

Abstract
Advanced paternal age has been associated with an increased risk for spontaneous congenital disorders and common complex diseases (such as some cancers, schizophrenia, and autism), but the mechanisms that mediate this effect have been poorly understood. A small group of disorders, including Apert syndrome (caused by FGFR2 mutations), achondroplasia, and thanatophoric dysplasia (FGFR3), and Costello syndrome (HRAS), which we collectively term “paternal age effect” (PAE) disorders, provides a good model to study the biological and molecular basis of this phenomenon. Recent evidence from direct quantification of PAE mutations in sperm and testes suggests that the common factor in the paternal age effect lies in the dysregulation of spermatogonial cell behavior, an effect mediated molecularly through the growth factor receptor-RAS signal transduction pathway. The data show that PAE mutations, although arising rarely, are positively selected and expand clonally in normal testes through a process akin to oncogenesis. This clonal expansion, which is likely to take place in the testes of all men, leads to the relative enrichment of mutant sperm over time—explaining the observed paternal age effect associated with these disorders—and in rare cases to the formation of testicular tumors. As regulation of RAS and other mediators of cellular proliferation and survival is important in many different biological contexts, for example during tumorigenesis, organ homeostasis and neurogenesis, the consequences of selfish mutations that hijack this process within the testis are likely to extend far beyond congenital skeletal disorders to include complex diseases, such as neurocognitive disorders and cancer predisposition.
Main Text
Introduction
It has long been recognized that older parents are at increased risk of having children with genetic disorders. Much work has gone into studying congenital anomalies in the offspring of aging mothers, and the best documented example of a strong maternal age effect occurs in Down syndrome (trisomy 21).1 However, growing evidence suggests that, independently of maternal age, the progeny of older fathers are more susceptible to a wide range of conditions, including spontaneous dominant disorders,2 congenital anomalies,3 childhood cancers,4 acute lymphoblastic leukemia,5 breast cancer,6 increased telomere length,7 autism,8 schizophrenia,9 bipolar disorder,10 and reduced neurocognitive abilities in childhood and infancy.11 Although the association between advanced paternal age and disease risk is not always consistent or reproducible,12,13 it is clear that it has become a concern, as witnessed by the precautionary measures taken for sperm donation in the case of assisted conception, for which it is now commonly advised to limit the age of donors to below 40 years.14 Yet, there is a notable lack of understanding of the mechanisms involved or even a general consensus on how to define what constitutes advanced paternal age.14,15 It is apparent that the average paternal age at conception is subject to demographic variation and fluctuates considerably for different years of birth or countries.15 In England and Wales, for instance, it has increased steadily from 29.06 years in the mid-1970s to 32.75 years by 2008, and today over one-third of children are born to fathers over the age of 35 years (England and Wales Birth Census data series FM1). Given that this trend is also observed in many other developed countries, it would be timely to re-evaluate the impact that paternal age has on genetic risks and predict its effects both on individual risk and on public health. However, this is hampered by lack of understanding of the mechanisms involved and the extent of the effects—which are likely to vary considerably for different disorders.
Although in some cases the evidence for an association with advanced paternal age is ill-defined or controversial, the situation is very different for a small group of congenital disorders, which we call “paternal age effect” (PAE) disorders and for which work performed over the last decade is reaching a consensus that allows a clearer picture of the biological mechanisms associated with some paternal age effects. The best known examples of PAE disorders involve specific point mutations in fibroblast growth factor receptor genes, FGFR2 (mutations of which cause Apert [MIM 101200], Crouzon [MIM 123500], and Pfeiffer[(MIM 101600] syndromes) and its paralog, FGFR3 (mutations of which cause achondroplasia [MIM 100800] and thanatophoric dysplasia [MIM 187601 and 187602], hypochondroplasia [MIM 146000], and Muenke syndrome [MIM 602849]). These disorders occur spontaneously with a remarkably high apparent rate, reaching 1 in 30,000 births for achondroplasia,16,17 and are associated with an increased paternal age, relative to the general population.2,18–21 Additionally, mutations in RET (causing multiple endocrine neoplasia types 2A22 [MIM 171400] and 2B23 [MIM 162300]), PTPN11 (encoding SHP2 and causing Noonan syndrome24 [MIM 163950]), and HRAS (Costello syndrome25–27 [MIM 218040]) are also associated with advanced paternal age, allowing a better understanding of the mechanisms involved in this process. Indeed there are striking parallels between all these PAE disorders that extend beyond the epidemiological observations of a paternal age effect: all are caused by a small number of dominantly-acting point mutations in key developmental regulators, which cluster within the growth factor receptor-RAS signaling pathway; moreover, the causative point mutations originate almost exclusively from the unaffected fathers, indicating that the original mutational events are taking place during spermatogenesis. In turn, these observations have led to efforts to quantify these mutations directly in the sperm and testes of men of different ages, which has now been accomplished for a handful of PAE mutations. The picture emerging from these technically challenging studies is that the paternal age effect associated with these conditions can be explained by a mechanism, which we term “selfish selection,” whereby rare spermatogonial cells bearing mutations are positively selected, leading to their progressive clonal expansion. This process of clonal expansion in the testis, which is associated with testicular tumors in rare cases, is likely to be universal and to affect all men as they age.
In the first half of this review, we will examine the evidence gathered from the small number of canonical PAE disorders. We will describe the characteristic features of the FGFR2 and FGFR3 mutations and their cognate disorders to show how these syndromes illustrate the mechanisms and principles involved in the paternal age effect. We will then collate information on mutations in the other PAE genes, describing how they provide important information on the molecular pathways (in the case of the RASopathies such as Costello and Noonan syndromes) and the biological mechanisms involved (in the case of RET-associated disorders). Later, we will consider the possibility that selfish selection is a common mechanism operating during male gametogenesis that has potentially far-reaching consequences for genetic variation, disease, and evolution. We will argue that the contributing mutational processes are likely not limited to single-base-pair substitutions in protein-coding genes but include regulatory mutations and copy-number variations and that the phenotypic consequences extend beyond certain rare monogenic congenital disorders to include common complex diseases. Recently acquired rare mutations have been postulated to contribute to some of the “missing heritability” that has been associated with complex disorders.28 As selfish selection is predicted to generate recurrent functional alleles acting on pleiotropic signaling, such mechanisms might contribute significantly to the mutational burden associated with complex phenotypes.
Spectrum and Characteristics of PAE Mutations and Associated Disorders
We define PAE disorders as a small class of syndromes caused by spontaneous dominant heterozygous mutations that present with a triad of unusual features comprising (1) an extreme bias in paternal origin of mutations (defined as the ratio of male-to-female mutations, α > 20), (2) a strong paternal age effect (i.e., the fathers of affected children are on average more than 2 years older than the matched general population), and (3) a high apparent germline mutation rate (>10−6 for particular individual mutations). Nine autosomal-dominant disorders (Apert, Crouzon, Pfeiffer, and Muenke syndromes; achondroplasia; Costello and Noonan syndromes; and multiple endocrine neoplasia types 2A and 2B), corresponding to specific point mutations within five genes (FGFR2, FGFR3, HRAS, PTPN11, and RET) can be identified that strictly fulfill all three criteria (Table 1).
Table 1.
PAE Genes and Associated Disorders
| Gene Symbol | Gene | DNA Mutationa(Amino Acid)b | Clinical Disorder | OMIM Reference Number | Mode of Transmission | Mutation Mechanism | Informative Paternal Mutations | Informative Maternal Mutations | Paternal Origin | Average Paternal Agec(Years) | Average Excess in Paternal Aged(Years) | Estimated Birth Prevalence for New Mutations | Sperm Study | Cancer Predisposition in Patients? | Reported as Somatic Mutation?e | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FGFR2 | fibroblast growth factor receptor 2 | c.755C>G (p.Ser252Trp) c.758C>G (p.Pro253Arg) | Apert syndrome | 101200 | autosomal dominant | GOF | 75 | 0 | 100% | 32.5; 34.1 | 2.5 (1.7–3.2) | ∼1/65,000 | yes | not reported | endometrial cancer | Cohen et al.18, Tolarova et al.19, Moloney et al.45, Wilkie et al.55, Goriely et al.56, Yoon et al.58, Hansen161 |
| FGFR2 | fibroblast growth factor receptor 2 | >50 different mutations, 11 used for paternal origin studies | Crouzon/Pfeiffer syndrome | 123500/101600 | autosomal dominant | GOF | 22 | 0 | 100% | 34.5 | 4.1 | ∼1/50,000–1/100,000 | yes, c.755C > T | not reported | lung stomach endometrial cancer | Goriely et al.56, Glaser et al162 |
| FGFR3 | fibroblast growth factor receptor 3 | c.1138G>A (p.Gly380Arg) c.1138G>C (p.Gly380Arg) | achondroplasia | 100800 | autosomal dominant | GOF | 40 | 0 | 100% | 35.9; 36.3; 37.2 | 7.1 (4.6–9.6) | ∼1/30,000 | yes | not reported | bladder cancer | Orioli et al.16,20, Waller et al.17, Tiemann-Boege et al.53, Wilkin et al.163 |
| FGFR3 | fibroblast growth factor receptor 3 | 749C>G (p.Pro250Arg) | Muenke syndrome | 602849 | autosomal dominant | GOF | 10 | 0 | 100% | 35.1 | 4.1 | ∼1/130,000 | no | not reported | no | Rannan-Eliya et al.21 |
| HRAS | Harvey rat sarcoma viral oncogene homolog | various mutations in a few codons, most commonly c.34G>A (p.Gly12Ser) | Costello syndrome | 218040 | autosomal dominant | GOF | 43 | 2 | 96% | 37.3; 38.0; 36.2 | 5.5 (4–7) | raref ∼1/300,000 | no | bladder rhabdo-myosarcoma | many cancer types | Sol-Church et al.25, Zampino et al26, Schulz et al.27, Søvik et al.164, Gripp et al.165 |
| PTPN11 | protein tyrosine phosphatase, nonreceptor type 11 (encoding SHP2) | many mutations, 5 used for paternal origin studies | Noonan syndrome | 163950 | autosomal dominant | GOF | 14 | 0 | 100% | 35.6 | 6.1 | ∼1/10,000f caused by various mutations | no | juvenile myelomonocytic leukemia (JMML) | JMML and other hematologic malignancies | Tartaglia et al.24 |
| RET | ret proto-oncogene | various mutations clustering at 6 cysteine codons, most commonly at p.Cys634 | multiple endocrine neoplasia type 2A | 171400 | autosomal dominant | GOF | 11 | 0 | 100% | 39.3 | 8.7 | ∼1/70,000 | no | medullary thyroid carcinoma (MTC) | thyroid cancer | Schuffenecker et al.22, Wohllk et al.166, Mulligan et al.167 |
| RET | ret proto-oncogene | c.2753T>C (p.Met918Thr) | multiple endocrine neoplasia type 2B | 162300 | autosomal dominant | GOF | 25 | 1 | 96% | 33 | 3.1 | ∼1/150,000 | no | medullary thyroid carcinoma (MTC) | thyroid cancer | Carlson et al.23, Kitamura et al.168 |
GOF is used as an abbreviation for gain-of-function.
The Reference sequences (RefSeq) are: NM_000141 for FGFR2, NM_000142 for FGFR3, NM_001130442 for HRAS, NM_002834 for PTPN11 and NM_020630 for RET.
The RefSeq are: NP_000132.3 for FGFR2, NP_000133.1 for FGFR3, NP_001123914.1 for HRAS, NP_002825.3 for SHP2, NP_066124.1 for RET (long isoform).
The different ages represent data obtained from independent studies. To establish statistical significance, these data are compared to matched population data to take into account the natural variation in average paternal age for different countries and years of birth.
The data given represent the average paternal age excess obtained from different studies (with data range given in brackets).
Reference: COSMIC database.
Collectively RASopathies have an incidence estimated to be 1/2,000 to 1/5,000 births.169 However, because of the large diagnostic overlap, precise epidemiological data on the prevalence of each mutation are scarce.
For historical and practical reasons, the best studied examples of PAE disorders are Apert syndrome, characterized by craniosynostosis (premature fusion of the cranial sutures) and severe syndactyly of both hands and feet, and achondroplasia, the most common cause of short-limbed dwarfism. Both conditions are fully penetrant and clinically homogeneous monogenic disorders that allow complete ascertainment and exhibit a straightforward genotype-phenotype correlation: about 99% of individuals with Apert syndrome carry either of two transversions (c.755C>G or c.758C>G), encoding substitutions in two adjacent amino acids (p.Ser252Trp or p.Pro253Arg, respectively) located within the extracellular region of the receptor tyrosine kinase protein fibroblast growth factor receptor-2 (FGFR2),29 whereas substitutions at a single nucleotide (encoding a p.Gly380Arg mutant protein) in its paralog FGFR3 cause more than 95% of achondroplasia cases;30 in the FGFR3 c.1138G>A transition outweighs the c.1138G>C transversion by a factor of 35-fold.30,31 Among the other FGFR-associated disorders belonging to the PAE class, Crouzon and Pfeiffer syndromes are clinically overlapping conditions typically caused by any of more than 50 specific activating point mutations in FGFR2; craniosynostosis occurs as in Apert syndrome, but limb abnormalities are milder.32 A single c.749C>G transversion in FGFR3 (resulting in a p.Pro250Arg substitution equivalent to the FGFR2 Apert-causing p.Pro253Arg) is responsible for all cases of Muenke syndrome, the most common genetic cause of coronal craniosynostosis, and makes this disorder allelic to, but clinically distinct from, achondroplasia.33
Of the other PAE disorders, Costello and Noonan syndromes are part of a larger family of neuro-cardio-facial-cutaneous syndromes or RASopathies, the latter term describing their collective dysregulation of the RAS signaling pathway.34 Patients with these conditions present with variable combinations of distinctive craniofacial features; short stature; failure to thrive; developmental delay; and skin, cardiac, and skeletal abnormalities. A few Noonan syndrome cases have also been associated with craniosynostosis.35 Up to 90% of Costello syndrome patients harbor the c.34G>A transition in HRAS (encoding p.Gly12Ser) at a well-known mutation hotspot in tumorigenesis, whereas about half of Noonan syndrome mutations are detected within the PTPN11 gene (encoding SHP2-containing tyrosine phosphatase). However, specific point mutations in other components of the RAS pathway (i.e., BRAF, CBL, CRAF, KRAS, NRAS, SHOC2, and SOS1) have also been described in Noonan patients.34,36
The last two PAE disorders, multiple endocrine neoplasia types 2A (Men2A) and 2B (Men2B), are caused by allelic mutations within the RET receptor tyrosine kinase and are complex syndromes of multiple endocrine neoplasms, characteristically including medullary thyroid carcinoma (MTC). Men2A might also be associated with pheochromocytoma and parathyroid adenomas, whereas in Men2B, the onset can be congenital with marfanoid habitus and mucosal neuromas (involving lips, tongue, eyebrows, and intestine) as well as skin hyperpigmentation and café-au-lait spots on the hands and feet. Although most cases of Men2A are caused by a handful of activating mutations at crucial cysteine residues located in the extracellular portion of RET (i.e., p.Cys609, p.Cys611, p.Cys618, p.Cys620, p.Cys630, and p.Cys634), Men2B is a more aggressive syndrome mostly caused by a single p.Met918Thr (c.2753T>C) substitution at a residue located in the catalytic core of the tyrosine kinase domain.37
Although each PAE syndrome is a clearly distinct and well-defined complex pathological entity, there is a noticeable phenotypic overlap between them; all involve combinations of congenital skeletal abnormalities (for example craniosynostosis, hypertelorism, syndactyly, coarse facies, marfanoid habitus, or short stature), in some cases associated with developmental delay, cardiac malformation, skin hyperpigmentation, and cancer predisposition. These complex phenotypes highlight the pleiotropic role played by the PAE genes during development, whereas the clinical overlap points to the fact that these genes are required in common cellular contexts in shared molecular pathways. Several closely related conditions, including thanatophoric dysplasia (caused by FGFR3 mutations) and cardio-facio-cutaneous syndrome ([MIM 115150] associated with BRAF, MAP2K1, and MAP2K2 mutations) that exhibit a large phenotypic and molecular overlap with the disorders described above, are likely to belong to the PAE class, although some of the experimental evidence for meeting all three PAE criteria is still lacking (Table 2). Significantly, at a biochemical level, all PAE mutations confer gain-of-function properties to the encoded mutant proteins through a wide diversity of mechanisms (Table S1, available online).
