

Abstract
Congenital stationary night blindness (CSNB) is a heterogeneous retinal disorder characterized by visual impairment under low light conditions. This disorder is due to a signal transmission defect from rod photoreceptors to adjacent bipolar cells in the retina. Two forms can be distinguished clinically, complete CSNB (cCSNB) or incomplete CSNB; the two forms are distinguished on the basis of the affected signaling pathway. Mutations in NYX, GRM6, and TRPM1, expressed in the outer plexiform layer (OPL) lead to disruption of the ON-bipolar cell response and have been seen in patients with cCSNB. Whole-exome sequencing in cCSNB patients lacking mutations in the known genes led to the identification of a homozygous missense mutation (c.1807C>T [p.His603Tyr]) in one consanguineous autosomal-recessive cCSNB family and a homozygous frameshift mutation in GPR179 (c.278delC [p.Pro93Glnfs∗57]) in a simplex male cCSNB patient. Additional screening with Sanger sequencing of 40 patients identified three other cCSNB patients harboring additional allelic mutations in GPR179. Although, immunhistological studies revealed Gpr179 in the OPL in wild-type mouse retina, Gpr179 did not colocalize with specific ON-bipolar markers. Interestingly, Gpr179 was highly concentrated in horizontal cells and Müller cell endfeet. The involvement of these cells in cCSNB and the specific function of GPR179 remain to be elucidated.
Main Text
Congenital stationary night blindness (CSNB) comprises a group of genetically and clinically heterogeneous retinal disorders. The associated genes encode proteins that are confined to the phototransduction cascade or are important in retinal signaling from photoreceptors to adjacent bipolar cells.1 Most of the patients with mutations in these genes show a typical electrophysiological phenotype characterized by an electronegative waveform of the dark-adapted bright flash electroretinogram (ERG), in which the amplitude of the b-wave is smaller than that of the a-wave.2 This so-called Schubert-Bornschein-type ERG response can be divided in two subtypes, incomplete CSNB ([icCSNB] CSNB2A [MIM 300071], CSNB2B [MIM 610427]) and complete CSNB ([cCSNB] CSNB1A [MIM 310500], CSNB1B [MIM 257270] and CSNB1C [MIM 613216]).3 icCSNB has been characterized by both a reduced rod b-wave and substantially reduced cone responses because of both ON- and OFF-bipolar cell dysfunction, whereas the complete type is associated with a drastically reduced rod b-wave response because of ON-bipolar cell dysfunction but largely normal cone b-wave amplitudes.4 icCSNB has been associated with mutations in CACNA1F [MIM 300110], CABP4 [MIM 608965], and CACNA2D4 [MIM 608171], whereas cCSNB has been associated with mutations in NYX [MIM 300278], GRM6 [MIM 604096], and TRPM1 [MIM 603576]. So far more than 280 mutations have been identified in these genes by us and others via direct sequencing of candidate genes (unpublished data) or microarray analysis.5 Prevalence studies determined that CACNA1F, NYX, and TRPM1 mutations leading to incomplete and complete CSNB occur more frequently (unpublished data). Genotyping studies of our CSNB cohort, comprising 160 patients, reveal that in ∼13% of cases mutations in known genes underlying CSNB were not identified. This is a strong indication that mutations in other genes remain to be discovered or that mutations in unscreened regions, that is regulatory elements and introns, might be involved. Mutations in many genes leading to CSNB have been identified through a candidate gene approach by comparing the human phenotype to similar phenotypes observed in knockout or naturally occurring animal models.6–13 The bottleneck of this approach is the size of a cohort and the identification of the “right” patient harboring the mutation in such a candidate gene. Novel techniques that use massively parallel sequencing of all human exons have recently been successful in identifying mutations in novel genes in other heterogeneous diseases such as Leber congenital amaurosis.14,15 To rapidly identify the missing mutations in our CSNB cohort after whole-exome enrichment (IntegraGen, Evry, France), we sequenced four exomes from a consanguineous autosomal-recessive cCSNB family (that included parents who were first cousins and two of three affected children) and from a sporadic male cCSNB patient of Portuguese origin (Figure S1A and S2, available online, shows the typical cCSNB ERG of patient CIC02756). One index patient from each family was previously excluded by Sanger sequencing for mutations in GRM6 and TRPM1. In addition, the