

Abstract
X-linked megalocornea (MGC1) is an ocular anterior segment disorder characterized by an increased cornea diameter and deep anterior chamber evident at birth and later onset of mosaic corneal degeneration (shagreen), arcus juvenilis, and presenile cataracts. We identified copy-number variation, frameshift, missense, splice-site and nonsense mutations in the Chordin-like 1 gene (CHRDL1) on Xq23 as the cause of the condition in seven MGC1 families. CHRDL1 encodes ventroptin, a bone morphogenic protein antagonist with a proposed role in specification of topographic retinotectal projections. Electrophysiological evaluation revealed mild generalized cone system dysfunction and, in one patient, an interhemispheric asymmetry in visual evoked potentials. We show that CHRDL1 is expressed in the developing human cornea and anterior segment in addition to the retina. We explored the impact of loss of ventroptin function on brain function and morphology in vivo. CHRDL1 is differentially expressed in the human fetal brain, and there is high expression in cerebellum and neocortex. We show that MGC1 patients have a superior cognitive ability despite a striking focal loss of myelination of white matter. Our findings reveal an unexpected requirement for ventroptin during anterior segment development and the consequences of a lack of function in the retina and brain.
Introduction
Anterior segment dysgenesis describes a range of phenotypes caused by failure of normal development of the anterior segment of the eye.1 These anomalies are often associated with an increased risk of glaucoma, corneal opacity, and loss of vision. X-linked megalocornea (MGC1 [MIM 309300]), a bilateral and developmental anomaly of the anterior segment of the eye, is characterized by a thin cornea of increased diameter (generally >12.5 mm at birth) and a very deep anterior chamber but no increase in intraocular pressure.2 In older patients, MGC1 is associated with distinctive secondary changes of mosaic corneal degeneration (shagreen) and corneal arcus juvenilis. Pathological changes also include mild iris atrophy with pigment dispersion, lens dislocation, and cataracts.2,3 Corneal endothelial cells have a normal morphology and cell density, which suggests that corneal endothelial cell hyperplasia occurs concurrently with excessive corneal growth.4
MGC1 was mapped over 20 years ago to Xq12-q26; however, disease gene identification was hampered because of the size of the interval (∼24.3 Mb).2,5,6 Here we describe identification of the causative gene, CHRDL1 (MIM 300350), which reveals an unexpected role for the protein (ventroptin) in anterior segment development.
Subjects and Methods
X-Linked Megalocornea Families and Ophthalmic Clinical Assessment
Genomic DNA was isolated from peripheral blood from the affected individuals and their family members. Informed consent was obtained from all participating families and subjects. This study followed the tenets of the Declaration of Helsinki (1983 revision) and was approved by the local ethics committees. Studies for families 1, 2, 3, 4, and 5 were approved by Moorfields Eye Hospital Research ethics committee, for family 6 by the Cleveland Clinic, and for family 7 by the Royal Victorian Eye and Ear Hospital. Informed consent, including permission to publish photographs, was obtained from all subjects. Patients and their relatives were clinically assessed by experienced ophthalmologists. Standard evaluation consisted of detailed ophthalmic examination and the additional measurement of the axial length of the eye and imaging of the anterior segment of the eye performed with ocular coherence tomography (OCT; Visante, Carl Zeiss Meditec), b-scan ultrasonography, and optical interferometry (IOLMaster, Carl Zeiss Meditec). The details of the clinical assessment of family 7 are described elsewhere.2,4 All available adult members of the UK families had detailed retinal and optic nerve head imaging. Color fundus photography, spectral domain optical coherence tomography (Spectralis OCT Heidelberg Retina Angiograph), scanning laser polarimetry (GDxPRO, Carl Zeiss Meditec), and confocal scanning laser ophthalmoscopy (Heidelberg Retina Tomograph) were undertaken in individuals II:1 and II:3 in family 1, and II:1 in family 5. Electrophysiological assessment included full-field and pattern electroretinography and visual evoked cortical potentials. Full-field electroretinography (ERG) was performed with protocols that exceeded the minimum standards of the International Society for Clinical Electrophysiology of Vision. Cortical visual evoked potentials (VEPs) were recorded to flash stimuli and high-contrast checkerboard reversal (check size 0.83 degrees) with multichannel recordings that otherwise followed the international standards.7,8