Table 2.
Disorders Sharing Several Features with PAE Disorders but for which Some Data Are Still Missing
| Gene symbol | Gene | DNA Mutation (Amino Acid)a | Clinical Disorder | OMIM Reference Number | Mode of Transmission | Mutation Mechanism | Informative Paternal Mutations | Informative Maternal Mutations | Paternal Origin Percentage | Average Paternal Ageb(Years) | Average Excess in Paternal Agec(Years) | Estimated Birth Prevalence for New Mutations | Sperm Study | Cancer Predisposition in Patients? | Reported as Somatic mutation?d | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BRAF | v-RAF murine sarcoma viral oncogene homolog-B1 | various mutations | cardio-facio-cutaneous syndrome | 115150 | autosomal dominant | GOF | n/a | n/a | n/a | 35.8 | 4.8 | raree ∼1/200,000–1/300,000 | no | not reported | many cancer types | Schulz et al.27 |
| MAP2K1 MAP2K2 | mitogen-activated protein kinase kinase 1/2 | various mutations | cardio-facio-cutaneous syndrome | 115150 | autosomal dominant | GOF | n/a | n/a | n/a | melanoma | Schulz et al.27 | |||||
| FGFR3 | fibroblast growth factor receptor 3 | mutations at six residues, most commonly c.742C>T (p.Arg248Cys) | thanatophoric dysplasia type I | 187600 | autosomal dominant | GOF | n/a | n/a | n/a | 33.6; 36.41 | 5.1 (2.5–7.6) | ∼1/60,000 | no | n.a. | bladder, skin, myeloma | Orioli et al.20, Goriely et al.62, Tavormina et al.63 |
| FGFR3 | fibroblast growth factor receptor 3 | c.1948A>G (p.Lys650Glu) | thanatophoric dysplasia type II | 187601 | autosomal dominant | GOF | n/a | n/a | n/a | bladder, skin, testis | Orioli et al.20, Goriely et al.62, Tavormina et al.63 |
The following abbreviations are used: GOF, gain-of-function; n/a, not available; n.a., not applicable (TD is a neonatal lethal disorder).
The RefSeq used for numbering are given in footnotes a and b of Table 1.
The different ages represent data obtained from independent studies. To establish statistical significance, these data are compared to matched population data to take into account the natural variation in average paternal age for different countries and years of birth.
The data given represent the average paternal age excess obtained from different studies (with data range given in brackets).
Reference: COSMIC database.
Collectively RASopathies have an incidence estimated to be 1/2,000 to 1/5,000 births.169 However, because of the large diagnostic overlap, precise epidemiological data on the prevalence of each mutation are scarce.
Paternal Origin of Mutations, Paternal Age Effect, and High Rate of Spontaneous Mutations
Each of the properties used to define PAE mutations will be considered separately, and we will discuss why they are relevant to our understanding of paternal age effects. We will argue that although a paternal bias in mutation origin is observed for many spontaneous dominant disorders, and likely results from a universal replication-based process that generates DNA copy errors during male gametogenesis, the extreme paternal bias observed for PAE mutations is caused by the distinct phenomenon of clonal expansion of spermatogonial cells expressing proteins with gain-of-function properties. This selective process taking place in the testis leads to the relative enrichment of mutant sperm over the course of time, which accounts for the epidemiological paternal age effect and the high de novo birth rates associated with the PAE disorders.
Gender Bias in the Origin of Spontaneous Mutations
Studies of mutations in humans have long established that there is a gender-specific signature in the origin of different types of genetic alterations. Although chromosomal nondisjunctions originate mainly in the female germline1 and large genomic rearrangements might show either a male or female bias in origin, the majority of point mutations and small deletions or insertions tend to be paternal in origin. This gender bias is generally explained by fundamental differences in germ cell biology in the female and male lineages.38,39 By the time of birth, germ cells in the developing ovary have already completed their proliferative phase; because all postnatal phases of germ cell development are meiotic, maternal mutagenic events involve mostly recombination-related mechanisms. By contrast, spermatogenesis requires regular mitotic divisions of spermatogonial stem cells (SSCs) throughout male reproductive life. From puberty onward, cells in the SSC lineage replicate at a rate of 23 divisions per year (in synchrony with the epithelial cycle, i.e., every 16 days in humans) to produce different types of cells; some maintain the stem cell population, whereas others follow a differentiative pathway and produce mature sperm after approximately five further divisions. Because this process involves recurrent rounds of DNA replications, random copy-error mutational events are predicted to arise mainly in the male germline,40 explaining the elevated male-to-female mutation ratio (α) of between 2 and 7, which is observed for the majority of spontaneous point mutations and small deletions41,42 (Table 3). In contrast to this general situation, PAE mutations show much more extreme bias in mutation origin with values of α > 20 (Table 1).
Table 3.
Examples of Dominant Disorders Associated with Biased Paternal Origin of Mutations but No Significant Paternal Age Effect
| Clinical Disorder | OMIM Reference Number | Gene Symbol | Number of Mutations Reported | Mode of Transmission | Mutation Mechanism | Informative Paternal Mutations | Informative Maternal Mutations | Paternal Origin | Average Paternal Age (Years) | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Alexander disease | 203450 | GFAP | ∼35, two recurrent | autosomal dominant | structural alteration | 24 | 4 | 86% | 32.3 NS | Li et al.170 |
| CHARGE (coloboma, heart anomaly choanal atresia, retardation, genital and ear anomalies) | 214800 | CHD7 | >200 | autosomal dominant | LOF haploinsufficency | 12 | 1 | 93% | 32.92 NS(a) | Pauli et al.171 |
| Craniofrontonasal syndrome | 304110 | EFNB1 | >100 | X linked dominant | LOF cellular interference | 15 | 2 | 88% | 32.1 NS | Twigg et al.172 |
| Dravet syndrome or severe myoclonic epilepsy of infancy | 607208 | SCN1A | >400 | autosomal dominant | LOF haploinsufficency | 43 | 13 | 77% | 33.4 NS | Heron et al.173, Sun et al.174 |
| Rett syndrome | 312750 | MECP2 | >300, eight recurrent | X linked dominant | LOF(b) | 53 | 5 | 91% | 31.3 NS | Trappe et al.175, Girard et al.176, Zhu et al.177 |
| Townes-Brocks syndrome | 107480 | SALL1 | >40 | autosomal dominant | LOF haploinsufficency | 14 | 2 | 88% | 29.9 NS | Böhm et al.178 |
| Treacher Collins syndrome | 154500 | TCOF1 | >120 | autosomal dominant | LOF haploinsufficency | 7 | 3 | 70% | 27.8 NS | Splendore et al.179 |
The following abbreviations are used: NS, not significantly different from control population on statistical testing; LOF, loss-of-function; GOF, gain-of-function.
Note that in this study paternal age is given for the whole cohort rather than for paternally derived mutations only.
Rett syndrome mostly affects females. Because of random X inactivation, the clinical presentation of Rett syndrome shows considerable heterogeneity.
Paternal Age Effect
For most spontaneous dominant disorders, despite the expectation of the copy-error hypothesis,39 epidemiological data concur that the bias in paternally originating mutations is not associated with a marked paternal age effect, showing that paternal bias and paternal age effect are not equivalent and can be dissociated. The paternal age effect is an epidemiological concept describing the fact that some spontaneous disorders tend to arise more frequently in the progeny of older men. As early as 1912, Wilhelm Weinberg, while studying sporadic cases of achondroplasia, noted a higher incidence of the disease in the last-born children of a sibship.43 In 1955, Lionel Penrose suggested that rather than birth order itself, the key risk factor associated with sporadic achondroplasia was advanced paternal age.40 Although the notion of paternal age effect is easy to grasp, confounding with correlated maternal age made its study problematic in the premolecular era because the parental origin of each individual mutation could not be deduced.
The concept of paternal age effect received further recognition after the comprehensive study by Risch et al.,2 which systematically analyzed the distribution of parental ages for spontaneous cases of 17 different autosomal-dominant disorders. Among these, the authors compared parental age profiles to the age distribution in the general population (obtained from year-matched population census data) for seven disorders and derived an observed/expected (O/E) ratio that gave a direct measurement of the age-specific incidence excess for each disorder. This approach suggested that the disorders fell into two main categories. Some syndromes showed no, or a small, linear increase with maternal and paternal ages; whereas for others, including Apert, Pfeiffer, and Crouzon syndromes and achondroplasia, the age excess was more pronounced and the relationship with paternal age was nonlinear (proposed to be exponential), rising sharply as the father's age increased even after correction for maternal age. Although these data singled out the paternal age as the major risk factor in this latter group of genetic disorders, the authors suggested that maternal age provided an independent but lesser contribution. Because we now know (from parent-of-origin studies) that maternal age is irrelevant to PAE disorders, this illustrates the inherent ambiguity arising from epidemiological studies when unsupported by molecular data. Moreover, because the magnitude of paternal age effects is of similar order (∼2–7 years) to natural variation in average paternal age attributable to place (country), year of birth, and whether the birth occurred within or outside marriage, case observations need to be carefully compared to control data sets matched for all these variables—for which information is often difficult to obtain.
The traditional explanation for these paternal age effects involved a model of age-dependent accumulation of recurrent mutations taking place within localized hypermutable DNA hotspots (the copy-error hypothesis).39,40 However, it was noted that a simple additive model of replication errors could not explain either the magnitude of some of the most extreme paternal age effects or the nonlinear shape of the observed epidemiological data.2,39,44 To account for this, it was suggested that other age-dependent factors, such as reduced DNA replication fidelity, inefficiency of repair mechanisms, or repeated mutagenic exposures would have to contribute to the accumulation of copy errors.39
Today, molecular characterization of the causative mutations allows refined documentation of paternal age effects by including only those cases shown to have a paternal origin, most commonly by determining the phase of the mutation with respect to nearby polymorphisms or haplotypes segregating in parent-child trios.45 This approach has established that the disorders associated with strong paternal age effects (∼2–7 years) also exhibit an extreme paternal bias in the origin of the causative mutations (Table 1). The bias observed for PAE mutations (α > 20) is more pronounced than would be expected from gender differences in the number of germ cell divisions.
High Germline Mutation Rate
The third defining characteristic of PAE disorders is the high absolute rates observed for specific de novo point mutations (Table 1 and Figure 1). The average background rate of spontaneous point mutations in the human genome, estimated by several different methods, including recent direct transgenerational measurement of mutational load, is ∼1.1 × 10−8 (range: 0.4 − 16 × 10−8, depending on the type of mutation) per nucleotide per generation46–52 (Figure 1A). Given the very narrow mutational spectrum of several sporadic PAE disorders, it can be calculated that the causative mutations comprise the most frequent germline nucleotide substitutions in the entire genome, occurring at levels up to 1,000-fold higher than the background nucleotide substitution frequencies (Table 1 and Figure 1B). Simple mechanisms of mutation accumulation, such as copy error, are difficult to reconcile with the very high α values and absolute rates of spontaneous PAE mutations, casting doubt on the notion that copy errors at localized mutation hotspots are the sole driver of this process. Experimental study of these mutations directly in sperm and testes has provided insights into their mechanisms of origin.
Figure 1.
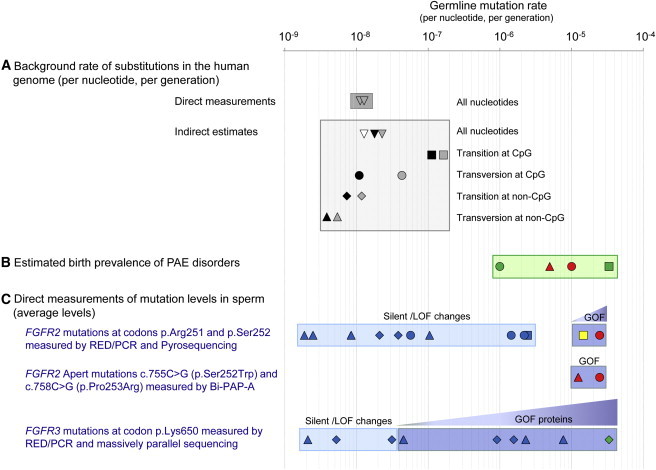
Rate of Germline Mutations in the Human Genome on a Logarithmic Scale
(A) Background rate of nucleotide substitution quantified by direct measurements of transgenerational mutational load48,50,52 (unfilled inverted triangles in dark gray box) and indirect estimates (lighter gray box) for all substitutions (inverted triangles), transitions at CpG dinucleotides (squares), transversions at CpG dinucleotides (circles), transitions at non-CpG dinucleotides (diamonds), and transversions at non-CpG dinucleotides (triangles). Data source indicated by symbol shading as follows: black,46 gray,47 and white.49
(B) Estimated birth prevalence for de novo mutations in two PAE syndromes (green box). Apert syndrome FGFR2 mutations caused by c.755C>G transversion at a CpG dinucleotide account for 66% of cases (red circle) and mutations caused by c.758C>G transversion at a non-CpG dinucleotide for 33% of cases (red triangle). Achondroplasia FGFR3 mutations caused by c.1138G>A transition or c.1138G>C transversion at a CpG dinucleotide are marked by the green square and circle, respectively. Note that these figures assume that all mutations are paternal in origin.
(C) Direct measurements of mutation levels in human sperm at specific locations in the genome. Symbols for each mutation (square, circle, diamond, and triangle) refer to mutation types as in (A). Top blue boxes show average levels for the FGFR2 c.752–755 positions (encompassing codons p.Arg251 and p.Ser252) measured by RED/PCR/pyrosequencing in the sperm of 99 healthy men. The strongly activating c.755C>G (p.Ser252Trp) Apert transversion (red circle in dark blue box) and the c.755C>T (p.Ser252Leu) transition (yellow square in dark blue box) are associated with gain-of-function (GOF) properties; these two substitutions are found at significantly higher levels in sperm than any of the ten other mutations encoding silent or loss-of-function (LOF) mutations (light blue box). The average age of the sperm donors was 37.4 years.56,57 Middle blue box shows average levels of the two Apert mutations measured in the sperm of 323 healthy men by Bi-PAP-A. The average age of the sperm donors was 38.7 years.58 Note that the very similar estimate for the FGFR2 mutation c.755C>G (red circles) was obtained by different methods. Prevalence in sperm exceeds birth prevalence (B) because sperm donors were older on average than the population of fathers. Bottom blue boxes show average levels at the FGFR3 codon p.Lys650 in the sperm of 78 men via RED/PCR/massively parallel sequencing. The substitutions encoding proteins with gain-of-function properties (dark blue box), and in particular the c.1948A>G transition (p.Lys650Glu, causing TDII, green diamond), are found at higher levels in sperm than the changes associated with silent/loss-of-function substitutions (light blue box). The average age of the sperm donors was 40.2 years.62
Direct Quantification of FGFR2- and FGFR3-Associated PAE Mutations in Sperm and Testes
The unusual characteristics of the Apert and achondroplasia point mutations have led several groups to develop methods to identify these mutations directly in sperm and testes of normal individuals. The sperm studies are a powerful complement to the epidemiological analysis of disease prevalence and allow direct estimates of mutation levels at specific positions within the genome. Another advantage of this approach is the ability to study a wider age range and, in particular, to assess the paternal age effect at older ages, when there are too few paternities for epidemiological data to be accurate.