sporadic male patient was also excluded for mutations in NYX. Research procedures were conducted in accordance with institutional guidelines and the Declaration of Helsinki. Prior to genetic testing, informed consent was obtained from all patients and their family members. Ophthalmic examination included best corrected visual acuity, slit lamp examination, fundoscopy, perimetry, full-field (ERG) incorporating the International Society for Clinical Electrophysiology of Vision (ISCEV) standards,16 fundus autofluorescence (FAF), and optical coherence tomography (OCT) (the extent of investigation depended on the referring center). Exons of DNA samples were captured with in-solution enrichment methodology (SureSelect Human All Exon Kits Version 3, Agilent, Massy, France) with the company's biotinylated oligonucleotide probe library (Human All Exon v3 50 Mb, Agilent). Each genomic DNA was then sequenced on a sequencer as paired-end 75 bases (Illumina HISEQ, Illumina, San Diego, USA). Image analysis and base calling were performed with Real Time Analysis (RTA) Pipeline version 1.9 with default parameters (Illumina). The bioinformatic analysis of sequencing data was based on a pipeline (Consensus Assessment of Sequence and Variation [CASAVA] 1.8, Illumina). CASAVA performs alignment, calls the SNPs based on the allele calls and read depth, and detects variants (SNPs and indels). Genetic variation annotation was performed by an in-house pipeline (IntegraGen) and results were provided per sample or family in tabulated text files. After excluding variants observed in dbSNP 132, data were further filtered to keep only variants in coding and splice regions that were present in a homozygous state in the affected children and in a heterozygous state in the parents from the consanguineous family. This allowed us to reduce the number of variants from 5,901 indels to 1 and from 66,621 SNPs to 7. The observed deletion represented a repeat deletion in the penultimate exon of VSIG10 and was therefore unlikely to be a disease-causing variant. However, three missense mutations predicted to be probably or possibly damaging were identified in three different genes (KIAA0753, CRHR1 [MIM 122561], and GPR179 [G protein-coupled receptor 179]) on chromosome 17. The p.Arg518Cys variant found in KIAA0753 was considered unlikely to be disease causing because this arginine residue is not evolutionarily conserved. On the other hand, both the p.Arg259Gln substitution in CRHR1 and the p.His603Tyr in GPR179 affected highly evolutionary conserved amino acid residues (Figure 1 and Figure S1B). Interestingly, the other cCSNB patient (CIC02756), also studied by whole-exome sequencing, carried a homozygous 1 bp deletion, resulting in a frameshift and premature termination (p.Pro96Glnfs∗57) in exon 1 of GPR179. These data strongly support the finding that mutations in GPR179 lead to CSNB found in both families (Table 1). For the c.1807C>T (p.His603Tyr) mutation, both parents were found to be heterozygous because the nucleotide A was read 11 times and 7 times in the father and mother, respectively, whereas the G was found 13 times and 11 times, respectively (reverse strand). The two affected children (patients CIC3308 and CIC04005) showed 26 times and 14 times the nucleotide A. The c.278delC deletion detected in the sporadic cCSNB patients was detected 22 times; 20 other reads of unknown type were also indicated. This might be due to the fact that at this position multiple Cs are present, and thus different reads might occur. Sanger sequencing confirmed the mutations in the index patients of each family. Both mutations cosegregated with the phenotype within the respective family (Figure S1A). In addition, next-generation sequencing data were used to analyze homozygous regions in the affected siblings (patients CIC03308 and CIC04005) of the consanguineous family. The analysis revealed seven major homozygous regions (>0.5 Mb), which were exclusively present on chromosome 17. GPR179 was present in the second largest homozygous region (10.8 Mb), whereas CRHR1 was present in a smaller region (1.3 Mb). In the other sporadic cCSNB patient, GPR179 was not present in any major homozygous region; this can be explained by the fact that the parents were only distant cousins.
Figure 1.
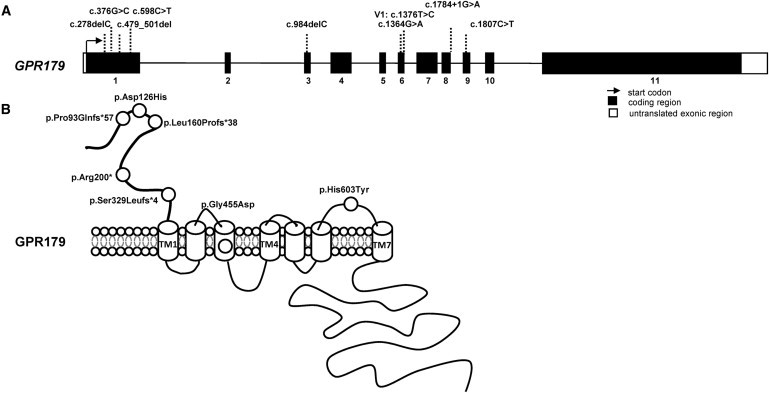
GPR179 mutations in CSNB.
(A) GPR179 structure containing 11 coding exons (NM_001004334.2). Different mutations identified in cCSNB patients are depicted.
(B) The specific domains for GPR179 were estimated by a prediction program (UniProtKB/Swiss-Prot).
Table 1.