Comparative Genomic Hybridization and Mutation Analysis
DNA for affected individual II:1 from family 1 was hybridized (labeled with Cy3) to a dense X chromosome array (median probe spacing of 1 per 340 bp, NimbleGen Systems) with a reference male DNA sample (labeled with Cy5). The data were visualized with SignalMap software (NimbleGen Systems). Primers were designed to amplify and bidirectionally sequence exons and exon/intron boundaries of the CHRDL1 gene (primers available on request). ReddyMix PCR Master Mix (Abgene, Thermo Scientific) was used for amplification with 2 pmol of each primer and 150 ng DNA with standard cycling conditions. PCR products were purified with ExoSAP- IT (USB) according to the manufacturer's protocols. Purified products were bidirectionally sequenced with ABI BigDye terminator cycle sequencing chemistry version 3.1 on a 3730 ABI Analyzer (Applied Biosystems) following the manufacturer's protocols. Patient DNA sequences were analyzed with Lasergene software (DNASTAR). None of the mutations identified were listed in either dbSNP or the 1000 Genomes Project database. To further verify that the mutations identified were not polymorphisms, 220 control male DNA samples were tested with specific primer pairs incorporating the mutation detected (primers available on request). The breakpoints of the segmental deletion detected by aCGH for family 1 were identified by PCR amplification across the deletion and direct sequencing (primers available on request). The extent of the deletion encompassing the CHRDL1 gene in family 7 was refined by targeted proximal and distal PCR (primers available on request). Genetic locations are based on hg19 genome assembly build (UCSC). Protein sequence alignments were performed by ClustalW.
Reverse Transcription PCR
RNA was extracted from microdissected human adult corneas and trabecular meshwork and fetal cornea and whole eyes with Trizol reagent (Invitrogen) according to the manufacturer's protocol. First-strand cDNA synthesis was performed with 50 ng total RNA via M-MLV Reverse Transcriptase (Promega) and random hexamer oligonucleotide primers. Second strand synthesis was performed with gene-specific primers from exon 3 to exon 4 (flanking an intron) for CHRDL1, 5′-AGATGGCATCCTTACCTGGA-3′ and 5′-ATGCACAGGAGAAAGGCAAT-3′, and exon 9 to exon 10 for PAX6, 5′- CACCAGTGTCTACCAACCAA-3′ and 5′- TTGCATAGGCAGGTTATTTG-3′.
In Situ Hybridization
Human fetal eyes were fixed in 4% (w/v) phosphate-buffered formaldehyde solution and equilibrated in 30% sucrose solution for cryoprotection prior to freezing in an optimal cutting temperature compound (RA Lamb). Sections were cut on a Leica CM1900 UV cryostat to 14 μm thickness and collected on Superfrost-Plus glass slides (VWR). In situ hybridization was performed in 200 mM NaCl, 5mM EDTA, 10mM Tris-HCl, 5mM sodium phosphate, 0.1mg/ml yeast tRNA, 10% dextran sulfate, 1× Denhardt's reagent, and 50% formamide with digoxigenin-incorporated riboprobes. Riboprobes were generated from a 627 bp fragment of the 3′untranslated region of human CHRDL1 amplified with the primers 5′-CTGGACTGCAGGCTTATTTT-3′ and 5′-ATTTACTCTCCTGGGCACAA-3′ (probe 1) or with a 777 bp fragment of the 3′ end of the coding region amplified with the primers 5′-GCATGGACAAGTGTGTGTTT −3′ and 5′-ACACACCACTCCACACAAAG −3′ (probe 2) and cloned into pGEMT-Easy (Promega). Sections from five eyes at Carnegie stage fetal week 1 (F1) were analyzed. Both riboprobes gave the same results; data are shown for probe 1 only. For antibody detection, slides were incubated with anti-digoxigenin conjugated with alkaline phosphatase (Roche) diluted 1:1,000 in 2% sheep serum. Expression patterns were visualized with a Nitro-Blue Tetrazolium Chloride/5-Bromo-4-Chloro-3′-Indolyphosphate p-Toluidine Salt (NBT/BCIP) system (Roche). Sections were mounted in DPX mounting medium (RA Lamb) and viewed on a Zeiss Axioplan 2 and images were captured with a Jenoptik C14 digital camera (OpenLab, Improvision).