The development of sensitive and reproducible methods to assay these nucleotide substitutions constitutes a major technical challenge. The main problem lies in the extreme rarity of the mutations to study, which, on the basis of observed birth prevalence of the cognate disorders, are expected to be present at levels around 1 in 30,000 to 1 in 100,000 at best (Figure 1B). At such low levels, the detection of single-nucleotide substitutions via direct PCR amplification is not feasible because several million genome copies are necessary to isolate only a few mutant molecules. For example, at a mutation level of 10−5, 10 μg of human genomic DNA—typically more than 100-fold in excess of the usual input for a single PCR reaction—corresponds to three million copies of the haploid genome and would therefore contain only ∼30 mutant molecules. In addition, the potential impact of the false-positive rate caused by in vitro PCR-induced copy errors, oxidative stress-induced mutations (for example, those arising during DNA extraction), or inadvertent contamination require careful consideration in the experimental design. Partly for these reasons, two early papers, on measurement of the achondroplasia53 and the Apert54 mutations are, in our opinion, subject to technical or interpretational flaws that call into question the conclusions of these studies. These issues have been discussed previously55 and hence are not considered further here.
In 2003, our group developed a sensitive and reproducible assay for the most common Apert mutation (c.755C>G in FGFR2).56 This approach involved a combination of restriction enzyme digestion ([RED] specifically targeting and digesting away the large excess of wild-type sequences around the position of interest) with PCR (RED/PCR) to enrich for mutant sequences, and pyrosequencing technology to quantify the resulting complex mixture of nucleotides. Measurements of this C>G transversion in 99 healthy men revealed elevated levels (average: 2.3 × 10−5; range: < 10−6 − 1.6 × 10−4) in the sperm of most men (Figure 1C) and a significant positive correlation (r = 0.39) with age; by contrast, the mutation levels measured in blood DNA from 11 individuals were much lower (an average of 0.28 × 10−6, i.e., below the limit of accurate detection) and did not increase with the donor's age. The levels of mutation measured in sperm accounted for the reported birth prevalence of Apert patients and the age increase mirrored the epidemiological observation of the paternal age effect described for Apert syndrome.56 Mutation levels in the sperm of six men who had fathered a child with the c.755C>G Apert mutation were within the range of values observed for controls of similar age, suggesting that these men were sampled from the general population and that their risk of fathering another affected child was extremely low.56
The RED/PCR approach enabled quantification of other mutations occurring around position c.755 of FGFR2, which revealed a surprising result. Another substitution, c.755C>T, encoding a p.Ser252Leu change that has been described occasionally in patients with mild Crouzon syndrome, was also present at unexpectedly high frequencies (on average only 1.6-fold lower than the c.755C>G levels) in sperm.57 This effect was not observed for c.755C>A (which encodes a change to a stop codon at position p.252) or for any of the nine nucleotide substitutions involving changes at FGFR2 positions c.752–754 (Figure 1C).57 Key evidence was also provided by a phasing assay that exploited a single-nucleotide polymorphism (SNP) located 118 bp from the c.755C nucleotide (Figures 2A and 2B). Instead of the c.755C>G mutations being distributed roughly equally on the two chromosomes in men heterozygous for the SNP—as would be predicted from a model of mutation accumulation at a recurrent hotspot—even in sperm with high mutation levels (corresponding to more than 1,000-fold excess over background), the majority of mutations were usually present on one or the other allele. This skewing effect was much more pronounced for the c.755C>G than the less abundant c.755C>T mutation, an observation incompatible with a neutral model of mutation accumulation, in which the more prevalent mutation is expected always to exhibit less skewing56 (Figure 2B). Collectively, these findings led us to propose56,57 that, rather than resulting from accumulation of multiple independent replication errors during spermatogenesis, the originating FGFR2 mutational events in Apert syndrome are rare, but once they occur, the mutations become enriched because of a selective advantage conferred by the mutant FGFR2 protein, leading to clonal expansion of the mutant SSC (Figure 2A). This process in turn results in a relative enrichment of mutant sperm over time (Figure 2B), accounting for the observed paternal age effect and the high birth rate associated with this specific mutation. To distinguish this mechanism from the previously proposed copy-error process involving the simple accumulation of mutational hits over time, we named this mechanism protein-driven selfish selection.57
Figure 2.
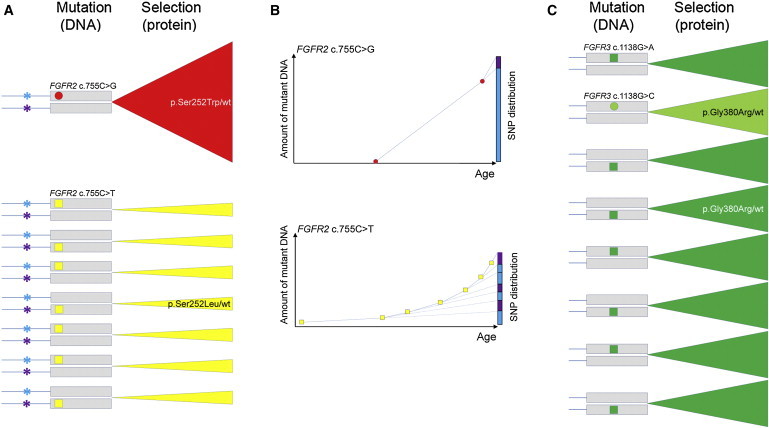
Relative Prevalence of FGFR2 and FGFR3 Mutations Explained by Combined Effects of Copy Error, at the DNA level, and Selfish Selection, at the Protein Level, in SSC
(A) Mutational events taking place at FGFR2 position c.755C. Each pair of lines with gray boxes denotes the two FGFR alleles (box represents the exon and lines represent the intron. Asterisks in (A) represent a SNP located in the intron upstream of FGFR2 position c.755; blue and purple denote the two different alleles of the SNP. Circles and squares represent single events of transversion and transition at CpG dinucleotide sites, respectively. Triangular sectors indicate expansion over time of clones carrying the resulting mutant protein (selection). wt is used as an abbreviation for wild-type. The c.755C>G transversion (red circle) occurs rarely, but the resulting p.Ser252Trp substitution (causing Apert syndrome in the germline) confers a strong selective advantage to the mutant SSC (red triangular sector), leading to clonal expansion over time. The background mutation rate for the c.755C>T transition at this CpG dinucleotide (yellow square) is higher (∼3.3- to 10-fold),46,47 but the resulting mutant protein (p.Ser252Leu associated with Crouzon syndrome) confers a weaker selective advantage compared to p.Ser252Trp, leading to slower clonal expansion (yellow triangular sectors). The original mutation events occur randomly on either FGFR2 allele (blue or purple asterisk in men heterozygous for the SNP) at different times within the testis of an aging man.
(B) Accumulation of c.755C mutations in FGFR2 in sperm over time. Because of the rarity of the mutational events, the distribution of the linked SNP (graphs, blue and purple columns on the right) is more skewed for the c.755C>G (above) than the c.755C>T mutation (below); however, the prevalence of c.755C>G is greater than c.755C>T because of the greater selective advantage conferred by the p.Ser252Trp compared to the p.Ser252Leu mutant protein.
(C) Mutational events taking place at FGFR3 position c.1138G cause achondroplasia. The c.1138G>A transition (green square) is more frequent than the c.1138G>C transversion (light green circle) at this CpG dinucleotide site. Both resulting proteins encode p.Gly380Arg that confers the same selective advantage to the mutant SSC (green triangular sectors). The difference in birth prevalence of achondroplasia caused by the two mutations is likely to be explained by the relative frequency of the original mutational events at FGFR3 position c.1138. Although selection for the FGFR3 achondroplasia mutation is probably weaker than for the FGFR2 Apert mutation, nevertheless achondroplasia mutations occur more frequently because of the high intrinsic rate of the c.1138G>A transition.
The main conclusions of these studies were confirmed and extended by independent work that exploited a different method of mutation detection involving a sensitive allele-specific PCR assay termed bidirectional pyrophosphorolysis-activated polymerization allele-specific amplification (Bi-PAP-A).58 The levels of both FGFR2 mutations associated with Apert syndrome (c.755C>G and c.758C>G) were estimated in the sperm of a large cohort of men and showed a similar average level and age effect for the c.755C>G mutation (2.4 × 10−5; range: < 10−6 − 7.25 × 10−4) to that previously reported56 (Figure 1C). Although the technique used was not able to partition mutations on the two FGFR2 alleles, the same group used their Bi-PAP-A assay to quantify the spatial distribution of mutations within whole human testes dissected into ∼200 pieces. This demonstrated a small number of localized mutation foci (0–∼8 per testis) exhibiting elevated mutation levels, up to 6% of the haploid genome in individual testicular pieces; rather than being distributed randomly at low level over the whole testis (as would be expected from a copy-error model), 95% of the Apert mutations clustered within fewer than 5% of the testis pieces and accounted for the mutation load present in epididymal sperm DNA from the same individual. Furthermore, mathematical modeling provided additional evidence for a mechanism involving infrequent mutational events followed by a process of localized clonal expansion of mutant SSC with weak selective advantage (∼0.01 per cell generation).59,60 A study of the FGFR3 achondroplasia mutation in testes,61 although relying on the same protocol and hence subject to the same technical limitations as earlier work in sperm,53 obtained a similar picture of localized mutational clusters in testicular biopsies of older men. Although experimental confirmation is awaited, we anticipate that the FGFR3 achondroplasia mutation is likely to show similar behavior to the Apert mutation in sperm (Figure 2C). This hypothesis is supported by another sperm study62 targeting an allelic FGFR3 mutation (c.1948A>G [encoding p.Lys650Glu]) associated with the germline disorder thanatophoric dysplasia (TD) type II,63 a severe neonatal lethal skeletal condition that shares many diagnostic features with the other PAE disorders (Table 2 and Figure 1C) and will be reviewed in the next section.
The emerging consensus is that the major determinant of the paternal age effect is not the hypermutability of specific nucleotides located at genomic hotspots but rather the effect of expressing the resulting mutant protein in SSC, which is associated with positive selection resulting in localized clonal expansion. Certainly, initial copy errors have to take place during spermatogenesis for this process to occur; therefore, the endogenous mutability and local sequence context64 of a nucleotide contribute to this process. As experimental data have shown that the rate of spontaneous transitions at CpG dinucleotides is up to ∼45 times higher than transversions at non-CpG sites46,47,65 (Figure 1A), the relative contribution of this effect (compared to selection) is likely to be small, although not insignificant. This is illustrated by the c.1138G>A transition in FGFR3, which is ∼35 times more commonly reported in de novo cases of achondroplasia than the c.1138G>C transversion;30,31 although both substitutions encode the same p.Gly380Arg change in FGFR3 and are therefore predicted to exhibit the same strength of positive selection at the protein level, a difference in background rates of mutability at this CpG dinucleotide is likely to explain the relative abundance of each substitution observed in patients with achondroplasia (Figure 2C). The relative contributions of intrinsic mutation rate and selection also explain the differing abundance of the mutations observed at FGFR2 position c.755C, again part of a CpG dinucleotide (Figures 2A and 2B). Similar to the achondroplasia situation, the endogenous rate of the c.755C>T transition is predicted to be higher than that of the c.755C>G transversion; however, at the protein level, the resulting gain-of-function associated with the c.755C>G, which encodes a stronger activating change (p.Ser252Trp) in FGFR2—as exemplified by the more severe phenotypic characteristics of Apert syndrome—than the c.755C>T (p.Ser252Leu) change associated with mild Crouzon syndrome, is likely to result in a more significant clonal expansion of the mutant SSC.56 By superimposing these two components, both the higher absolute mutation levels and the stronger allelic bias observed in sperm for the c.755C>G compared to the c.755C>T substitution can be explained (Figures 2A and 2B).
In summary, by combining the processes of background variation in copy-errors with selfish selection in the testis, the cardinal properties of PAE mutations (the extreme male mutation bias, the paternal age effect and the high germline mutation rate) are all accounted for. Conceptually, the mechanisms involved in protein-driven selfish selection of PAE mutations are not new but are more familiar in the context of somatic mutations occurring during neoplasia rather than in association with germline disorders. These links are explored in the next section.
Selfish Selection in the Testis: Spermatocytic Seminoma and Other Links with Oncogenesis
In agreement with their highly localized mutational spectrum, biochemical evidence concurs that all known PAE mutations result in activating proteins with gain-of-function properties (Table S1). Consistent with this, comparison of the spectrum of PAE mutations observed in the germline to that of somatic mutations found in malignancies shows that the majority of them have been reported as (or are allelic to) oncogenic mutations that, in different cellular contexts, have been associated with various tumor types (Table 1 and Figure 3), including endometrial cancer (FGFR2); multiple myeloma and transitional cell carcinoma of the bladder (FGFR3); hematological malignancies, most often juvenile myelomonocytic leukemia (JMML) (PTPN11/SHP2); and medullary thyroid carcinoma (RET). Somatic HRAS mutations have been described in many tumors, including cancers of the bladder, cervix, thyroid, prostate, and breast; mutations at three hotspots encoding (p.Gly12, p.Gly13, and p.Gln61) in the paralogous RAS proteins (HRAS, KRAS, and NRAS) are found in ∼30% of all malignancies (Catalogue of Somatic Mutations In Cancer [COSMIC] database).
Figure 3.
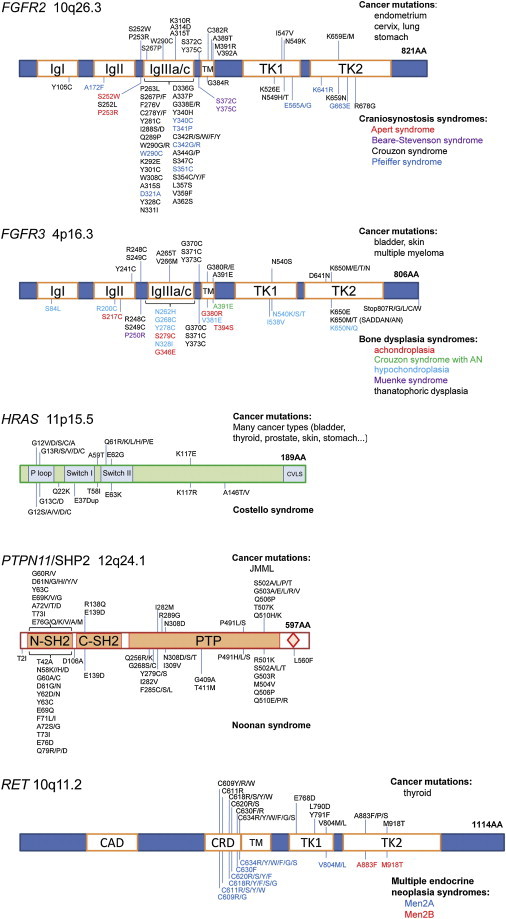
Somatic and Germline Gain-Of-Function Mutations Associated with the Five PAE Genes
In each case, the name of the gene or protein, its genomic location, and the functional protein domains (see abbreviations below) are indicated; the most common mutations (single-letter amino acid codes) associated with cancer (COSMIC database) are indicated above the schematic of the protein along with some of the cancers for which the mutations have been described. The germline mutations associated with congenital syndromes (Human Gene Mutation Database [HGMD] database) are indicated below the protein and are color coded according to the associated disorders (key on the figure). A few complex rearrangements and splice-site mutations are omitted for clarity. Proteins are numbered and drawn to scale (except for HRAS, for which the scale is 3:1, and RET, for which the scale is 0.9:1) according to the reference sequences (for RefSeq, see footnote b in Table 1); the length in amino acids (AA) is indicated in bold. The following abbreviations are used: Ig, immunoglobulin-like domain; TM, transmembrane domain; TK, tyrosine kinase domain; P loop, phosphate-binding loop; Switch, switch domain; CVLS, prenylation signal sequence; SH2, Src Homology 2 domain; PTP, phosphotyrosine domain; CAD, cadherin domain; CRD, cysteine-rich domain; SADDAN, severe achondroplasia with developmental delay and acanthosis nigricans; AN, acanthosis nigricans; and JMML, juvenile myelomonocytic leukemia.
