genetic variation in the context of somatic mosaicism. In: Feuk L., editor. Genomic Structural Variation. Humana Press; New York: 2012. [DOI] [PubMed] [Google Scholar]
- 19.Rodríguez-Santiago B., Malats N., Rothman N., Armengol L., Garcia-Closas M., Kogevinas M., Villa O., Hutchinson A., Earl J., Marenne G. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010;87:129–138. doi: 10.1016/j.ajhg.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piotrowski A., Bruder C.E., Andersson R., Diaz de Ståhl T., Menzel U., Sandgren J., Poplawski A., von Tell D., Crasto C., Bogdan A. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 21.Bruder C.E., Piotrowski A., Gijsbers A.A., Andersson R., Erickson S., Diaz de Ståhl T., Menzel U., Sandgren J., von Tell D., Poplawski A. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 2008;82:763–771. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steemers F.J., Chang W., Lee G., Barker D.L., Shen R., Gunderson K.L. Whole-genome genotyping with the single-base extension assay. Nat. Methods. 2006;3:31–33. doi: 10.1038/nmeth842. [DOI] [PubMed] [Google Scholar]
- 23.Olshen A.B., Venkatraman E.S., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 24.Conlin L.K., Thiel B.D., Bonnemann C.G., Medne L., Ernst L.M., Zackai E.H., Deardorff M.A., Krantz I.D., Hakonarson H., Spinner N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Razzaghian H.R., Shahi M.H., Forsberg L.A., de Ståhl T.D., Absher D., Dahl N., Westerman M.P., Dumanski J.P. Somatic mosaicism for chromosome X and Y aneuploidies in monozygotic twins heterozygous for sickle cell disease mutation. Am. J. Med. Genet. A. 2010;152A:2595–2598. doi: 10.1002/ajmg.a.33604. [DOI] [PubMed] [Google Scholar]
- 26.R_Development_Core_Team. (2010). R: A language and environment for statistical computing. In. (Vienna, Austria). URL: http://www.R-project.org/
- 27.Workman C., Jensen L.J., Jarmer H., Berka R., Gautier L., Nielser H.B., Saxild H.H., Nielsen C., Brunak S., Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-9-research0048. research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunnarsson R., Staaf J., Jansson M., Ottesen A.M., Göransson H., Liljedahl U., Ralfkiaer U., Mansouri M., Buhl A.M., Smedby K.E. Screening for copy-number alterations and loss of heterozygosity in chronic lymphocytic leukemia—a comparative study of four differently designed, high resolution microarray platforms. Genes Chromosomes Cancer. 2008;47:697–711. doi: 10.1002/gcc.20575. [DOI] [PubMed] [Google Scholar]
- 29.Gunnarsson R., Isaksson A., Mansouri M., Göransson H., Jansson M., Cahill N., Rasmussen M., Staaf J., Lundin J., Norin S. Large but not small copy-number alterations correlate to high-risk genomic aberrations and survival in chronic lymphocytic leukemia: A high-resolution genomic screening of newly diagnosed patients. Leukemia. 2010;24:211–215. doi: 10.1038/leu.2009.187. [DOI] [PubMed] [Google Scholar]
- 30.González J.R., Rodríguez-Santiago B., Cáceres A., Pique-Regi R., Rothman N., Chanock S.J., Armengol L., Pérez-Jurado L.A. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12:166. doi: 10.1186/1471-2105-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schunkert H., König I.R., Kathiresan S., Reilly M.P., Assimes T.L., Holm H., Preuss M., Stewart A.F., Barbalic M., Gieger C., Cardiogenics. CARDIoGRAM Consortium Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Assimes T.L., Knowles J.W., Basu A., Iribarren C., Southwick A., Tang H., Absher D., Li J., Fair J.M., Rubin G.D. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum. Mol. Genet. 2008;17:2320–2328. doi: 10.1093/hmg/ddn132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagenkord J.M., Monzon F.A., Kash S.F., Lilleberg S., Xie Q., Kant J.A. Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: Performance comparison of Affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays. J. Mol. Diagn. 2010;12:184–196. doi: 10.2353/jmoldx.2010.090118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernasconi P., Boni M., Cavigliano P.M., Calatroni S., Giardini I., Rocca B., Zappatore R., Dambruoso I., Caresana M. Clinical relevance of cytogenetics in myelodysplastic syndromes. Ann. N Y Acad. Sci. 2006;1089:395–410. doi: 10.1196/annals.1386.034. [DOI] [PubMed] [Google Scholar]
- 35.Haase D. Cytogenetic features in myelodysplastic syndromes. Ann. Hematol. 2008;87:515–526. doi: 10.1007/s00277-008-0483-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiu R.V., Gondek L.P., O'Keefe C.L., Elson P., Huh J., Mohamedali A., Kulasekararaj A., Advani A.S., Paquette R., List A.F. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011;117:4552–4560. doi: 10.1182/blood-2010-07-295857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braun T., de Botton S., Taksin A.L., Park S., Beyne-Rauzy O., Coiteux V., Sapena R., Lazareth A., Leroux G., Guenda K. Characteristics and outcome of myelodysplastic syndromes (MDS) with isolated 20q deletion: A report on 62 cases. Leuk. Res. 2011;35:863–867. doi: 10.1016/j.leukres.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Bejar R., Levine R., Ebert B.L. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J. Clin. Oncol. 2011;29:504–515. doi: 10.1200/JCO.2010.31.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrad D.F., Bird C., Blackburne B., Lindsay S., Mamanova L., Lee C., Turner D.J., Hurles M.E. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Surveillance Epidemiology and End Results (SEER) Program. Fast stats. Bethesda, MD, National Cancer Institute, NIH, USA (2011) URL: http://seer.cancer.gov/faststats/
- 41.Mori H., Colman S.M., Xiao Z., Ford A.M., Healy L.E., Donaldson C., Hows J.M., Navarrete C., Greaves M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. USA. 2002;99:8242–8247. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naylor K., Li G., Vallejo A.N., Lee W.W., Koetz K., Bryl E., Witkowski J., Fulbright J., Weyand C.M., Goronzy J.J. The influence of age on T cell generation and TCR diversity. J. Immunol. 2005;174:7446–7452. doi: 10.4049/jimmunol.174.11.7446. [DOI] [PubMed] [Google Scholar]
- 43.Gibson K.L., Wu Y.C., Barnett Y., Duggan O., Vaughan R., Kondeatis E., Nilsson B.O., Wikby A., Kipling D., Dunn-Walters D.K. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell. 2009;8:18–25. doi: 10.1111/j.1474-9726.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.