Patients with Pathogenic GPR179 Mutations
| Patient Number | Relationship to Index Patient | Sex | Mutations Excluded in Following Genes | Ethnicity and Location | Exon | Nucleotide Exchange (RNA or Protein Effect) | Allele State |
Control Alleles (Mutated or WT) |
Phenotype Index |
|---|---|---|---|---|---|---|---|---|---|
| CIC02756a | – | male | NYX, GRM6, TRPM1 | Portuguese-French; Paris, France | 1 | c.278delC (p.Pro93Glnfs∗57) | homozygous | 0/366 | cCSNB, high myopia, nystagmus, moderate decreased visual acuity |
| CIC02757 | unaffected father | male | – | 1 | c.278delC (p.Pro93Glnfs∗57) | heterozygous | |||
| CIC02758 | unaffected mother | female | – | 1 | c.278delC (p.Pro93Glnfs∗57) | heterozygous | |||
| CIC03631 | – | female | GRM6, TRPM1 | French; Lille, France | 1, 3 | c.376G>C (p.Asp126His), c.984delC (p.Ser329Leufs∗4) | compound heterozygous | 0/366 and 0/372 | cCSNB, high myopia, strabismus, micronystagmus |
| 7699 | – | female | GRM6, TRPM1 | Tübingen, Germany | 1, 6 | c.479_501del (Leu160Profs∗38), c.1364G>A (p.Gly455Asp) | compound heterozygous | 0/366 and 0/384 | cCSNB, strabismus, minimal rotational nystagmus, normal visual field |
| 7692 | unaffected father | male | – | 1 | c.479_501del (p.Leu160Profs∗38) | heterozygous | |||
| 7697 | unaffected mother | female | – | 6 | c.1364G>A (p.Gly455Asp) | heterozygous | |||
| Y1049 | – | female | GRM6, TRPM1 | Lille, France | 1, IVS8 | c.598C>T (p.Arg200∗), c.1784+1G>A (r.spl?) | compound heterozygous | 0/378 0/378 |
cCSNB |
| Y1166 | unaffected father | male | GRM6, TRPM1 | IVS8 | c.1784+1G>A (r.spl) | heterozygous | |||
| Y1167 | unaffected mother | female | GRM6, TRPM1 | 1 | c.598C>T (p.Arg200∗) | heterozygous | |||
| Y1048 | affected sister | female | GRM6, TRPM1 | 1, IVS8 | c.598C>T (p.Arg200∗), c.1784+1G>A (r.spl) | compound heterozygous | |||
| 26985b,c | – | male | GRM6, TRPM1 | Lebanon; Freiburg, Germany | 9 | c.1807C>T (p.His603Tyr) | homozygous | 0/366 | cCSNB, left exotropia, until age of 2 nystagmus |
| CIC03306 | father | male | – | 9 | c.1807C>T (p.His603Tyr) | heterozygous | ERG b-wave were slightly reduced for high flash strength | ||
| CIC03307 | unaffected mother | female | – | 9 | c.1807C>T (p.His603Tyr) | heterozygous | - | ||
| CIC03308 | affected sister | female | – | 9 | c.1807C>T (p.His603Tyr) | homozygous | cCSNB | ||
| CIC04005 | affected sister | female | – | 9 | c.1807C>T (p.His603Tyr) | homozygous | cCSNB, visual acuity reduced | ||
CSNB mutations are annotated according to the recommendation of the Human Genome Variation Society, with nucleotide position +1 corresponding to the A of the translation-initiation codon ATG in the cDNA nomenclature RefSeq NM_001004334.2.
The diagnostic for GRM6 for this patient was performed in Zurich, Switzerland.
For this family consanguinity has been reported (Figure S1A).
We screened 40 CSNB patients (cCSNB and unclassified CSNB) of various origins and from different clinical centers in Europe, the United States, Canada, and Israel by using Sanger sequencing for 27 fragments covering the 11 coding exons and flanking intronic regions of GPR179 (NM_001004334.2). These were amplified by PCR in the presence of 1.5 mM MgCl2 at an annealing temperature of 60°C. For one of the fragments a specific solution (solution S, 3×, fragment exon 11 m, Hot Fire Polymerase, Solis BioDyne, Tartu, Estonia, and primers; Table S1) was used. The PCR products were sequenced with a sequencing mix (BigDyeTerm v1.1 CycleSeq kit, Applied Biosystems, Courtabœuf, France), analyzed on an automated 48-capillary sequencer (ABI 3730 Genetic analyzer, Applied Biosystems), and the results interpreted by applying SeqScape software (Applied Biosystems). We detected three additional cCSNB patients who carried compound heterozygous disease-causing mutations (Table 1). The mutation spectrum identified herein comprises missense, splice-site, and nonsense mutations and deletions. None of these changes were present in control chromosomes (≥366 chromosomes). For patients whose family members could be investigated, the mutations cosegregated with the cCSNB phenotype, and the genotypes were indicative of an autosomal-recessive mode of inheritance (Table 1 and Figure S1A). Missense mutations were predicted to be pathogenic by PolyPhen and SIFT programs and were also found to affect evolutionarily conserved amino acid residues (Figure S1B). On the basis of all of the above evidence, we conclude that mutations in GPR179 lead to cCSNB. Interestingly, we found four cCSNB patients with no mutations in GRM6, TRPM1, NYX, or GPR179, indicating that mutations in additional genes probably remain to be identified to explain these cases of cCSNB. In addition, a few rare variants (Table S2) in GPR179 were identified in patients screened by Sanger sequencing and were classified as variants of unknown pathogenicity because only one mutation was observed or they did not affect conserved amino acid residues. The frequencies of GPR179 polymorphisms found in our patients are provided in Table S3.