The link between PAE genes implicated in selfish selection and their involvement in tumorigenesis led us to propose that the clonal expansion associated with the selective advantage conferred to the mutant SSC by the FGFR2 Apert mutation could contribute to the formation of testicular tumors.56,66 Although no somatic FGFR2 mutations were identified in a panel of cancer cell lines and common testicular tumors,66 we found mutations in other PAE genes in a distinct testicular neoplasm termed spermatocytic seminoma (SPS).62 SPS is a rare germ cell tumor comprising less than 5% of seminomas that is found exclusively in the testis (with no ovarian equivalent), where it presents as a slow growing well-circumscribed mass that rarely metastasizes. Unlike classical seminomas that mostly affect young adults and originate during embryonic gonadal development, SPSs derive from adult spermatogonia and are found specifically in older men; the mean age at diagnosis is around 54 years (age range: 19–92 years).67–69 Screening of 30 SPS identified two tumors with an p.Lys650Glu alteration in FGFR3, identical to that causing the lethal germline disorder TDII, whereas five further samples carried a mutation within the Costello syndrome-associated gene, HRAS.62 The HRAS alterations all occurred at position p.Gln61 (three p.Gln61Arg and two p.Gln61Lys), substitutions that confer stronger transforming activity in vitro than the common p.Gly12Ser Costello syndrome mutation.70 It is anticipated that germline alterations at HRAS, which have never been reported, will be analogous to the strongly activating p.Lys650Glu in FGFR3 and might result in prenatal lethal phenotypes.26,71
Another illustration of the link between selfish selection in the testis and the principles of oncogenesis is provided by FGFR3 mutations altering p.Lys650, which were studied in the sperm of 78 healthy donors following identification of the c.1948A>G (p.Lys650Glu) mutations in SPS. This study utilized RED/PCR coupled with massively parallel sequencing62 to quantify each of the nine possible substitutions occurring within the codon encoding p.Lys650. Among these, six missense germline mutations have been reported in association with four distinct clinical phenotypes72,73 of variable severity, ranging from the lethal condition TDII, to the much milder disorders hypochondroplasia and acanthosis nigricans (MIM 100600). In agreement with the proposal that oncogenic and selfish properties depend on the biochemical strength conferred by the encoded gain of function to the altered protein, there was a strong correlation between the severity of the associated clinical phenotype, the documented degree of receptor activation,72 and the relative enrichment of mutations in sperm observed for each of the mutations encompassing codon p.Lys650 (Figure 1C).62 Moreover, the relative abundance of each mutation in sperm mirrored the distribution of somatic mutations altering p.Lys650 reported in bladder cancers,62 further suggesting that common molecular pathways and similar oncogenic mechanisms operate in these two different cellular contexts.
The selfish selection process also shares many similarities with the formation of seborrheic keratoses (SK), benign skin tumors that present as brown plaques with a verrucous surface. The prevalence of these lesions increases with age and are present in more than 80% of individuals over age 50; some people have many lesions of variable size (up to a few centimeters). Analysis of multiple SK within individuals reveals that these lesions frequently carry somatic FGFR3 oncogenic mutations, which in the germline are associated with TD.74 These similarities provide a useful analogy whereby PAE mutations can be viewed as monoclonal expansion events promoting the formation of “mole-like” lesions within the testes of aging men. As with SKs, for most PAE mutations, the clonal expansion of mutant SSC appears to be subexponential,56 suggesting that it is limited by growth arrest or activation of a cellular senescence response that restrains tumor progression.75,76
Taken together this evidence bolsters the view that PAE mutations should essentially be considered as somatic mutations with oncogenic properties and that selfish selection in the testis operates by the same general principles that have been described in tumorigenesis. However, these oncogenic mutations are unique because they are taking place within germ cells rather than somatic cells. Because mutant SSCs are able to produce sperm that can transmit the genetic lesion (as a germline mutation) to the offspring, the consequences of this process extend to the next generation. As a result, a single mutational event results both in a somatic phenotype that causes localized cellular growth—and in extreme cases leads to the formation of a testicular tumor—as well as a germline phenotype associated with a specific syndrome, uniquely unifying the concepts of germline and somatic mutation to an event taking place within a single cell, the SSC.
Multiple Nucleotide Substitutions Are a Frequent Feature of Selfish Selection
By analogy with tumorigenesis, where accumulation of sequential independent mutations is an important process contributing to oncogenesis and tumor progression,77,78 there is an enrichment of PAE mutations caused by multiple nucleotide substitutions (Table 4). Many of these mutations encode amino acid substitutions conferring strong and/or unique gain-of-function properties that, owing to the specific features of the genetic code, could not arise as single-nucleotide substitutions, whereas others are caused by mutations located in different codons that synergize with one another and potentiate the activity of the mutant protein. On the basis of the estimated background rate of double mutations (∼10−11),46 if these multiple substitutions had only arisen by chance, we would anticipate finding less than one case of any double substitution in the entire human population. Consistent with progressive enrichment for selectively advantageous mutations, we showed in the case of an Apert syndrome patient with a rare p.Ser252Phe missense change in FGFR2 (c.755_756delCGinsTC), that this double substitution had arisen sequentially over two generations.57 A similar process has been reported for double substitutions in RET causing Men2B.79,80
Table 4.
Multiple Nucleotide Substitutions Identified in PAE Disorders
| Germline Substitutiona | Protein Changea | Associated Disorder | Number of Cases Reported | References |
|---|---|---|---|---|
| FGFR2 | ||||
| c.514_515delGCinsTT | p.Ala172Phe | Pfeiffer syndrome | 1 | Kan et al.32, Ibrahimi et al.180 |
| c.755_756delCGinsTT | p.Ser252Phe | Apert syndrome | 2 | Lajeunie et al.181, Oldridge et al.182 |
| c.755_756delCGinsTC | p.Ser252Phe | Apert syndrome | 1b | Goriely et al.57 |
| c.755_756delCGinsTC | p.Ser252Phe | Apert syndrome | sperm | Goriely et al.56 |
| c.755_756delCGinsAC | p.Ser252Tyr | not known | sperm | Goriely et al.57 |
| c.755_757delCGCinsTCT | p.Ser252_Pro253 delinsPheArg | Pfeiffer syndrome | 1 | Oldridge et al.182 |
| c.[755C>T; 943G>T] | p.[Ser252Leu; Ala315Ser] | syndactyly | 1 | Goriely et al.57, Wilkie et al.183, Ibrahimi et al.184 |
| c.1024_1025delGCinsCT | p.Cys342Ser | Pfeiffer syndrome | 1 | Cornejo-Roldan et al.185 |
| FGFR3 | ||||
| c.[1130T>G; 1138G>A] | p.[Leu377Arg; Gly380Arg] | severe form of achondroplasia | 1 | Rump et al.186 |
| c.1138_1139delGGinsAA | p.Gly380Lys | hypochondroplasia | 1 | Santos et al.187 |
| c.[1454A>G; 1620C>A] | p.[Gln485Arg; Asn540Lys] | thanatophoric dysplasia | 1 | Pannier et al.188 |
| HRAS | ||||
| c.35_36delGCinsAA | p.Gly12Glu | Costello syndrome | 1 | Kerr et al.189 |
| c.35_36delGCinsTT | p.Gly12Val | Costello with cardiomyopathy | 2 | Aoki et al.71, van der Burgt et al.190 |
| RET | ||||
| c.[1895_1897delGCinsCG; 1900T>C] | p.[Glu632_Cys634 delinsAspValArg] | Men2A | 1 | Mulligan et al.191 |
| c.1902_1903delCCinsGG | p.[Cys634_Arg635 delinsTrpGly] | Men2A | 1 | Lips et al.192 |
| c.[2332G>A; 2411G>A] | p.[Val778Ile; Val804Met] | FMTC | 1 | Kasprzak et al.193 |
| c.[2411G>A; 2417A>G] | p.[Val804Met; Tyr806Cys] | Men2B | 1b | Miyauchi et al.79, Iwashita et al.80 |
| c.[2411G>A; 2531G>T] | p.[Val804Met; Arg844Leu] | FMTC | 1 | Bartsch et al.194 |
| c.[2411G>A; 2714A>G] | p.[Val804Met; Ser904Cys] | Men2B | 1 | Menko et al.195 |
| c.2647_2648delGCinsTT | p.Ala883Phe | Men2B | 5 | Gimm et al.196, Smith et al.197, Jasim et al.198 |
FMTC is used as an abbreviation for familial medullary thyroid carcinoma.
The RefSeq used for numbering are given in footnotes a and b of Table 1.
Cases where the second “hit” is documented to have been acquired de novo on the paternal allele.
Dysregulation of Growth Factor Receptor-RAS Signaling Links All PAE Disorders
The quantitative data supporting selfish spermatogonial selection have emerged from studies of FGFR2 and FGFR3 mutations, but the same principles are likely to extend to the other PAE disorders. By considering Men2A/B and Noonan and Costello syndromes, we can gather further clues about the likely biological and molecular mechanisms underlying selfish selection. Indeed, of the associated mutated genes, RET encodes a well-characterized regulator of SSC proliferative behavior (see next section), whereas PTPN11/SHP2 and HRAS encode key signaling transduction components located downstream of FGFR2, FGFR3, and RET, positioning all five PAE genes within a single molecular pathway, the growth factor receptor-RAS pathway.
As summarized in Figure 4, the RAS signaling cascade is activated upon binding of specific ligands to their cognate cell surface growth factor receptor tyrosine kinases (FGFRs and RET). This signaling cascade controls many different cellular functions, including proliferation, differentiation, survival, and apoptosis. The three RAS genes (HRAS, KRAS, and NRAS) encode small GTPases that act as key signal regulators, a role mediated by their ability to cycle between an active guanosine triphosphate (GTP)-bound and an inactive guanosine diphosphate (GDP)-bound conformation. In their active state, RAS proteins transduce signals through different effectors, such as the mitogen-activated protein kinases (MAPK) or phosphoinositide 3-kinase (PI3K). The MAPK pathway is activated by recruitment of the RAF serine threonine kinases (ARAF, BRAF, and CRAF) to the plasma membrane where they phosphorylate the MEK1 (MAP2K1) and MEK2 (MAP2K2) kinases, which in turn phosphorylate the extracellular signal-regulated kinases (ERK) ERK1 (MAPK3) and ERK2 (MAPK1). Upon phosphorylation, ERKs translocate to the nucleus where they modulate the activity of various transcription factors. Alternatively, activated GTP-bound RAS also interacts with the catalytic subunits of PI3K, which lead to production of phosphatidylinositol 3,4,5,-triphosphate (PIP3) at the plasma membrane. This signal is then transduced through PIP3 targets by phosphorylation of AKT (also known as protein kinase B [PKB]) serine/threonine kinases (pAKT) or stimulation of mammalian target of rapamycin (mTOR). In addition, the PI3K-AKT pathway can bypass RAS and be directly activated in response to growth factor stimulation.
Figure 4.
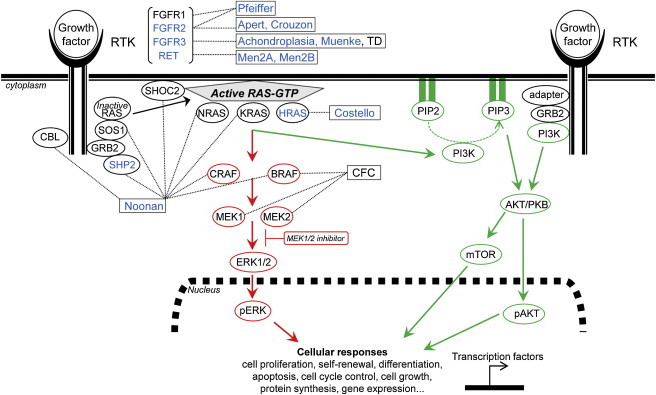
PAE Disorders Cluster within the Receptor Tyrosine Kinase (RTK)-RAS Signaling Pathway
The known PAE disorders (boxes) and the five PAE proteins that fulfill all three criteria to belong to the PAE class (as defined in the text) are indicated in blue. Other candidate PAE disorders (Table 2) are in black within the boxes. Signaling downstream of RAS involves the RAF/MEK/ERK (MAPK-ERK branch) (in red) and the PI3K-AKT-mTOR (in green) pathways. The MEK1/2 inhibitor (in red box) specifically blocks the phosphorylation of ERK1/2. For details and abbreviations see the main text.
Two lines of evidence suggest that dysregulation of the growth factor receptor-RAS pathway leading to selfish selection of PAE mutations is mediated by downstream activation of the MAPK-ERK branch. As well as the established PAE genes PTPN11/SHP2 (an upstream regulator of RAS) and HRAS itself, paternal age effects (but in the absence of formal demonstration of the parental origin of the causative de novo mutations) have also been described for cardio-facio-cutaneous (CFC) syndrome (Table 2) caused by mutations in downstream components of the RAS-(MAPK) pathway (BRAF, MEK1, and MEK2), directly implicating MAPK-ERK in the pathophysiology of these disorders. More indirectly, the phenotypic rescue of mouse models for Apert81 and Noonan82,83 syndromes, through treatment with specific pathway inhibitors (MEK1/MEK2 inhibitor) (Figure 4) or by genetic means84 provides strong evidence that these particular phenotypes, which include craniosynostosis, growth delay, facial dysmorphia, cardiac defects, and/or pulmonary valve stenosis, are caused by abnormal signal transduction through the MAPK-ERK branch.
Notwithstanding these observations, there is also indirect evidence implicating the PI3K-AKT cascade in selfish selection. For example, fibroblasts from Costello syndrome patients show an increased level of phosphorylated AKT compared with the level in controls, whereas MEK/ERK phosphorylation remains unchanged.85 Functional data (discussed in the next section) also support a key role for the PI3K/AKT pathway in SSC self-renewal.