Association Study
To determine whether gender or common variants within the CHRDL1 gene influence corneal diameter in the general population, we examined a cohort of 20- to 21-year-old young adults, of whom 88% were white, ascertained for the Raine Eye Health Study in Western Australia (n = 830; males n = 427) and a Chinese population cohort (n = 1,345; males n = 693) ascertained in Singapore. White-to-white (WTW) measurements for the study group were taken with an IOLMaster (Carl Zeiss Meditec) in the Raine cohort and by Visante measures of anterior of chamber width, defined as the horizontal distance between the scleral spurs in the Chinese cohort. WTW measurements and GWAS data were available for all participants. Individual genotype data for CHRDL1 were extracted from the genome-wide Illumina 660 Quad Array or 610 K array data. Genotyping was performed on an Illumina BeadArray Reader. Samples with call rate below 97% were discarded from further analysis (Raine cohort 16 samples). SNPs had a call rate >95%, a Hardy-Weinberg p value less than 5.7 × 10−7 or had a minor allele frequency less than 1%. CHRDL1 SNPs assessed were rs197023, rs12689346, rs5943057, rs12857107. Cross-sectional analyses of SNP allelic dosage on the WTW measurements were performed with multivariate linear regression adjusting for gender for CHRDL1 SNPs with PLINK version 1.07. Outcomes were all approximately normally distributed. For nonnormal outcomes, they were log-transformed to normality. Analyses were performed with the statistical graphics software R version 2.11.1.
Fetal Brain Expression Analysis
Raw gene expression data from human fetal brain tissue were gathered from a published study.9 Postmortem specimens from four late mid-fetal human brains (18, 19, 21, and 23 weeks of gestation) were collected from the Human Fetal Tissue Repository at the Albert Einstein College of Medicine (AECOM). Details of specimens, tissue processing, microdissection, and neuropathological assessment were described elsewhere.9 All samples were originally analyzed by these authors with Affymetrix Exon 1.0 ST Arrays. The data set included regional expression data for the striatum, thalamus, hippocampus, and nine areas of neocortex, including four areas of the prefrontal cortex (orbital, dorsolateral, medial, and ventrolateral prefrontal neocortex), one motor-somatosensory area, two areas of the temporal cortex (auditory and association temporal neocortices) and areas from parietal association and occipital visual neocortices. For our study, cell files from human fetal specimens were downloaded from the NCBI Gene Expression Omnibus (accession GSE13344). Background correction, quantile normalization, log2 transformation and median polishing for summarization were performed. A full set of probe sets from the exon-array including core and noncore probe sets were used. Brain region, date of hybridization and variations due to individual genetic differences were identified as greater sources of variability by PCA and therefore included as covariates in the analysis of variance (ANOVA) to investigate whether CHRDL1 (13 probe sets) and BMP4 (13 probe sets) were differentially expressed across brain regions. All exon array data were analyzed with Affymetrix Power Tools software package (Affymetrix) and Partek Genomics Suite (Partek).
Cognitive Tests
Neuropsychological tests employed were as described previously.10 All of the standardized tests selected only require verbal interaction. Intellectual level and executive and memory function were tested. Details are given in the Supplemental Data.
Magnetic Resonance Imaging Acquisition
Control (26 healthy volunteers; 14 males and 12 females, mean age 42 years, range 22–62 years) and MGC1 subject (two individuals) scans were acquired on the same 3T GE Excite II scanner (General Electric Medical Systems) with identical scan acquisition sequences. Standard imaging gradients with a maximum strength of 40 mT m−1 and slew rate of 150 T m−1 were used. All data were acquired with a body coil for transmission and an eight channel phased array coil for reception. The scanning protocol included whole-brain T1-weighted and diffusion-weighted scanning. The T1-weighted scan was performed with the following parameters: two shots, echo time (TE) = 30 ms, repetition time (TR) = 4,500 ms, matrix 256 × 256, 88 contiguous 1.5-mm slices). Diffusion-weighted images were obtained via spin-echo planar imaging (EPI) sequence (TE = 73 ms, 60 contiguous 2.4-mm thick axial slices, matrix size = 96 × 96, voxel size = 1.875 × 1.875 × 2.4 mm3) with 52 noncollinear directions at a b value of 1,200 mm2s-1 (δ = 21 ms, Δ = 29 ms, with full gradient strength of 40 mT m−1) and six nondiffusion-weighted (b = 0) scans. The gradient directions were calculated and ordered as described elsewhere.11 The diffusion tensor imaging (DTI) acquisition time for a total of 3,480 image slices was 25 min, depending on subject heart rate.