To date no information is available on the functional characterization of GPR179. To predict the protein structure and the influence of the mutations identified herein, we created homology models. The human GPR179 sequence (UniProtKB identifier Q6PRD1) was used as a probe for similarity searches in the UniProtKB database with the use of the BlastP program.17,18 In total, more than 100 metazoan sequences (excluding fragments) that were annotated or predicted as GPR179- or GPR158-like were highlighted and aligned with a customized version of the PipeAlign program.19–21 GPR179 codes for a protein with 2,367 amino acids that can be divided into four main regions corresponding to a small signal peptide (positions 1–25), the N-terminal extracellular region (position 26-381), the seven transmembrane (7TM)-spanning region (position 382-628), and the intracellular C-terminal region (position 629-2367) (Figure 1B). Sequence analysis predicted that the N-terminal extracellular region contains a calcium-binding EGF-like domain (position 278-324), whereas the C-terminal intracellular region is characterized by the presence of a short motif centered on the sequence CPWE, which is repeated at least 22 times in the GPR179-related proteins. GPR179 proteins are present in all vertebrates and are closely related to GPR158 and GPR158-like proteins. It is noteworthy that the major differences between GPR179 and the closely related GPR158 proteins rely on the absence of the calcium-binding EGF-like domain at the N-terminal part and a reduced number of CPWE motifs (up to three) in all GPR158 homologs. Interestingly, three other molecules, the regulator of G protein signaling 9 (RGS9 [MIM 604067]), the retinal rod rhodopsin-sensitive cGMP 3,5-cyclic phosphodiesterase subunit gamma (PDE6G [MIM 180073]) and the retinal cone rhodopsin-sensitive cGMP 3,5-cyclic phosphodiesterase subunit gamma (PDE6H [MIM 601190]) share the same protein motif CPWE. These molecules have been implicated in the inhibition of the G protein or amplification of the signal in the phototransduction cascade. Mutations in those genes lead to different retinal disorders, including bradyopsia [MIM 608415],22 rod-cone dystrophy [MIM 613582],23 and cone dystrophy [MIM 610024].24
Based on their seven transmembrane domain regions, both proteins (GPR179 and GPR158) belong to the glutamate receptor or class C GPCR proteins. This class includes, among others, metabotropic glutamate receptors (GRMs), two γ-aminobutyric acid B receptor (GABABR), the calcium-sensing receptor (CASR), the sweet and umami taste receptors and various orphan receptors.25 The different deletions and the early termination mutation in GPR179 identified in our patients are located in exons 1 and 3 and are predicted to lead to nonsense-mediated mRNA decay, which might result in the absence of a protein product. Alternatively, if a protein is formed, only the first extracellular part would be present but would lack all transmembrane domains of GPR179, resulting in truncated protein (Figure 1B). The missense alterations (p.Asp126His, p.Gly455Asp, and p.His603Tyr) affect evolutionarily conserved amino acid residues, which are predicted to be part of the first extracellular domain, within the third transmembrane domain, and in the last extracellular domain (Figure 1B). Multiple alignment analysis of more than 100 metazoan GPR179-related sequences shows strict conservation of the asparagine at position 126 (Asp126), the glycine at position 455 (Gly455), and the histidine at position 603 (His603) in vertebrate sequences. PolyPhen and SIFT programs annotated the three amino acid substitutions to be possibly pathogenic.26 These programs use conservation among species and homologs to predict the pathogenic character of a mutation. In addition, an inductive logic programming prediction web server27 predicted p.Gly455Asp and p.His603Tyr to be pathogenic. This program uses available 3D structures to predict the influence of a mutation. To date, no model of the 3D structure of the amino acid residues <300 is available, therefore the possible pathogenic effect of p.Asp126His could not be predicted with this program. To further gain insight into the deleterious effect of the missense mutations, we generated 3D models of the seven transmembrane (7TM)-spanning region of the human wild-type GPR179 and of the two altered proteins (p.Gly455Asp and p.His603Tyr) by homology modeling with MODELER software (Figure 2).20 Two known 7TM templates, the bovine taste receptor (PDB 1F88) and the squid rhodopsine (PDB 2ZIY), were used to construct the homology models. For each 3D model construction, ten homology models were constructed, and the models with the best normalized discrete optimized potential energy (DOPE) score were selected.28 The homology 3D models were visualized and analyzed by means of the SM2PH-db,28 and figures were constructed with PyMOL software (version 0.99). Multilevel characterization of the mutants (physico-chemical changes and structural modifications induced by the substitution, as well as functional and structural features related to the mutated position) can be visualized and analyzed by the MSV3d web server. Structural analysis of the 3D homology models based on the squid RHO 3D model (2ZIY) localized the His603 in the external loop bridging the sixth and seventh transmembranes, whereas the Gly455 is localized within the third transmembrane helix, which is part of a binding pocket·(Figures 1B and 2). Our homology model predicts that the amino acid exchange p.Gly455Asp introduces a long negatively charged side chain that might point toward the cavity of the binding pocket. This suggests that the phenotypic consequences observed for this mutation might be related to some steric constraints hampering the normal functioning of the receptor. The steric constraints in respect to the p.His603Tyr mutation are less obvious. However, strong conservation across species and homologs are indicative of an important role for the histidine at position 603. Although, for the moment, the 3D structure of the amino acid residues <300 of GPR179 is not available, we know from other receptors that the N terminus of such proteins is important for ligand binding, and thus the p.Asp126His mutation might be associated with loss of this binding. On the other hand, the amino acids that are mutated in our patients also might be important for structural properties of the protein in the endoplasmic reticulum. Thus a misfolded protein is likely to be excluded from the strictly regulated transport to the membrane. Similar findings were observed for mutations in GRM6, identified in cCSNB patients. Mutated metabotropic glutamate receptor 6 could not reach the membrane, leading to cCSNB.29 Further functional analysis of the mutant variants is needed to determine whether these mutations lead to downregulation of the GPR179 transcript, trafficking problems, abolishment of ligand binding, or interactions with other proteins involved in signal transmission from photoreceptors to the adjacent bipolar cells.
Figure 2.

3D Model of the Transmembrane Region of GPR179
(A) 3D homology model based on the 3D model of the wild-type squid rhodopsin (2ZIY). Wild-type Gly455 and His603 residues are indicated in green and the mutated Asp455 and Tyr603 in orange.
(B) Superimposition of 3D models of the wild-type residues and the Gly455Asp alteration (aspartate in orange).
As mentioned above, little is known about the structure and function of GPR179. However, an EST profile is available (Unigene database) and shows a restricted pattern of expression in the human eye, heart, and brain. Furthermore, transcriptomic data of whole retina from rd1 mice revealed increased expression of GPR179 compared to the expression in wild-type mice starting from postnatal day 12 (Figure 3). The rd1 mouse, carrying Pde6b mutations, is a naturally occurring model with progressive rod photoreceptor degeneration, leading to a complete loss of all rods by post natal day 36, and preserved inner retina.30 This would suggest that GPR179 is expressed in the inner nuclear layer of the retina. Interestingly, Nyx, another gene with mutations leading to cCSNB, shows a similar expression profile in the rd1 mouse (Figure 3).