PAE Genes Regulate Spermatogonial Self-Renewal
Although orthologs of all five PAE genes are expressed during mammalian spermatogenesis,57,62,69,86–89 the functional role of RET has been most extensively characterized in mice. Although Ret is expressed on the surface of SSCs,90 its ligand Glial-derived neurotrophic factor (Gdnf) is expressed by the supporting Sertoli cells, from where it regulates dose-dependent self-renewal and differentiation of SSCs. Heterozygous knock-out of Gdnf is characterized by a progressive loss of undifferentiated spermatogonia that results in infertility and Sertoli cell-only seminiferous tubules in older animals,91 whereas Gdnf overexpression in SSC produces clusters of spermatogonia that are unable to differentiate and eventually develop into seminomatous tumors.92
The crucial role of Gdnf in controlling the balance between SSC proliferation and differentiation during spermatogenesis is also illustrated by in vitro analysis. Long-term culture of murine SSC can only be supported in minimal serum-free media conditions in the presence of GDNF and FGF-2 (fibroblast growth factor-2); addition of FGF-2 to the culture medium is essential and acts synergistically with GDNF to promote in vitro expansion of SSCs.93 Under these conditions, isolated SSCs proliferate over long periods (>2 years) and can be used in functional transplantation assays in which the derived cells produce germ cell colonies, reconstituting the entire spermatogenic process in recipient testes from infertile mice.93,94 Multiple signaling pathways are activated in response to GDNF/FGF-2 stimulation; murine SSCs cultured with small molecule inhibitors of Akt showed significant impairment of SSC maintenance following transplantation,95 suggesting a critical role for the PI3K-AKT pathway in promoting SSC survival and/or proliferation. However, AKT activation alone was not sufficient to drive SSC self-renewal, which could only be achieved upon costimulation with FGF-2.96 Notably, Lee et al.97 showed that the minimal culture requirements for GDNF/FGF-2 could be bypassed by transfecting SSCs with an activated form of Hras (HrasGly12Val). Hras activation promoted long-term self-renewal of SSCs that underwent spermatogenesis after transplantation and, as in the case of Gdnf overexpression, HrasGly12Val-transfected SSCs produced seminomatous tumors in the mouse testis. The use of specific pathway inhibitors showed that this effect was mainly mediated through the PI3K-AKT signal transduction pathway, although an intact MAPK-ERK cascade also facilitated proliferation.97
The detailed molecular machinery and cellular mechanisms involved in controlling SSC self-renewal and differentiation during spermatogenesis are still poorly understood: the extent to which homeostasis is maintained through control of self-renewal, proliferation, differentiation, and/or survival of cells in the SSC lineage remains unclear.98,99 Moreover, the data reviewed above have been generated with rodent SSCs, which share some but not all of their features with their human counterparts. Although research on human SSCs has been limited, culture of spermatogonia derived from testes of freshly deceased donors showed that upregulation of the MAPK-ERK pathway was involved in their proliferation.100
In summary, these in vitro and in vivo data concur that the activity of the growth factor receptor-RAS signaling pathway is integral to the regulation of SSCs and relies on a complex crosstalk among multiple effectors, including the PI3K-AKT and MAPK-ERK transduction cascades. Strikingly, all known PAE disorders are caused by mutations in genes involved in this process, supporting the idea that the paternal age effect is mediated through dysregulation of RAS signaling and alteration of the growth and proliferative properties of SSCs.
Does Selfish Selection Contribute to the Origins of Complex Diseases?
Thus far, we have focused on established PAE disorders and have highlighted how advanced paternal age and selfish selection can be explained in relation to the biological and molecular mechanisms controlling population dynamics of SSCs. To what extent is this interpretation relevant to paternal age effects associated with more common disorders that have a complex etiology?
It is apparent that spermatogonial populations are subject to regular turnover, and therefore, any mutation that happens to arise in, or near, any gene controlling the cellular and homeostatic properties of SSC is a potential target for selection and enrichment in SSCs. Clearly, many components of the growth factor receptor-RAS signaling pathway are predicted to be vulnerable to this process, but in theory, any pathway that has been implicated in either oncogenesis (COSMIC database) or cell competition101 and is expressed in the testis89,102,103 could also be a target. Based on this interpretation, the spectrum of mutations subject to selfish selection is unlikely to be limited to a handful of strongly activating missense substitutions in known oncogenes. Selection could also involve loss-of-function mechanisms (that might result in haploinsufficiency104), changes in regulatory regions (resulting in altered gene expression105–107), differences in gene dosage (leading to cell competition108), or copy-number variations (CNV) in genes controlling SSC proliferative behavior.
The case of CNVs is of particular interest because genomic deletions and duplications are known to affect a wide range of human phenotypes including many Mendelian traits and complex disorders such as autism and schizophrenia.109 Recurrent CNVs mediated by unequal crossing-over between large near-identical segmental duplications are recombination-dependent (meiotic) events.110–112 In contrast, rare CNVs generated by nonrecurrent rearrangements mostly originate through replication-based processes, involving different molecular mechanisms (e.g., fork stalling and template switching [FoSTeS] and/or microhomology-mediated break-induced replication [MMBIR]).113–115 Because of this mitotic origin, it is anticipated that nonrecurrent CNVs should preferentially arise in male germ cells. Investigations into the parental origin of de novo nonrecurrent CNVs are under way116 but are laborious and require a careful separation of spontaneous genomic rearrangements into likely recombination- and replication-based events.117,118
To appreciate the long-term consequences for genome evolution, it is important to consider the interplay between the strength of selfish selection occurring in the testis and the impact of these mutations on the fitness of the offspring. Given the severe phenotypes they cause, the pathogenic alleles associated with classical PAE disorders are unlikely to segregate for many generations and therefore are predicted to have little impact on overall disease burden (Figure 5, red mutations). However, selfish mutations associated with weaker selective advantage in the testis (and leading to lower levels of enrichment in sperm) that are anticipated to cause milder or incompletely penetrant phenotypes are a potential source of heritable genetic variation that might contribute both to adaptive evolution and to the genetic burden of disease (Figure 5, orange mutations). Given that both MAPK-ERK and PI3K-AKT signaling play important roles in brain development, learning, memory, synaptic plasticity, and cognition,119,120 subtle alterations of these pathways are likely to contribute to the pathology of learning disability and neurocognitive disorders, as well as cancer. In this context it is relevant that several epidemiological studies have reported an association between advanced paternal age and predisposition to neurocognitive disorders such as schizophrenia9,121,122 and autism.8,123,124 Although there are several confounding factors that could explain these observations,13 selfish selection provides one plausible mechanism. Notably, recent reports have highlighted the importance of rare (or private) de novo mutations (both point mutations and CNVs) in the causation of common disorders such as learning disabilities,125 autism,126–128 and schizophrenia.129–132 Information on parental origin and paternal age is not available in most of these studies; it will be important to collect these data systematically.
Figure 5.
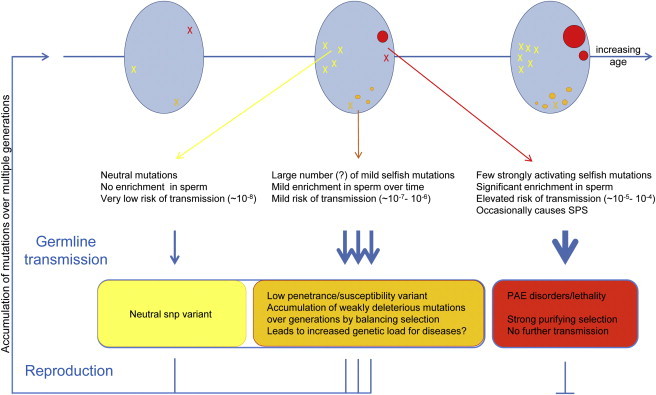
Long-Term Consequences of Selfish Selection in the Testis
Blue ovals represent the testis at three ages from puberty (left) to senescence (right). During the recurrent rounds of replication required for spermatogenesis and SSC self-renewal, stochastic mutations (represented by X) occur randomly in the testis. Depending on the functional consequence of the resulting mutation for the SSC, three scenarios are illustrated. Functionally neutral mutations (in yellow; left part of each testis) do not accumulate and are associated with a very low risk of individual transmission (∼10−8, the background rate of nucleotide substitution in the genome). In red (right part of the testis) are typical PAE mutations that confer a strong selective advantage to the mutant SSC, leading over time to the formation of large “mole-like” clones (ovals) and an increased risk of transmission in older men (up to 1,000-fold higher than the background mutation rate). In rare cases, these mutations are associated with spermatocytic seminoma (SPS). In orange (bottom part of the testis) is depicted an intermediate scenario for mutations with milder selective advantage (i.e., weak gain-of-function, change in copy number or regulation of expression) that are enriched over time in SSC to a lesser extent (>1- to 100-fold). Many such mutational targets could potentially exist. Although strongly activating mutations associated with classical PAE disorders are deleterious and will be rapidly eliminated because of low reproductive fitness, neutral and mildly pathogenic mutations are potentially transmissible over many generations, contributing to genetic heterogeneity. Some mildly pathogenic mutations, although associated with deleterious phenotypes, might be maintained in the population either by recurrent mutation or because they provide a beneficial fitness trait during spermatogenesis.
The emerging picture in neurocognitive disorders suggests that the genetic component of disease risk arises both from de novo mutations and from segregating variants that are individually mildly deleterious and/or show clinical variability in expressivity. In most cases, the disease manifests through an adverse combination of mild-risk alleles (in familial cases) and/or when de novo mutations are acquired in a general sensitized background that is already loaded with low-penetrant pathogenic variants (in simplex cases).133 Notably, comprehensive unbiased screens for pathogenic CNVs associated with autism have highlighted the importance of genomic regions containing functional gene sets involved in cell proliferation and in GTPase/RAS signaling.126,134 Similarly, FGFR135 and the AKT-mTOR136–138 signaling pathways have been implicated in the pathology of schizophrenia. In other words, the pathways implicated in the pathogenesis of these neurocognitive disorders overlap substantially with those subject to selfish selection in the testis and are therefore potential targets for this process (Figure 5, orange mutations).139
Spermatogenesis, Weakly Pathogenic Variants, and Evolution
Aside from selfish selection promoting clonal expansion of mutant SSCs in the context in which mutant cells are surrounded by wild-type counterparts (i.e., mosaic—involving the interaction between two genetically distinct cell populations), mutations in components of spermatogonial signaling pathways might also be advantageous in a constitutive state (i.e., when all the cells are genotypically identical). So long as they are not overly deleterious to the organism, low-penetrance pathogenic alleles could segregate over many generations and be maintained, or even become fixed in the population, through a mechanism of balancing selection (i.e., as a trade-off between a beneficial fitness trait during spermatogenesis that compensates for the deleterious effect in other cellular contexts) (Figure 5). Several lines of evidence are consistent with this interpretation: (1) comparative evolutionary studies suggest that a large proportion of genes expressed in spermatogenesis (including genes that promote germ cell development, apoptosis and tumor suppression) are under positive selection;102 (2) the intriguing case of a patient diagnosed with both Klinefelter syndrome (a condition normally associated with male infertility and azoospermia) and achondroplasia who fathered a child suggests that the presence of the activating FGFR3 mutation might have rescued the testicular function in this individual;140 (3) several genome-wide association studies have identified common variants close to PAE genes associated with increased disease risk, including SNPs in or near FGFR2 and MAP3K1 in breast cancer,141,142 FGFR1 and FGFR2 in schizophrenia,143,144 FGFR3 in bladder cancer,145 HRAS in several cancer types,146–148 and RET in Hirschsprung disease105 or genomic regions associated with schizophrenia149 and other complex diseases150 showing signatures of positive selection.
Perspectives and Conclusions
Driven by recognition of epidemiological paternal age effects, targeted molecular approaches in a handful of monogenic disorders have led to a clearer understanding of the molecular mechanisms and principles underlying this phenomenon. Owing to the technical challenges, only a few base pairs in the human genome have been systematically interrogated to date. Nevertheless, the data suggest that the sperm of all men are progressively enriched for PAE mutations as they age and highlight that selfish selection is likely to be a universal process. A major challenge will be to establish how many sites in our genome are subject to selfish selection with the eventual aim of obtaining a frequency spectrum of point mutation (as well as CNV-based mutational processes) at every genomic position. This will establish whether the few documented PAE disorders are indeed extreme examples of a much more common process. So far, whole-genome sequences of a few family trios48,50 have shown that the transgenerational mutation load and parental origin for each de novo mutation52 can be directly determined. As much larger such data sets become available, it should be possible to establish the relative contribution of selfish selection to genetic heterogeneity and to disease risk by ascertaining whether paternally-derived mutations are truly random or, as selfish selection predicts, whether there is a significant enrichment of functional alleles in or near genes involved in the control of spermatogonial self-renewal. Given that selfish selection might also be responsible for the emergence of advantageous traits involving cognition, increased brain size, or adaptation to new environments, these mechanisms might have contributed to human evolution.103,151
Current data already provide valuable new evidence for genetic counseling of couples having children affected with spontaneous PAE disorders. Although high-level somatic and/or germline mosaicism has very occasionally been observed,152,153 such cases are swamped out by the much more prevalent selfish selection mechanism. Provided that genetic testing on parents' blood shows no evidence of mosaic mutation, the recurrence risk for further affected children is likely to be extremely low (below 1 in 1,000, for all paternal ages). This information could influence parental decisions on whether to opt for invasive prenatal testing in future pregnancies. The localized nature of PAE mutations, and their paternal origin, make these disorders very good candidates for prenatal screening via free fetal DNA from maternal plasma; because average paternal age at conception has been steadily increasing,15 it could become cost-effective to devise a systematic screen targeting common PAE mutations in the case of older fathers.154–156
New technologies, such as the so-called third generation of single-molecule sequencing157 that does not rely on PCR amplification prior to sequencing, are anticipated to allow further elucidation of the impact of the selfish selection mechanism. Whole-genome sequencing of single-cells might soon be used to directly quantify the mutational load carried by individual sperm cells.158,159 Whether applied to single sperm cells, fetal cells in maternal plasma, or oncogenic mutations in the circulation,160 we are bound to learn more about the contribution of selfish selection to human health and evolution. Unusually, humans represent in many ways the ideal “model” organism to study these ultrarare mutational processes.
Acknowledgments
This review is dedicated to the memory of James F. Crow (1916–2012), a pioneer in the study of paternal age effects. We thank Gil McVean (Oxford, UK) and Ewa Rajpert-DeMeyts (Copenhagen, Denmark) for their long-term collaboration in this work, members of the Wilkie lab for critical reading of the manuscript and the Wellcome Trust (091182) for financial support.
Contributor Information
Anne Goriely, Email: anne.goriely@imm.ox.ac.uk.
Andrew O.M. Wilkie, Email: awilkie@hammer.imm.ox.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
England and Wales Birth Census data series FM1, http://www.ons.gov.uk/ons/statbase
National Center for Biotechnology Information Reference Sequence (RefSeq) database, http://www.ncbi.nlm.nih.gov/RefSeq/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Hassold T., Hunt P. Maternal age and chromosomally abnormal pregnancies: What we know and what we wish we knew. Curr. Opin. Pediatr. 2009;21:703–708. doi: 10.1097/MOP.0b013e328332c6ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Risch N., Reich E.W., Wishnick M.M., McCarthy J.G. Spontaneous mutation and parental age in humans. Am. J. Hum. Genet. 1987;41:218–248. [PMC free article] [PubMed] [Google Scholar]
- 3.Green R.F., Devine O., Crider K.S., Olney R.S., Archer N., Olshan A.F., Shapira S.K., National Birth Defects Prevention Study Association of paternal age and risk for major congenital anomalies from the National Birth Defects Prevention Study, 1997 to 2004. Ann. Epidemiol. 2010;20:241–249. doi: 10.1016/j.annepidem.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yip B.H., Pawitan Y., Czene K. Parental age and risk of childhood cancers: A population-based cohort study from Sweden. Int. J. Epidemiol. 2006;35:1495–1503. doi: 10.1093/ije/dyl177. [DOI] [PubMed] [Google Scholar]
- 5.Murray L., McCarron P., Bailie K., Middleton R., Davey Smith G., Dempsey S., McCarthy A., Gavin A. Association of early life factors and acute lymphoblastic leukaemia in childhood: Historical cohort study. Br. J. Cancer. 2002;86:356–361. doi: 10.1038/sj.bjc.6600012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J.Y., Lee K.M., Park S.K., Noh D.Y., Ahn S.H., Yoo K.Y., Kang D. Association of paternal age at birth and the risk of breast cancer in offspring: A case control study. BMC Cancer. 2005;5:143. doi: 10.1186/1471-2407-5-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Meyer T., Rietzschel E.R., De Buyzere M.L., De Bacquer D., Van Criekinge W., De Backer G.G., Gillebert T.C., Van Oostveldt P., Bekaert S., Asklepios investigators Paternal age at birth is an important determinant of offspring telomere length. Hum. Mol. Genet. 2007;16:3097–3102. doi: 10.1093/hmg/ddm271. [DOI] [PubMed] [Google Scholar]
- 8.Grether J.K., Anderson M.C., Croen L.A., Smith D., Windham G.C. Risk of autism and increasing maternal and paternal age in a large north American population. Am. J. Epidemiol. 2009;170:1118–1126. doi: 10.1093/aje/kwp247. [DOI] [PubMed] [Google Scholar]
- 9.Malaspina D. Paternal factors and schizophrenia risk: De novo mutations and imprinting. Schizophr. Bull. 2001;27:379–393. doi: 10.1093/oxfordjournals.schbul.a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frans E.M., Sandin S., Reichenberg A., Lichtenstein P., Långström N., Hultman C.M. Advancing paternal age and bipolar disorder. Arch. Gen. Psychiatry. 2008;65:1034–1040. doi: 10.1001/archpsyc.65.9.1034. [DOI] [PubMed] [Google Scholar]
- 11.Saha S., Barnett A.G., Foldi C., Burne T.H., Eyles D.W., Buka S.L., McGrath J.J. Advanced paternal age is associated with impaired neurocognitive outcomes during infancy and childhood. PLoS Med. 2009;6:e40. doi: 10.1371/journal.pmed.1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X.K., Wen S.W., Krewski D., Fleming N., Yang Q., Walker M.C. Paternal age and adverse birth outcomes: Teenager or 40+, who is at risk? Hum. Reprod. 2008;23:1290–1296. doi: 10.1093/humrep/dem403. [DOI] [PubMed] [Google Scholar]
- 13.Petersen L., Mortensen P.B., Pedersen C.B. Paternal age at birth of first child and risk of schizophrenia. Am. J. Psychiatry. 2011;168:82–88. doi: 10.1176/appi.ajp.2010.10020252. [DOI] [PubMed] [Google Scholar]
- 14.Toriello H.V., Meck J.M., Professional Practice and Guidelines Committee Statement on guidance for genetic counseling in advanced paternal age. Genet. Med. 2008;10:457–460. doi: 10.1097/GIM.0b013e318176fabb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bray I., Gunnell D., Davey Smith G. Advanced paternal age: How old is too old? J. Epidemiol. Community Health. 2006;60:851–853. doi: 10.1136/jech.2005.045179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orioli I.M., Castilla E.E., Barbosa-Neto J.G. The birth prevalence rates for the skeletal dysplasias. J. Med. Genet. 1986;23:328–332. doi: 10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waller D.K., Correa A., Vo T.M., Wang Y., Hobbs C., Langlois P.H., Pearson K., Romitti P.A., Shaw G.M., Hecht J.T. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am. J. Med. Genet. A. 2008;146A:2385–2389. doi: 10.1002/ajmg.a.32485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen M.M., Jr., Kreiborg S., Lammer E.J., Cordero J.F., Mastroiacovo P., Erickson J.D., Roeper P., Martínez-Frías M.L. Birth prevalence study of the Apert syndrome. Am. J. Med. Genet. 1992;42:655–659. doi: 10.1002/ajmg.1320420505. [DOI] [PubMed] [Google Scholar]
- 19.Tolarova M.M., Harris J.A., Ordway D.E., Vargervik K. Birth prevalence, mutation rate, sex ratio, parents' age, and ethnicity in Apert syndrome. Am. J. Med. Genet. 1997;72:394–398. doi: 10.1002/(sici)1096-8628(19971112)72:4<394::aid-ajmg4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 20.Orioli I.M., Castilla E.E., Scarano G., Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am. J. Med. Genet. 1995;59:209–217. doi: 10.1002/ajmg.1320590218. [DOI] [PubMed] [Google Scholar]
- 21.Rannan-Eliya S.V., Taylor I.B., De Heer I.M., Van Den Ouweland A.M., Wall S.A., Wilkie A.O.M. Paternal origin of FGFR3 mutations in Muenke-type craniosynostosis. Hum. Genet. 2004;115:200–207. doi: 10.1007/s00439-004-1151-5. [DOI] [PubMed] [Google Scholar]
- 22.Schuffenecker I., Ginet N., Goldgar D., Eng C., Chambe B., Boneu A., Houdent C., Pallo D., Schlumberger M., Thivolet C., Lenoir G.M. Prevalence and parental origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Le Groupe d'Etude des Tumeurs a Calcitonine. Am. J. Hum. Genet. 1997;60:233–237. [PMC free article] [PubMed] [Google Scholar]
- 23.Carlson K.M., Bracamontes J., Jackson C.E., Clark R., Lacroix A., Wells S.A., Jr., Goodfellow P.J. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am. J. Hum. Genet. 1994;55:1076–1082. [PMC free article] [PubMed] [Google Scholar]
- 24.Tartaglia M., Cordeddu V., Chang H., Shaw A., Kalidas K., Crosby A., Patton M.A., Sorcini M., van der Burgt I., Jeffery S., Gelb B.D. Paternal germline origin and sex-ratio distortion in transmission of PTPN11 mutations in Noonan syndrome. Am. J. Hum. Genet. 2004;75:492–497. doi: 10.1086/423493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sol-Church K., Stabley D.L., Nicholson L., Gonzalez I.L., Gripp K.W. Paternal bias in parental origin of HRAS mutations in Costello syndrome. Hum. Mutat. 2006;27:736–741. doi: 10.1002/humu.20381. [DOI] [PubMed] [Google Scholar]
- 26.Zampino G., Pantaleoni F., Carta C., Cobellis G., Vasta I., Neri C., Pogna E.A., De Feo E., Delogu A., Sarkozy A. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum. Mutat. 2007;28:265–272. doi: 10.1002/humu.20431. [DOI] [PubMed] [Google Scholar]
- 27.Schulz A.L., Albrecht B., Arici C., van der Burgt I., Buske A., Gillessen-Kaesbach G., Heller R., Horn D., Hübner C.A., Korenke G.C. Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello syndrome. Clin. Genet. 2008;73:62–70. doi: 10.1111/j.1399-0004.2007.00931.x. [DOI] [PubMed] [Google Scholar]
- 28.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilkie A.O.M., Slaney S.F., Oldridge M., Poole M.D., Ashworth G.J., Hockley A.D., Hayward R.D., David D.J., Pulleyn L.J., Rutland P. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat. Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 30.Rousseau F., Bonaventure J., Legeai-Mallet L., Pelet A., Rozet J.M., Maroteaux P., Le Merrer M., Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 31.Bellus G.A., Hefferon T.W., Ortiz de Luna R.I., Hecht J.T., Horton W.A., Machado M., Kaitila I., McIntosh I., Francomano C.A. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 1995;56:368–373. [PMC free article] [PubMed] [Google Scholar]
- 32.Kan S.H., Elanko N., Johnson D., Cornejo-Roldan L., Cook J., Reich E.W., Tomkins S., Verloes A., Twigg S.R.F., Rannan-Eliya S. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am. J. Hum. Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vajo Z., Francomano C.A., Wilkin D.J. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr. Rev. 2000;21:23–39. doi: 10.1210/edrv.21.1.0387. [DOI] [PubMed] [Google Scholar]
- 34.Aoki Y., Niihori T., Narumi Y., Kure S., Matsubara Y. The RAS/MAPK syndromes: Novel roles of the RAS pathway in human genetic disorders. Hum. Mutat. 2008;29:992–1006. doi: 10.1002/humu.20748. [DOI] [PubMed] [Google Scholar]
- 35.Kratz C.P., Zampino G., Kriek M., Kant S.G., Leoni C., Pantaleoni F., Oudesluys-Murphy A.M., Di Rocco C., Kloska S.P., Tartaglia M., Zenker M. Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations. Am. J. Med. Genet. A. 2009;149A:1036–1040. doi: 10.1002/ajmg.a.32786. [DOI] [PubMed] [Google Scholar]
- 36.Tartaglia M., Zampino G., Gelb B.D. Noonan syndrome: Clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1:2–26. doi: 10.1159/000276766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raue F., Frank-Raue K. Update multiple endocrine neoplasia type 2. Fam. Cancer. 2010;9:449–457. doi: 10.1007/s10689-010-9320-2. [DOI] [PubMed] [Google Scholar]
- 38.Vogel F., Rathenberg R. Spontaneous mutation in man. Adv. Hum. Genet. 1975;5:223–318. doi: 10.1007/978-1-4615-9068-2_4. [DOI] [PubMed] [Google Scholar]
- 39.Crow J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 40.Penrose L.S. Parental age and mutation. Lancet. 1955;269:312–313. doi: 10.1016/s0140-6736(55)92305-9. [DOI] [PubMed] [Google Scholar]
- 41.Makova K.D., Li W.H. Strong male-driven evolution of DNA sequences in humans and apes. Nature. 2002;416:624–626. doi: 10.1038/416624a. [DOI] [PubMed] [Google Scholar]
- 42.Taylor J., Tyekucheva S., Zody M., Chiaromonte F., Makova K.D. Strong and weak male mutation bias at different sites in the primate genomes: Insights from the human-chimpanzee comparison. Mol. Biol. Evol. 2006;23:565–573. doi: 10.1093/molbev/msj060. [DOI] [PubMed] [Google Scholar]
- 43.Weinberg W. Zur Vererbung des Zwergwuchses. Arch. Rassen-u Gesell. Biol. 1912;9:710–718. [Google Scholar]
- 44.Crow J.F. Age and sex effects on human mutation rates: An old problem with new complexities. J. Radiat. Res. (Tokyo) 2006;47(Suppl B):B75–B82. doi: 10.1269/jrr.47.b75. [DOI] [PubMed] [Google Scholar]
- 45.Moloney D.M., Slaney S.F., Oldridge M., Wall S.A., Sahlin P., Stenman G., Wilkie A.O.M. Exclusive paternal origin of new mutations in Apert syndrome. Nat. Genet. 1996;13:48–53. doi: 10.1038/ng0596-48. [DOI] [PubMed] [Google Scholar]
- 46.Kondrashov A.S. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum. Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- 47.Nachman M.W., Crowell S.L. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roach J.C., Glusman G., Smit A.F., Huff C.D., Hubley R., Shannon P.T., Rowen L., Pant K.P., Goodman N., Bamshad M. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kondrashov F.A., Kondrashov A.S. Measurements of spontaneous rates of mutations in the recent past and the near future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010;365:1169–1176. doi: 10.1098/rstb.2009.0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conrad D.F., Keebler J.E., DePristo M.A., Lindsay S.J., Zhang Y., Casals F., Idaghdour Y., Hartl C.L., Torroja C., Garimella K.V., 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tiemann-Boege I., Navidi W., Grewal R., Cohn D., Eskenazi B., Wyrobek A.J., Arnheim N. The observed human sperm mutation frequency cannot explain the achondroplasia paternal age effect. Proc. Natl. Acad. Sci. USA. 2002;99:14952–14957. doi: 10.1073/pnas.232568699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glaser R.L., Broman K.W., Schulman R.L., Eskenazi B., Wyrobek A.J., Jabs E.W. The paternal-age effect in Apert syndrome is due, in part, to the increased frequency of mutations in sperm. Am. J. Hum. Genet. 2003;73:939–947. doi: 10.1086/378419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilkie A.O.M. Bad bones, absent smell, selfish testes: The pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Goriely A., McVean G.A., Röjmyr M., Ingemarsson B., Wilkie A.O.M. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643–646. doi: 10.1126/science.1085710. [DOI] [PubMed] [Google Scholar]
- 57.Goriely A., McVean G.A., van Pelt A.M., O'Rourke A.W., Wall S.A., de Rooij D.G., Wilkie A.O.M. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc. Natl. Acad. Sci. USA. 2005;102:6051–6056. doi: 10.1073/pnas.0500267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoon S.R., Qin J., Glaser R.L., Jabs E.W., Wexler N.S., Sokol R., Arnheim N., Calabrese P. The ups and downs of mutation frequencies during aging can account for the Apert syndrome paternal age effect. PLoS Genet. 2009;5:e1000558. doi: 10.1371/journal.pgen.1000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin J., Calabrese P., Tiemann-Boege I., Shinde D.N., Yoon S.R., Gelfand D., Bauer K., Arnheim N. The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol. 2007;5:e224. doi: 10.1371/journal.pbio.0050224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi S.K., Yoon S.R., Calabrese P., Arnheim N. A germ-line-selective advantage rather than an increased mutation rate can explain some unexpectedly common human disease mutations. Proc. Natl. Acad. Sci. USA. 2008;105:10143–10148. doi: 10.1073/pnas.0801267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dakouane Giudicelli M., Serazin V., Le Sciellour C.R., Albert M., Selva J., Giudicelli Y. Increased achondroplasia mutation frequency with advanced age and evidence for G1138A mosaicism in human testis biopsies. Fertil. Steril. 2008;89:1651–1656. doi: 10.1016/j.fertnstert.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 62.Goriely A., Hansen R.M., Taylor I.B., Olesen I.A., Jacobsen G.K., McGowan S.J., Pfeifer S.P., McVean G.A., Rajpert-De Meyts E., Wilkie A.O.M. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat. Genet. 2009;41:1247–1252. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tavormina P.L., Shiang R., Thompson L.M., Zhu Y.Z., Wilkin D.J., Lachman R.S., Wilcox W.R., Rimoin D.L., Cohn D.H., Wasmuth J.J. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- 64.Hodgkinson A., Eyre-Walker A. The genomic distribution and local context of coincident SNPs in human and chimpanzee. Genome Biol Evol. 2010;2:547–557. doi: 10.1093/gbe/evq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorlov I.P., Gorlova O.Y., Amos C.I. Relative effects of mutability and selection on single nucleotide polymorphisms in transcribed regions of the human genome. BMC Genomics. 2008;9:292. doi: 10.1186/1471-2164-9-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hansen R.M.S., Goriely A., Wall S.A., Roberts I.S., Wilkie A.O.M. Fibroblast growth factor receptor 2, gain-of-function mutations, and tumourigenesis: Investigating a potential link. J. Pathol. 2005;207:27–31. doi: 10.1002/path.1816. [DOI] [PubMed] [Google Scholar]
- 67.Eble J.N. Spermatocytic seminoma. Hum. Pathol. 1994;25:1035–1042. doi: 10.1016/0046-8177(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 68.Rajpert-De Meyts E. Recent advances and future directions in research on testicular germ cell cancer. Int. J. Androl. 2007;30:192–197. doi: 10.1111/j.1365-2605.2007.00810.x. [DOI] [PubMed] [Google Scholar]
- 69.Lim J., Goriely A., Turner G.D., Ewen K.A., Jacobsen G.K., Graem N., Wilkie A.O.M., Rajpert-De Meyts E. OCT2, SSX and SAGE1 reveal the phenotypic heterogeneity of spermatocytic seminoma reflecting distinct subpopulations of spermatogonia. J. Pathol. 2011;224:473–483. doi: 10.1002/path.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fasano O., Aldrich T., Tamanoi F., Taparowsky E., Furth M., Wigler M. Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc. Natl. Acad. Sci. USA. 1984;81:4008–4012. doi: 10.1073/pnas.81.13.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aoki Y., Niihori T., Kawame H., Kurosawa K., Ohashi H., Tanaka Y., Filocamo M., Kato K., Suzuki Y., Kure S., Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 72.Bellus G.A., Spector E.B., Speiser P.W., Weaver C.A., Garber A.T., Bryke C.R., Israel J., Rosengren S.S., Webster M.K., Donoghue D.J., Francomano C.A. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am. J. Hum. Genet. 2000;67:1411–1421. doi: 10.1086/316892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berk D.R., Spector E.B., Bayliss S.J. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch. Dermatol. 2007;143:1153–1156. doi: 10.1001/archderm.143.9.1153. [DOI] [PubMed] [Google Scholar]
- 74.Hafner C., Hartmann A., Real F.X., Hofstaedter F., Landthaler M., Vogt T. Spectrum of FGFR3 mutations in multiple intraindividual seborrheic keratoses. J. Invest. Dermatol. 2007;127:1883–1885. doi: 10.1038/sj.jid.5700804. [DOI] [PubMed] [Google Scholar]
- 75.Ota S., Zhou Z.Q., Link J.M., Hurlin P.J. The role of senescence and prosurvival signaling in controlling the oncogenic activity of FGFR2 mutants associated with cancer and birth defects. Hum. Mol. Genet. 2009;18:2609–2621. doi: 10.1093/hmg/ddp195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krejci P., Prochazkova J., Smutny J., Chlebova K., Lin P., Aklian A., Bryja V., Kozubik A., Wilcox W.R. FGFR3 signaling induces a reversible senescence phenotype in chondrocytes similar to oncogene-induced premature senescence. Bone. 2010;47:102–110. doi: 10.1016/j.bone.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vogelstein B., Kinzler K.W. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 78.Nowell P.C. Tumor progression: A brief historical perspective. Semin. Cancer Biol. 2002;12:261–266. doi: 10.1016/s1044-579x(02)00012-3. [DOI] [PubMed] [Google Scholar]