Figure 3.

Indirect Expression Analysis of Gpr179 in rd1 and Wild-Type Mice
Expression of Gpr179 (1459268_at) compared to the expression of Nyx (1446344_at, a known molecule expressed in the inner nuclear layer, also implicated in cCSNB) during rod degeneration in the rd1 mouse. Neural retinas from rd1 and wild-type mice on identical genetic backgrounds47 were hybridized to the mouse genome 430 2.0 array (Affymetrix, High Wycombe, UK). The expression profiles are similar from postnatal day (PND) 5 to PND12. Thereafter, the expression of Gpr179, as well as Nyx, increases in the rd1 retina. This phenomenon correlates temporally with the loss of rod photoreceptor cells and is likely due to the unaffected inner retinal cells in the rd1 specimen at this age.
Real-time PCR experiments with two different primer sets (Table S4) confirmed the expression of GPR179 in human retina (commercially available cDNA from Clontech, Saint-Germain-en-Laye, France), giving a signal of ΔCT = 13.46 (CTGPR179 = 30.03) in relation to beta-actin (ACTB [MIM 102630]) (CT ACTB = 16.57) (primers Table S4). Sanger sequencing of the amplified RT-PCR products from retinas confirmed the presence of the GPR179 transcript. Using the same conditions, we could not detect the transcript in lymphocytes or HEK293 cells.
We investigated the localization of the Gpr179 protein in adult mouse retina by immunostaining coronal eye cryosections with a rabbit polyclonal antibody directed against GPR179 (Sigma-Aldrich, Saint-Quentin Fallavier, France). Bound primary antibody was detected with a secondary antibody (Alexa Fluor 488-conjugated, Invitrogen, Courtaboeuf, France), and the nuclei were counterstained (4′,6-diamidino-2-phenylindole [DAPI], Euromedex, Souffelweyersheim, France). Immunofluorescence was analyzed with a confocal microscope (FV1000 fluorescent, Olympus, Hamburg, Germany). Gpr179 expression could be detected in the outer plexiform layer (OPL) and in the inner limiting membrane (ILM), in close proximity to the ganglion cells (Figure 4, green). Colocalization studies with a mouse anti-Bassoon (Enzo Lifescienes, Lyon, France), a specific marker for ribbon synapse, excluded a close vicinity between Gpr179 and presynaptic terminals (Figure 4A). Furthermore, immunostaining with mouse antibodies against Goα (Millipore, Molsheim, France) and PKCα (Sigma-Aldrich), two specific ON-bipolar markers, demonstrated the absence of colocalization of Gpr179 with these proteins (Figures 4B and 4C). Instead, Gpr179 appears to be localized in a distinct compartment within bipolar cells or in other cells, such as horizontal cells (indicated by the arrow in Figure 4A). Interestingly, bipolar cell dendrites, stained with PKCα, seem to surround Gpr179 (indicated by the arrow in Figure 4C). Alternatively, the Gpr179 OPL staining could also be localized within Müller cell processes present within this layer. In addition, a mouse antibody against calretinin (Millipore), a specific marker for ganglion cells and their dendrites, was used and did not show colocalization with Gpr179 immunostaining (Figure 4D, an example of a ganglion cell dendrite is marked with an arrow). Instead, Gpr179 was highly expressed in Müller cell endfeet at the level of the ILM; similar results had previously been shown for the potassium channel Kir4 .1 macromolecular complex.31–33 Therefore, immunolocalization of Gpr179 suggests its localization in the OPL either in bipolar cells in a cellular compartment distinct from the synaptic membrane and cell body, and/or in horizontal cells, and/or in Müller cell processes as well as within the Müller cell endfeet.
Figure 4.