- 79.Miyauchi A., Futami H., Hai N., Yokozawa T., Kuma K., Aoki N., Kosugi S., Sugano K., Yamaguchi K. Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn. J. Cancer Res. 1999;90:1–5. doi: 10.1111/j.1349-7006.1999.tb00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iwashita T., Murakami H., Kurokawa K., Kawai K., Miyauchi A., Futami H., Qiao S., Ichihara M., Takahashi M. A two-hit model for development of multiple endocrine neoplasia type 2B by RET mutations. Biochem. Biophys. Res. Commun. 2000;268:804–808. doi: 10.1006/bbrc.2000.2227. [DOI] [PubMed] [Google Scholar]
- 81.Shukla V., Coumoul X., Wang R.H., Kim H.S., Deng C.X. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 2007;39:1145–1150. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- 82.Chen P.C., Wakimoto H., Conner D., Araki T., Yuan T., Roberts A., Seidman C.E., Bronson R., Neel B.G., Seidman J.G., Kucherlapati R. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome–associated Sos1 mutation. J. Clin. Invest. 2010;120:4353–4365. doi: 10.1172/JCI43910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu X., Simpson J., Hong J.H., Kim K.H., Thavarajah N.K., Backx P.H., Neel B.G., Araki T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Invest. 2011;121:1009–1025. doi: 10.1172/JCI44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krenz M., Yutzey K.E., Robbins J. Noonan syndrome mutation Q79R in Shp2 increases proliferation of valve primordia mesenchymal cells via extracellular signal-regulated kinase 1/2 signaling. Circ. Res. 2005;97:813–820. doi: 10.1161/01.RES.0000186194.06514.b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosenberger G., Meien S., Kutsche K. Oncogenic HRAS mutations cause prolonged PI3K signaling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum. Mutat. 2009;30:352–362. doi: 10.1002/humu.20855. [DOI] [PubMed] [Google Scholar]
- 86.Cancilla B., Davies A., Ford-Perriss M., Risbridger G.P. Discrete cell- and stage-specific localisation of fibroblast growth factors and receptor expression during testis development. J. Endocrinol. 2000;164:149–159. doi: 10.1677/joe.0.1640149. [DOI] [PubMed] [Google Scholar]
- 87.von Kopylow K., Kirchhoff C., Jezek D., Schulze W., Feig C., Primig M., Steinkraus V., Spiess A.N. Screening for biomarkers of spermatogonia within the human testis: A whole genome approach. Hum. Reprod. 2010;25:1104–1112. doi: 10.1093/humrep/deq053. [DOI] [PubMed] [Google Scholar]
- 88.Krajewska M., Banares S., Zhang E.E., Huang X., Scadeng M., Jhala U.S., Feng G.S., Krajewski S. Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am. J. Pathol. 2008;172:1312–1324. doi: 10.2353/ajpath.2008.070594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hermo L., Pelletier R.M., Cyr D.G., Smith C.E. Surfing the wave, cycle, life history, and genes/proteins expressed by testicular germ cells. Part 5: Intercellular junctions and contacts between germs cells and Sertoli cells and their regulatory interactions, testicular cholesterol, and genes/proteins associated with more than one germ cell generation. Microsc. Res. Tech. 2010;73:409–494. doi: 10.1002/jemt.20786. [DOI] [PubMed] [Google Scholar]
- 90.Naughton C.K., Jain S., Strickland A.M., Gupta A., Milbrandt J. Glial cell-line derived neurotrophic factor-mediated RET signaling regulates spermatogonial stem cell fate. Biol. Reprod. 2006;74:314–321. doi: 10.1095/biolreprod.105.047365. [DOI] [PubMed] [Google Scholar]
- 91.Meng X., Lindahl M., Hyvönen M.E., Parvinen M., de Rooij D.G., Hess M.W., Raatikainen-Ahokas A., Sainio K., Rauvala H., Lakso M. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287:1489–1493. doi: 10.1126/science.287.5457.1489. [DOI] [PubMed] [Google Scholar]
- 92.Meng X., de Rooij D.G., Westerdahl K., Saarma M., Sariola H. Promotion of seminomatous tumors by targeted overexpression of glial cell line-derived neurotrophic factor in mouse testis. Cancer Res. 2001;61:3267–3271. [PubMed] [Google Scholar]
- 93.Kubota H., Avarbock M.R., Brinster R.L. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA. 2004;101:16489–16494. doi: 10.1073/pnas.0407063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brinster R.L., Zimmermann J.W. Spermatogenesis following male germ-cell transplantation. Proc. Natl. Acad. Sci. USA. 1994;91:11298–11302. doi: 10.1073/pnas.91.24.11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oatley J.M., Avarbock M.R., Brinster R.L. Glial cell line-derived neurotrophic factor regulation of genes essential for self-renewal of mouse spermatogonial stem cells is dependent on Src family kinase signaling. J. Biol. Chem. 2007;282:25842–25851. doi: 10.1074/jbc.M703474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee J., Kanatsu-Shinohara M., Inoue K., Ogonuki N., Miki H., Toyokuni S., Kimura T., Nakano T., Ogura A., Shinohara T. Akt mediates self-renewal division of mouse spermatogonial stem cells. Development. 2007;134:1853–1859. doi: 10.1242/dev.003004. [DOI] [PubMed] [Google Scholar]
- 97.Lee J., Kanatsu-Shinohara M., Morimoto H., Kazuki Y., Takashima S., Oshimura M., Toyokuni S., Shinohara T. Genetic reconstruction of mouse spermatogonial stem cell self-renewal in vitro by Ras-cyclin D2 activation. Cell Stem Cell. 2009;5:76–86. doi: 10.1016/j.stem.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 98.Nakagawa T., Nabeshima Y., Yoshida S. Functional identification of the actual and potential stem cell compartments in mouse spermatogenesis. Dev. Cell. 2007;12:195–206. doi: 10.1016/j.devcel.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 99.Klein A.M., Nakagawa T., Ichikawa R., Yoshida S., Simons B.D. Mouse germ line stem cells undergo rapid and stochastic turnover. Cell Stem Cell. 2010;7:214–224. doi: 10.1016/j.stem.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 100.He Z., Kokkinaki M., Jiang J., Dobrinski I., Dym M. Isolation, characterization, and culture of human spermatogonia. Biol. Reprod. 2010;82:363–372. doi: 10.1095/biolreprod.109.078550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moreno E. Is cell competition relevant to cancer? Nat. Rev. Cancer. 2008;8:141–147. doi: 10.1038/nrc2252. [DOI] [PubMed] [Google Scholar]
- 102.Nielsen R., Bustamante C., Clark A.G., Glanowski S., Sackton T.B., Hubisz M.J., Fledel-Alon A., Tanenbaum D.M., Civello D., White T.J. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Khaitovich P., Enard W., Lachmann M., Pääbo S. Evolution of primate gene expression. Nat. Rev. Genet. 2006;7:693–702. doi: 10.1038/nrg1940. [DOI] [PubMed] [Google Scholar]
- 104.McGowan K.A., Li J.Z., Park C.Y., Beaudry V., Tabor H.K., Sabnis A.J., Zhang W., Fuchs H., de Angelis M.H., Myers R.M. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Emison E.S., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 106.De Gobbi M., Viprakasit V., Hughes J.R., Fisher C., Buckle V.J., Ayyub H., Gibbons R.J., Vernimmen D., Yoshinaga Y., de Jong P. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science. 2006;312:1215–1217. doi: 10.1126/science.1126431. [DOI] [PubMed] [Google Scholar]
- 107.To M.D., Perez-Losada J., Mao J.H., Hsu J., Jacks T., Balmain A. A functional switch from lung cancer resistance to susceptibility at the Pas1 locus in Kras2LA2 mice. Nat. Genet. 2006;38:926–930. doi: 10.1038/ng1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moreno E., Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 109.Zhang F., Gu W., Hurles M.E., Lupski J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lupski J.R., Stankiewicz P. Genomic disorders: Molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 114.Arlt M.F., Mulle J.G., Schaibley V.M., Ragland R.L., Durkin S.G., Warren S.T., Glover T.W. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009;84:339–350. doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang F., Seeman P., Liu P., Weterman M.A., Gonzaga-Jauregui C., Towne C.F., Batish S.D., De Vriendt E., De Jonghe P., Rautenstrauss B. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: Rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hehir-Kwa J.Y., Rodríguez-Santiago B., Vissers L.E., de Leeuw N., Pfundt R., Buitelaar J.K., Pérez-Jurado L.A., Veltman J.A. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet. 2011;48:776–778. doi: 10.1136/jmedgenet-2011-100147. [DOI] [PubMed] [Google Scholar]
- 117.Itsara A., Wu H., Smith J.D., Nickerson D.A., Romieu I., London S.J., Eichler E.E. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thomas N.S., Morris J.K., Baptista J., Ng B.L., Crolla J.A., Jacobs P.A. De novo apparently balanced translocations in man are predominantly paternal in origin and associated with a significant increase in paternal age. J. Med. Genet. 2010;47:112–115. doi: 10.1136/jmg.2009.069716. [DOI] [PubMed] [Google Scholar]
- 119.Krab L.C., Goorden S.M., Elgersma Y. Oncogenes on my mind: ERK and MTOR signaling in cognitive diseases. Trends Genet. 2008;24:498–510. doi: 10.1016/j.tig.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 120.Samuels I.S., Saitta S.C., Landreth G.E. MAP'ing CNS development and cognition: An ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Malaspina D., Corcoran C., Fahim C., Berman A., Harkavy-Friedman J., Yale S., Goetz D., Goetz R., Harlap S., Gorman J. Paternal age and sporadic schizophrenia: Evidence for de novo mutations. Am. J. Med. Genet. 2002;114:299–303. doi: 10.1002/ajmg.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Byrne M., Agerbo E., Ewald H., Eaton W.W., Mortensen P.B. Parental age and risk of schizophrenia: A case-control study. Arch. Gen. Psychiatry. 2003;60:673–678. doi: 10.1001/archpsyc.60.7.673. [DOI] [PubMed] [Google Scholar]
- 123.Tsuchiya K.J., Matsumoto K., Miyachi T., Tsujii M., Nakamura K., Takagai S., Kawai M., Yagi A., Iwaki K., Suda S. Paternal age at birth and high-functioning autistic-spectrum disorder in offspring. Br. J. Psychiatry. 2008;193:316–321. doi: 10.1192/bjp.bp.107.045120. [DOI] [PubMed] [Google Scholar]
- 124.Durkin M.S., Maenner M.J., Newschaffer C.J., Lee L.C., Cunniff C.M., Daniels J.L., Kirby R.S., Leavitt L., Miller L., Zahorodny W., Schieve L.A. Advanced parental age and the risk of autism spectrum disorder. Am. J. Epidemiol. 2008;168:1268–1276. doi: 10.1093/aje/kwn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 126.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.O'Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S., Karakoc E., Mackenzie A.P., Ng S.B., Baker C. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Levy D., Ronemus M., Yamrom B., Lee Y.H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 129.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 130.Awadalla P., Gauthier J., Myers R.A., Casals F., Hamdan F.F., Griffing A.R., Côté M., Henrion E., Spiegelman D., Tarabeux J. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet. 2010;87:316–324. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Girard S.L., Gauthier J., Noreau A., Xiong L., Zhou S., Jouan L., Dionne-Laporte A., Spiegelman D., Henrion E., Diallo O. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 132.Xu B., Roos J.L., Dexheimer P., Boone B., Plummer B., Levy S., Gogos J.A., Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Girirajan S., Rosenfeld J.A., Cooper G.M., Antonacci F., Siswara P., Itsara A., Vives L., Walsh T., McCarthy S.E., Baker C. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gilman S.R., Iossifov I., Levy D., Ronemus M., Wigler M., Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Klejbor I., Myers J.M., Hausknecht K., Corso T.D., Gambino A.S., Morys J., Maher P.A., Hard R., Richards J., Stachowiak E.K., Stachowiak M.K. Fibroblast growth factor receptor signaling affects development and function of dopamine neurons - inhibition results in a schizophrenia-like syndrome in transgenic mice. J. Neurochem. 2006;97:1243–1258. doi: 10.1111/j.1471-4159.2006.03754.x. [DOI] [PubMed] [Google Scholar]
- 136.Kalkman H.O. The role of the phosphatidylinositide 3-kinase-protein kinase B pathway in schizophrenia. Pharmacol. Ther. 2006;110:117–134. doi: 10.1016/j.pharmthera.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 137.Kim J.Y., Duan X., Liu C.Y., Jang M.H., Guo J.U., Pow-anpongkul N., Kang E., Song H., Ming G.L. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kéri S., Seres I., Kelemen O., Benedek G. Neuregulin 1-stimulated phosphorylation of AKT in psychotic disorders and its relationship with neurocognitive functions. Neurochem. Int. 2009;55:606–609. doi: 10.1016/j.neuint.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 139.Goriely A., Wilkie A.O.M. Missing heritability: Paternal age effect mutations and selfish spermatogonia. Nat. Rev. Genet. 2010;11:589. doi: 10.1038/nrg2809-c1. [DOI] [PubMed] [Google Scholar]
- 140.Juul A., Aksglaede L., Lund A.M., Duno M., Skakkebaek N.E., Rajpert-De Meyts E. Preserved fertility in a non-mosaic Klinefelter patient with a mutation in the fibroblast growth factor receptor 3 gene: Case report. Hum. Reprod. 2007;22:1907–1911. doi: 10.1093/humrep/dem126. [DOI] [PubMed] [Google Scholar]
- 141.Easton D.F., Pooley K.A., Dunning A.M., Pharoah P.D., Thompson D., Ballinger D.G., Struewing J.P., Morrison J., Field H., Luben R., SEARCH collaborators. kConFab. AOCS Management Group Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hunter D.J., Kraft P., Jacobs K.B., Cox D.G., Yeager M., Hankinson S.E., Wacholder S., Wang Z., Welch R., Hutchinson A. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.O'Donovan M.C., Norton N., Williams H., Peirce T., Moskvina V., Nikolov I., Hamshere M., Carroll L., Georgieva L., Dwyer S., Molecular Genetics of Schizophrenia Collaboration Analysis of 10 independent samples provides evidence for association between schizophrenia and a SNP flanking fibroblast growth factor receptor 2. Mol. Psychiatry. 2009;14:30–36. doi: 10.1038/mp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shi Y., Li Z., Xu Q., Wang T., Li T., Shen J., Zhang F., Chen J., Zhou G., Ji W. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat. Genet. 2011;43:1224–1227. doi: 10.1038/ng.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kiemeney L.A., Sulem P., Besenbacher S., Vermeulen S.H., Sigurdsson A., Thorleifsson G., Gudbjartsson D.F., Stacey S.N., Gudmundsson J., Zanon C. A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer. Nat. Genet. 2010;42:415–419. doi: 10.1038/ng.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Johne A., Roots I., Brockmöller J. A single nucleotide polymorphism in the human H-ras proto-oncogene determines the risk of urinary bladder cancer. Cancer Epidemiol. Biomarkers Prev. 2003;12:68–70. [PubMed] [Google Scholar]
- 147.Sathyan K.M., Nalinakumari K.R., Abraham T., Kannan S. Influence of single nucleotide polymorphisms in H-Ras and cyclin D1 genes on oral cancer susceptibility. Oral Oncol. 2006;42:607–613. doi: 10.1016/j.oraloncology.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Y., Jin M., Liu B., Ma X., Yao K., Li Q., Chen K. Association between H-RAS T81C genetic polymorphism and gastrointestinal cancer risk: A population based case-control study in China. BMC Cancer. 2008;8:256. doi: 10.1186/1471-2407-8-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Crespi B., Summers K., Dorus S. Adaptive evolution of genes underlying schizophrenia. Proc. Biol. Sci. 2007;274:2801–2810. doi: 10.1098/rspb.2007.0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lappalainen T., Salmela E., Andersen P.M., Dahlman-Wright K., Sistonen P., Savontaus M.L., Schreiber S., Lahermo P., Kere J. Genomic landscape of positive natural selection in Northern European populations. Eur. J. Hum. Genet. 2010;18:471–478. doi: 10.1038/ejhg.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dorus S., Vallender E.J., Evans P.D., Anderson J.R., Gilbert S.L., Mahowald M., Wyckoff G.J., Malcom C.M., Lahn B.T. Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell. 2004;119:1027–1040. doi: 10.1016/j.cell.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 152.Gripp K.W., Stabley D.L., Nicholson L., Hoffman J.D., Sol-Church K. Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am. J. Med. Genet. A. 2006;140:2163–2169. doi: 10.1002/ajmg.a.31456. [DOI] [PubMed] [Google Scholar]
- 153.Goriely A., Lord H., Lim J., Johnson D., Lester T., Firth H.V., Wilkie A.O.M. Germline and somatic mosaicism for FGFR2 mutation in the mother of a child with Crouzon syndrome: Implications for genetic testing in “paternal age-effect” syndromes. Am. J. Med. Genet. A. 2010;152A:2067–2073. doi: 10.1002/ajmg.a.33513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tang D.L., Li Y., Zhou X., Li X., Zheng F. Multiplex fluorescent PCR for noninvasive prenatal detection of fetal-derived paternally inherited diseases using circulatory fetal DNA in maternal plasma. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009;144:35–39. doi: 10.1016/j.ejogrb.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 155.Chitty L.S., Griffin D.R., Meaney C., Barrett A., Khalil A., Pajkrt E., Cole T.J. New aids for the non-invasive prenatal diagnosis of achondroplasia: Dysmorphic features, charts of fetal size and molecular confirmation using cell-free fetal DNA in maternal plasma. Ultrasound Obstet. Gynecol. 2011;37:283–289. doi: 10.1002/uog.8893. [DOI] [PubMed] [Google Scholar]
- 156.Lim J.H., Kim M.J., Kim S.Y., Kim H.O., Song M.J., Kim M.H., Park S.Y., Yang J.H., Ryu H.M. Non-invasive prenatal detection of achondroplasia using circulating fetal DNA in maternal plasma. J. Assist. Reprod. Genet. 2011;28:167–172. doi: 10.1007/s10815-010-9489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 158.Fan H.C., Wang J., Potanina A., Quake S.R. Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kalisky T., Quake S.R. Single-cell genomics. Nat. Methods. 2011;8:311–314. doi: 10.1038/nmeth0411-311. [DOI] [PubMed] [Google Scholar]
- 160.Chen Z., Feng J., Buzin C.H., Liu Q., Weiss L., Kernstine K., Somlo G., Sommer S.S. Analysis of cancer mutation signatures in blood by a novel ultra-sensitive assay: Monitoring of therapy or recurrence in non-metastatic breast cancer. PLoS ONE. 2009;4:e7220. doi: 10.1371/journal.pone.0007220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hansen, R.M.S. (2006). Fibroblast growth factor receptor 2 mutations in human sperm: Biological implications. DPhil thesis, University of Oxford, Oxford, UK.