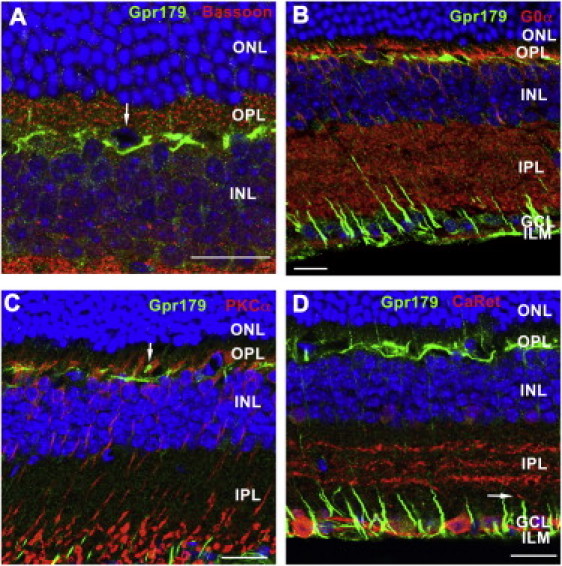
Gpr179 Immunohistochemistry on Retinal Sections of Wild-Type Mice
GPR179 signal (green) in the OPL and ILM double labeled with other retinal markers (red) were detected in wild-type mice by confocal microscopy. The scale bar represents 20 μm. The following abbreviations are used: ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; GCL, ganglion cell layer; and ILM, inner limiting membrane. Gpr179 did not colocalize with the presynaptic marker Basoon (A), the ON-bipolar cell markers Goα (B) and PKCα (C), or with the ganglion cell labeled with calretinin (D). Some cells in the upper part of INL were surrounded by specific Gpr179 staining (arrow in A). The shape and the localization might indicate that these cells represent horizontal cells. Bipolar cell dendrites, stained with PKCα seem to surround Gpr179 (arrow in C). Calretinin antibody labeled specifically the ganglion cells, and their dendrites did not colocalize with Gpr179 (arrow in D). Instead, it seems that Gpr179 is highly concentrated in Müller cell endfeet (D); similar results have been previously shown for a potassium channel Kir4.1.31
The OPL localization of Gpr179 and the same associated ON-bipolar dysfunction phenotype as for Grm6, Nyx, or Trpm1 alterations34–38 would suggest that GPR179 is part of the same transduction pathway and could directly interact with any of these proteins. However, immunolocalization studies are not in keeping with this hypothesis. Instead, immunostaining suggests Müller cell localization and could place the Gpr179 functional role within these cells, possibly through the Kir4.1 macromolecular complex. This complex was shown to involve at least the potassium channel Kir4.1, the water channel aquaporin-4 (AQP4), and the dystrophin isoform Dp71. Interestingly, although Kir4.1 and Aqp4 knockout mice do not show Schubert- Bornschein ERG abnormalities,39,40 a subset of dystrophin mutations, responsible for Duchenne muscular dystrophy (DMD [MIM 310200]) are associated with such ERG abnormalities.41–44 Therefore, one hypothesis would be that Gpr179 is part of the Kir4.1 macromolecular complex. Gpr179 might directly interact with dystrophin isoforms, and its dysfunction would lead to cCSNB in a similar mechanism as in DMD. In order to reconcile the Grm6/Nyx/Trpm1-signaling pathway within bipolar cells and Gpr179 within Muller cells, one might hypothesize that Gpr179 could be involved in an as-yet unknown interaction between ON-bipolar cells and Müller cells that would be essential for ON-bipolar cell depolarization resulting in b-wave formation. On the other hand, our immunostaining studies could also suggest specific localization of Gpr179 within horizontal cells. Therefore, another hypothesis could be that ON-bipolar cells directly interact with horizontal cells. Lack of this interaction due to Gpr179 dysfunction could lead to the reduced b-wave observed in patients with cCSNB.
Including the current study, mutations in four genes (NYX, GRM6, TRPM1, and GPR179) have been implicated in cCSNB (Figure 5).6,7,10–13,45,46 These genes code for nyctalopin, metabotropic glutamate receptor 6, transient receptor potential cation melastatin 1 channel, and G protein-coupled receptor 179, respectively. All but GPR179 localize postsynaptically to the photoreceptors in the retina in ON-bipolar cells.35 Further functional studies will eventually clarify the exact role of this novel protein within the ON-bipolar cells pathway, which will also improve our understanding of the overall visual signal transduction through the retina.
Figure 5.
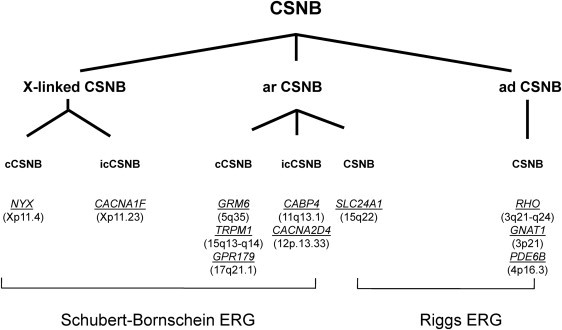
Genes Underlying CSNB
Different forms of CSNB in human are classified according to their electroretinographic feature, mode of inheritance, clinical phenotype, and mutated genes. Patients discussed herein show a complete Schubert-Bornschein type of ERG. The following abbreviations are used: cCSNB, complete CSNB; icCSNB, incomplete CSNB; ar, autosomal recessive; ad, autosomal dominant. Genes are indicated in italics and underlined. Chromosomal location is given between brackets. The phenotype of patients with mutations in icCSNB is more variable and can even lead to progressive cone or cone-rod dystrophy.1
Acknowledgments
The authors are grateful to the families described in this study; to Dominique Santiard-Baron and Christine Chaumeil for their help in DNA collection; to the clinical staff; and to Olivier Goreau, Serge Picaud, and Alvaro Rendon for antibodies used in the colocalization experiments. The project was supported by GIS-maladies rares (C.Z.), Retina France ([part of the 100-Exome Project] I.A., C.P.H., J.-A.S., H.D. and C.Z.), Foundation Voir et Entendre (C.Z.), Agence National de la Recherche (S.S.B), Foundation Fighting Blindness (FFB) grant CD-CL-0808-0466-CHNO (I.A. and the CIC503, recognized as an FFB center), FFB grant C-CMM-0907-0428-INSERM04, Ville de Paris and Région Ille de France, the French Association against Myopathy (AFM) grant KBM-14390 (O.P.), and National Institutes of Health grant 1R01EY020902-01A1 (K.B.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ESEfinder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
GenCards, http://www.genecards.org/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
Sm2ph Central, http://decrypthon.igbmc.fr/sm2ph/cgi-bin/home
National Center for Biotechnology Information, http://ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Pymol, http://pymol.org/
SIFT (Sorting Intolerant From Tolerant), http://blocks.fhcrc.org/sift/SIFT.html
UniProtKB/Swiss-Prot, http://www.uniprot.org
USCS Human Genome Browser, http://genome.ucsc.edu/
References
- 1.Zeitz C. Molecular genetics and protein function involved in nocturnal vision. Expert Rev. Ophthalmol. 2007;2:467–485. [Google Scholar]
- 2.Schubert G., Bornschein H. Analysis of the human electroretinogram.Ophthalmologica. 1952;123:396–413. doi: 10.1159/000301211. [DOI] [PubMed] [Google Scholar]
- 3.Miyake Y., Yagasaki K., Horiguchi M., Kawase Y., Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986;104:1013–1020. doi: 10.1001/archopht.1986.01050190071042. [DOI] [PubMed] [Google Scholar]
- 4.Audo I., Robson A.G., Holder G.E., Moore A.T. The negative ERG: Clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008;53:16–40. doi: 10.1016/j.survophthal.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Zeitz C., Labs S., Lorenz B., Forster U., Uksti J., Kroes H.Y., De Baere E., Leroy B.P., Cremers F.P., Wittmer M. Genotyping microarray for CSNB-associated genes. Invest. Ophthalmol. Vis. Sci. 2009;50:5919–5926. doi: 10.1167/iovs.09-3548. [DOI] [PubMed] [Google Scholar]
- 6.Dryja T.P., McGee T.L., Berson E.L., Fishman G.A., Sandberg M.A., Alexander K.R., Derlacki D.J., Rajagopalan A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA. 2005;102:4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeitz C., van Genderen M., Neidhardt J., Luhmann U.F., Hoeben F., Forster U., Wycisk K., Mátyás G., Hoyng C.B., Riemslag F. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest. Ophthalmol. Vis. Sci. 2005;46:4328–4335. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]
- 8.Zeitz C., Kloeckener-Gruissem B., Forster U., Kohl S., Magyar I., Wissinger B., Mátyás G., Borruat F.X., Schorderet D.F., Zrenner E. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 2006;79:657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wycisk K.A., Zeitz C., Feil S., Wittmer M., Forster U., Neidhardt J., Wissinger B., Zrenner E., Wilke R., Kohl S., Berger W. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am. J. Hum. Genet. 2006;79:973–977. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Audo I., Kohl S., Leroy B.P., Munier F.L., Guillonneau X., Mohand-Saïd S., Bujakowska K., Nandrot E.F., Lorenz B., Preising M. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:720–729. doi: 10.1016/j.ajhg.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z., Sergouniotis P.I., Michaelides M., Mackay D.S., Wright G.A., Devery S., Moore A.T., Holder G.E., Robson A.G., Webster A.R. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am. J. Hum. Genet. 2009;85:711–719. doi: 10.1016/j.ajhg.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Genderen M.M., Bijveld M.M., Claassen Y.B., Florijn R.J., Pearring J.N., Meire F.M., McCall M.A., Riemslag F.C., Gregg R.G., Bergen A.A., Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:730–736. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura M., Sanuki R., Yasuma T.R., Onishi A., Nishiguchi K.M., Koike C., Kadowaki M., Kondo M., Miyake Y., Furukawa T. TRPM1 mutations are associated with the complete form of congenital stationary night blindness. Mol. Vis. 2010;16:425–437. [PMC free article] [PubMed] [Google Scholar]
- 14.Sergouniotis P.I., Davidson A.E., Mackay D.S., Li Z., Yang X., Plagnol V., Moore A.T., Webster A.R. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 2011;89:183–190. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 16.Marmor M.F., Fulton A.B., Holder G.E., Miyake Y., Brigell M., Bach M., International Society for Clinical Electrophysiology of Vision ISCEV Standard for full-field clinical electroretinography (2008 update) Doc. Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 17.UniProt Consortium The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38(Database issue):D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plewniak F., Bianchetti L., Brelivet Y., Carles A., Chalmel F., Lecompte O., Mochel T., Moulinier L., Muller A., Muller J. PipeAlign: A new toolkit for protein family analysis. Nucleic Acids Res. 2003;31:3829–3832. doi: 10.1093/nar/gkg518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eswar N., Eramian D., Webb B., Shen M.Y., Sali A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 21.Eramian D., Eswar N., Shen M.Y., Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17:1881–1893. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishiguchi K.M., Sandberg M.A., Kooijman A.C., Martemyanov K.A., Pott J.W., Hagstrom S.A., Arshavsky V.Y., Berson E.L., Dryja T.P. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature. 2004;427:75–78. doi: 10.1038/nature02170. [DOI] [PubMed] [Google Scholar]
- 23.Dvir L., Srour G., Abu-Ras R., Miller B., Shalev S.A., Ben-Yosef T. Autosomal-recessive early-onset retinitis pigmentosa caused by a mutation in PDE6G, the gene encoding the gamma subunit of rod cGMP phosphodiesterase. Am. J. Hum. Genet. 2010;87:258–264. doi: 10.1016/j.ajhg.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piri N., Gao Y.Q., Danciger M., Mendoza E., Fishman G.A., Farber D.B. A substitution of G to C in the cone cGMP-phosphodiesterase gamma subunit gene found in a distinctive form of cone dystrophy. Ophthalmology. 2005;112:159–166. doi: 10.1016/j.ophtha.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 25.Lagerström M.C., Schiöth H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 26.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luu, T., Nguyen, N., Friedrich, A., Muller, J., Moulinier, L., and Poch, O. (2011). Extracting knowledge from a mutation database related to human monogenic disease using inductive logic programming. International Conference on Bioscience, Biochemistry and Bioinformatics; Singapore. IACSIT 5, 83–100.