- 162.Glaser R.L., Jiang W., Boyadjiev S.A., Tran A.K., Zachary A.A., Van Maldergem L., Johnson D., Walsh S., Oldridge M., Wall S.A. Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. Am. J. Hum. Genet. 2000;66:768–777. doi: 10.1086/302831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wilkin D.J., Szabo J.K., Cameron R., Henderson S., Bellus G.A., Mack M.L., Kaitila I., Loughlin J., Munnich A., Sykes B. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am. J. Hum. Genet. 1998;63:711–716. doi: 10.1086/302000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Søvik O., Schubbert S., Houge G., Steine S.J., Norgård G., Engelsen B., Njølstad P.R., Shannon K., Molven A. De novo HRAS and KRAS mutations in two siblings with short stature and neuro-cardio-facio-cutaneous features. J. Med. Genet. 2007;44:e84. doi: 10.1136/jmg.2007.049361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gripp K.W., Hopkins E., Sol-Church K., Stabley D.L., Axelrad M.E., Doyle D., Dobyns W.B., Hudson C., Johnson J., Tenconi R. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am. J. Med. Genet. A. 2011;155A:706–716. doi: 10.1002/ajmg.a.33884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wohllk N., Cote G.J., Bugalho M.M., Ordonez N., Evans D.B., Goepfert H., Khorana S., Schultz P., Richards C.S., Gagel R.F. Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 1996;81:3740–3745. doi: 10.1210/jcem.81.10.8855832. [DOI] [PubMed] [Google Scholar]
- 167.Mulligan L.M., Eng C., Healey C.S., Clayton D., Kwok J.B., Gardner E., Ponder M.A., Frilling A., Jackson C.E., Lehnert H. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat. Genet. 1994;6:70–74. doi: 10.1038/ng0194-70. [DOI] [PubMed] [Google Scholar]
- 168.Kitamura Y., Scavarda N., Wells S.A., Jr., Jackson C.E., Goodfellow P.J. Two maternally derived missense mutations in the tyrosine kinase domain of the RET protooncogene in a patient with de novo MEN 2B. Hum. Mol. Genet. 1995;4:1987–1988. doi: 10.1093/hmg/4.10.1987. [DOI] [PubMed] [Google Scholar]
- 169.Sharland M., Burch M., McKenna W.M., Paton M.A. A clinical study of Noonan syndrome. Arch. Dis. Child. 1992;67:178–183. doi: 10.1136/adc.67.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li R., Johnson A.B., Salomons G.S., van der Knaap M.S., Rodriguez D., Boespflug-Tanguy O., Gorospe J.R., Goldman J.E., Messing A., Brenner M. Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum. Genet. 2006;119:137–144. doi: 10.1007/s00439-005-0116-7. [DOI] [PubMed] [Google Scholar]
- 171.Pauli S., von Velsen N., Burfeind P., Steckel M., Mänz J., Buchholz A., Borozdin W., Kohlhase J. CHD7 mutations causing CHARGE syndrome are predominantly of paternal origin. Clin. Genet. 2012;81:234–239. doi: 10.1111/j.1399-0004.2011.01701.x. [DOI] [PubMed] [Google Scholar]
- 172.Twigg S.R.F., Matsumoto K., Kidd A.M., Goriely A., Taylor I.B., Fisher R.B., Hoogeboom A.J., Mathijssen I.M., Lourenco M.T., Morton J.E. The origin of EFNB1 mutations in craniofrontonasal syndrome: Frequent somatic mosaicism and explanation of the paucity of carrier males. Am. J. Hum. Genet. 2006;78:999–1010. doi: 10.1086/504440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Heron S.E., Scheffer I.E., Iona X., Zuberi S.M., Birch R., McMahon J.M., Bruce C.M., Berkovic S.F., Mulley J.C. De novo SCN1A mutations in Dravet syndrome and related epileptic encephalopathies are largely of paternal origin. J. Med. Genet. 2010;47:137–141. doi: 10.1136/jmg.2008.065912. [DOI] [PubMed] [Google Scholar]
- 174.Sun H., Zhang Y., Liu X., Ma X., Yang Z., Qin J., Jiang Y., Qi Y., Wu X. Analysis of SCN1A mutation and parental origin in patients with Dravet syndrome. J. Hum. Genet. 2010;55:421–427. doi: 10.1038/jhg.2010.39. [DOI] [PubMed] [Google Scholar]
- 175.Trappe R., Laccone F., Cobilanschi J., Meins M., Huppke P., Hanefeld F., Engel W. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 2001;68:1093–1101. doi: 10.1086/320109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Girard M., Couvert P., Carrié A., Tardieu M., Chelly J., Beldjord C., Bienvenu T. Parental origin of de novo MECP2 mutations in Rett syndrome. Eur. J. Hum. Genet. 2001;9:231–236. doi: 10.1038/sj.ejhg.5200618. [DOI] [PubMed] [Google Scholar]
- 177.Zhu X., Li M., Pan H., Bao X., Zhang J., Wu X. Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in 24 sporadic patients with Rett syndrome in China. J. Child Neurol. 2010;25:842–848. doi: 10.1177/0883073809350722. [DOI] [PubMed] [Google Scholar]
- 178.Böhm J., Munk-Schulenburg S., Felscher S., Kohlhase J. SALL1 mutations in sporadic Townes-Brocks syndrome are of predominantly paternal origin without obvious paternal age effect. Am. J. Med. Genet. A. 2006;140:1904–1908. doi: 10.1002/ajmg.a.31383. [DOI] [PubMed] [Google Scholar]
- 179.Splendore A., Jabs E.W., Félix T.M., Passos-Bueno M.R. Parental origin of mutations in sporadic cases of Treacher Collins syndrome. Eur. J. Hum. Genet. 2003;11:718–722. doi: 10.1038/sj.ejhg.5201029. [DOI] [PubMed] [Google Scholar]
- 180.Ibrahimi O.A., Yeh B.K., Eliseenkova A.V., Zhang F., Olsen S.K., Igarashi M., Aaronson S.A., Linhardt R.J., Mohammadi M. Analysis of mutations in fibroblast growth factor (FGF) and a pathogenic mutation in FGF receptor (FGFR) provides direct evidence for the symmetric two-end model for FGFR dimerization. Mol. Cell. Biol. 2005;25:671–684. doi: 10.1128/MCB.25.2.671-684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lajeunie E., Cameron R., El Ghouzzi V., de Parseval N., Journeau P., Gonzales M., Delezoide A.L., Bonaventure J., Le Merrer M., Renier D. Clinical variability in patients with Apert's syndrome. J. Neurosurg. 1999;90:443–447. doi: 10.3171/jns.1999.90.3.0443. [DOI] [PubMed] [Google Scholar]
- 182.Oldridge M., Lunt P.W., Zackai E.H., McDonald-McGinn D.M., Muenke M., Moloney D.M., Twigg S.R.F., Heath J.K., Howard T.D., Hoganson G. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum. Mol. Genet. 1997;6:137–143. doi: 10.1093/hmg/6.1.137. [DOI] [PubMed] [Google Scholar]
- 183.Wilkie A.O.M., Patey S.J., Kan S.H., van den Ouweland A.M., Hamel B.C. FGFs, their receptors, and human limb malformations: Clinical and molecular correlations. Am. J. Med. Genet. 2002;112:266–278. doi: 10.1002/ajmg.10775. [DOI] [PubMed] [Google Scholar]
- 184.Ibrahimi O.A., Zhang F., Eliseenkova A.V., Itoh N., Linhardt R.J., Mohammadi M. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum. Mol. Genet. 2004;13:2313–2324. doi: 10.1093/hmg/ddh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cornejo-Roldan L.R., Roessler E., Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum. Genet. 1999;104:425–431. doi: 10.1007/s004390050979. [DOI] [PubMed] [Google Scholar]
- 186.Rump P., Letteboer T.G., Gille J.J., Torringa M.J., Baerts W., van Gestel J.P., Verheij J.B., van Essen A.J. Severe complications in a child with achondroplasia and two FGFR3 mutations on the same allele. Am. J. Med. Genet. A. 2006;140:284–290. doi: 10.1002/ajmg.a.31084. [DOI] [PubMed] [Google Scholar]
- 187.Santos H.G., Almeida M., Fernandes H., Wilkie A.O.M. Clinical hypochondroplasia in a family caused by a heterozygous double mutation in FGFR3 encoding GLY380LYS. Am. J. Med. Genet. A. 2007;143:355–359. doi: 10.1002/ajmg.a.31556. [DOI] [PubMed] [Google Scholar]
- 188.Pannier S., Martinovic J., Heuertz S., Delezoide A.L., Munnich A., Schibler L., Serre V., Legeai-Mallet L. Thanatophoric dysplasia caused by double missense FGFR3 mutations. Am. J. Med. Genet. A. 2009;149A:1296–1301. doi: 10.1002/ajmg.a.32880. [DOI] [PubMed] [Google Scholar]
- 189.Kerr B., Delrue M.A., Sigaudy S., Perveen R., Marche M., Burgelin I., Stef M., Tang B., Eden O.B., O'Sullivan J. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J. Med. Genet. 2006;43:401–405. doi: 10.1136/jmg.2005.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.van der Burgt I., Kupsky W., Stassou S., Nadroo A., Barroso C., Diem A., Kratz C.P., Dvorsky R., Ahmadian M.R., Zenker M. Myopathy caused by HRAS germline mutations: Implications for disturbed myogenic differentiation in the presence of constitutive HRas activation. J. Med. Genet. 2007;44:459–462. doi: 10.1136/jmg.2007.049270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mulligan L.M., Kwok J.B., Healey C.S., Elsdon M.J., Eng C., Gardner E., Love D.R., Mole S.E., Moore J.K., Papi L. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 192.Lips C.J., Landsvater R.M., Höppener J.W., Geerdink R.A., Blijham G., van Veen J.M., van Gils A.P., de Wit M.J., Zewald R.A., Berends M.J. Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N. Engl. J. Med. 1994;331:828–835. doi: 10.1056/NEJM199409293311302. [DOI] [PubMed] [Google Scholar]
- 193.Kasprzak L., Nolet S., Gaboury L., Pavia C., Villabona C., Rivera-Fillat F., Oriola J., Foulkes W.D. Familial medullary thyroid carcinoma and prominent corneal nerves associated with the germline V804M and V778I mutations on the same allele of RET. J. Med. Genet. 2001;38:784–787. doi: 10.1136/jmg.38.11.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bartsch D.K., Hasse C., Schug C., Barth P., Rothmund M., Höppner W. A RET double mutation in the germline of a kindred with FMTC. Exp. Clin. Endocrinol. Diabetes. 2000;108:128–132. doi: 10.1055/s-2000-5806. [DOI] [PubMed] [Google Scholar]
- 195.Menko F.H., van der Luijt R.B., de Valk I.A., Toorians A.W., Sepers J.M., van Diest P.J., Lips C.J. Atypical MEN type 2B associated with two germline RET mutations on the same allele not involving codon 918. J. Clin. Endocrinol. Metab. 2002;87:393–397. doi: 10.1210/jcem.87.1.8136. [DOI] [PubMed] [Google Scholar]
- 196.Gimm O., Marsh D.J., Andrew S.D., Frilling A., Dahia P.L., Mulligan L.M., Zajac J.D., Robinson B.G., Eng C. Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J. Clin. Endocrinol. Metab. 1997;82:3902–3904. doi: 10.1210/jcem.82.11.4508. [DOI] [PubMed] [Google Scholar]
- 197.Smith D.P., Houghton C., Ponder B.A. Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene. 1997;15:1213–1217. doi: 10.1038/sj.onc.1201481. [DOI] [PubMed] [Google Scholar]
- 198.Jasim S., Ying A.K., Waguespack S.G., Rich T.A., Grubbs E.G., Jimenez C., Hu M.I., Cote G., Habra M.A. Multiple endocrine neoplasia type 2B with a RET proto-oncogene A883F mutation displays a more indolent form of medullary thyroid carcinoma compared with a RET M918T mutation. Thyroid. 2011;21:189–192. doi: 10.1089/thy.2010.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.