- 28.Friedrich A., Garnier N., Gagnière N., Nguyen H., Albou L.P., Biancalana V., Bettler E., Deléage G., Lecompte O., Muller J. SM2PH-db: An interactive system for the integrated analysis of phenotypic consequences of missense mutations in proteins involved in human genetic diseases. Hum. Mutat. 2010;31:127–135. doi: 10.1002/humu.21155. [DOI] [PubMed] [Google Scholar]
- 29.Zeitz C., Forster U., Neidhardt J., Feil S., Kälin S., Leifert D., Flor P.J., Berger W. Night blindness-associated mutations in the ligand-binding, cysteine-rich, and intracellular domains of the metabotropic glutamate receptor 6 abolish protein trafficking. Hum. Mutat. 2007;28:771–780. doi: 10.1002/humu.20499. [DOI] [PubMed] [Google Scholar]
- 30.Carter-Dawson L.D., LaVail M.M., Sidman R.L. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest. Ophthalmol. Vis. Sci. 1978;17:489–498. [PubMed] [Google Scholar]
- 31.Connors N.C., Kofuji P. Potassium channel Kir4.1 macromolecular complex in retinal glial cells. Glia. 2006;53:124–131. doi: 10.1002/glia.20271. [DOI] [PubMed] [Google Scholar]
- 32.Claudepierre T., Rodius F., Frasson M., Fontaine V., Picaud S., Dreyfus H., Mornet D., Rendon A. Differential distribution of dystrophins in rat retina. Invest. Ophthalmol. Vis. Sci. 1999;40:1520–1529. [PubMed] [Google Scholar]
- 33.Dalloz C., Sarig R., Fort P., Yaffe D., Bordais A., Pannicke T., Grosche J., Mornet D., Reichenbach A., Sahel J. Targeted inactivation of dystrophin gene product Dp71: Phenotypic impact in mouse retina. Hum. Mol. Genet. 2003;12:1543–1554. doi: 10.1093/hmg/ddg170. [DOI] [PubMed] [Google Scholar]
- 34.Vardi N., Duvoisin R., Wu G., Sterling P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J. Comp. Neurol. 2000;423:402–412. doi: 10.1002/1096-9861(20000731)423:3<402::aid-cne4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 35.Morgans C.W., Ren G., Akileswaran L. Localization of nyctalopin in the mammalian retina. Eur. J. Neurosci. 2006;23:1163–1171. doi: 10.1111/j.1460-9568.2006.04647.x. [DOI] [PubMed] [Google Scholar]
- 36.Gregg R.G., Kamermans M., Klooster J., Lukasiewicz P.D., Peachey N.S., Vessey K.A., McCall M.A. Nyctalopin expression in retinal bipolar cells restores visual function in a mouse model of complete X-linked congenital stationary night blindness. J. Neurophysiol. 2007;98:3023–3033. doi: 10.1152/jn.00608.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgans C.W., Zhang J., Jeffrey B.G., Nelson S.M., Burke N.S., Duvoisin R.M., Brown R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA. 2009;106:19174–19178. doi: 10.1073/pnas.0908711106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koike C., Obara T., Uriu Y., Numata T., Sanuki R., Miyata K., Koyasu T., Ueno S., Funabiki K., Tani A. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc. Natl. Acad. Sci. USA. 2010;107:332–337. doi: 10.1073/pnas.0912730107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kofuji P., Ceelen P., Zahs K.R., Surbeck L.W., Lester H.A., Newman E.A. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: Phenotypic impact in retina. J. Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J., Patil R.V., Verkman A.S. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Invest. Ophthalmol. Vis. Sci. 2002;43:573–579. [PubMed] [Google Scholar]
- 41.Cibis G.W., Fitzgerald K.M., Harris D.J., Rothberg P.G., Rupani M. The effects of dystrophin gene mutations on the ERG in mice and humans. Invest. Ophthalmol. Vis. Sci. 1993;34:3646–3652. [PubMed] [Google Scholar]
- 42.De Becker I., Riddell D.C., Dooley J.M., Tremblay F. Correlation between electroretinogram findings and molecular analysis in the Duchenne muscular dystrophy phenotype. Br. J. Ophthalmol. 1994;78:719–722. doi: 10.1136/bjo.78.9.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillers D.A., Bulman D.E., Weleber R.G., Sigesmund D.A., Musarella M.A., Powell B.R., Murphey W.H., Westall C., Panton C., Becker L.E. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat. Genet. 1993;4:82–86. doi: 10.1038/ng0593-82. [DOI] [PubMed] [Google Scholar]
- 44.Pillers D.A., Fitzgerald K.M., Duncan N.M., Rash S.M., White R.A., Dwinnell S.J., Powell B.R., Schnur R.E., Ray P.N., Cibis G.W., Weleber R.G. Duchenne/Becker muscular dystrophy: Correlation of phenotype by electroretinography with sites of dystrophin mutations. Hum. Genet. 1999;105:2–9. doi: 10.1007/s004399900111. [DOI] [PubMed] [Google Scholar]
- 45.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 46.Pusch C.M., Zeitz C., Brandau O., Pesch K., Achatz H., Feil S., Scharfe C., Maurer J., Jacobi F.K., Pinckers A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000;26:324–327. doi: 10.1038/81627. [DOI] [PubMed] [Google Scholar]
- 47.Viczian A., Sanyal S., Toffenetti J., Chader G.J., Farber D.B. Photoreceptor-specific mRNAs in mice carrying different allelic combinations at the rd and rds loci. Exp. Eye Res. 1992;54:853–860. doi: 10.1016/0014-4835(92)90148-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.