Image Analysis: Voxel-Based Morphometry
Differences in total white matter (WM) volume were determined via voxel-based morphometry (VBM) implemented with SPM5 (Wellcome Trust Center for Neuroimaging, London, UK) with default parameters. The images were bias-corrected, normalized, and segmented. Subsequently, the warped WM segments were affine-transformed into Montreal Neurological Institute (MNI) space and were scaled by the Jacobian determinants of deformations to account for the local compression and stretching that occurs as a consequence of the warping and affine transformation (modulated WM volumes). Finally the modulated volumes were smoothed with a Gaussian kernel of 4 mm full-width of half maximum. The volume maps were statistically analyzed with a general linear model based on Gaussian random field theory. Group analysis was performed with a voxel-wise two-sample t test. Age, gender, and total intracranial volume were included in the analysis as regressors of no interest.
Image Analysis: Tract-Based Spatial Statistics
All scans were transferred to a Linux-based Sun Ultra workstation. The DICOM files of each DTI acquisition were converted into a single multivolume ANALYZE 7.5 file and then corrected for eddy currents with the “eddy-correct” algorithm implemented in FSL v4.1.3.
Statistical analysis of the fractional anisotropy (FA) values utilized a tract-based spatial statistics (TBSS) technique from the Diffusion Toolbox implemented in the FMRIB software, as described previously.11,12
Briefly, FA maps from all subjects were aligned to the most representative map. A mean FA map was created and aligned into MNI152 standard space.11,12 The mean FA map was then thinned to create a mean FA skeleton, representing the main fiber tracts, and was thresholded at FA > 0.2. Two contrasts for each diffusivity index were estimated: subjects greater than controls and controls greater than subjects.
Results
Assessment of MGC1 Phenotype in Study Families
We ascertained six new families with MGC1 (Figure 1A). In family 1, affected individuals II:1 and II:3 had corneal diameters of 16.0 and 15.5 mm, respectively, and large anterior chamber depths of 6.3 and 7.0 mm (Figure 1E and Table 1). Central corneal thickness was also markedly reduced in both individuals (359 μm and 381 μm; Figure 1E and Table 1). Reported mean values for these ocular parameters in normal male adult eyes were 11.92 mm (SD = 0.40 mm) for corneal diameter, 3.15 mm (SD = 0.42 mm) for anterior chamber depth, and 534 μm (SD = 31 μm) for central corneal thickness (both sexes).13–15 In individuals II:1 and II:3, mosaic corneal degeneration and corneal arcus juvenilis were present and both had bilateral cataract extractions in their 50s (Figures 1B and 1C and Table 1). Families 2–5 contained a single affected male (age range 8–52 years, Figure 1 and Table 1). In this latter patient group, corneal diameters ranged from 15.0 to 16.0 mm, anterior chamber depth from 5.7 to 7.4 mm (Figure 1D), and central corneal thickness from 377 to 446 μm (not correlated with age). Mosaic corneal degeneration and corneal arcus were not present in the two youngest affected individuals, aged 8 and 9 years (II:1 in family 2 and II:2 in family 3, Figure 1 and Table 1). In family 6, affected individuals III:8 and V:2 had corneal diameters of 14.0 mm and central corneal thickness ranging from 437 to 443 μm (Figure 1 and Table 1). Mosaic corneal degeneration and corneal arcus were present, even in the youngest individual (V:2, age 16 years). Iris transillumination with pigment dispersion was detected in all individuals examined. No patient had glaucoma or a significant visual loss. Individual V:2 in family 6 had significant myopic astigmatism on refraction. No neurological or systemic abnormalities were detected for any affected individuals and carrier females had no clinical signs of MGC1.
Figure 1.
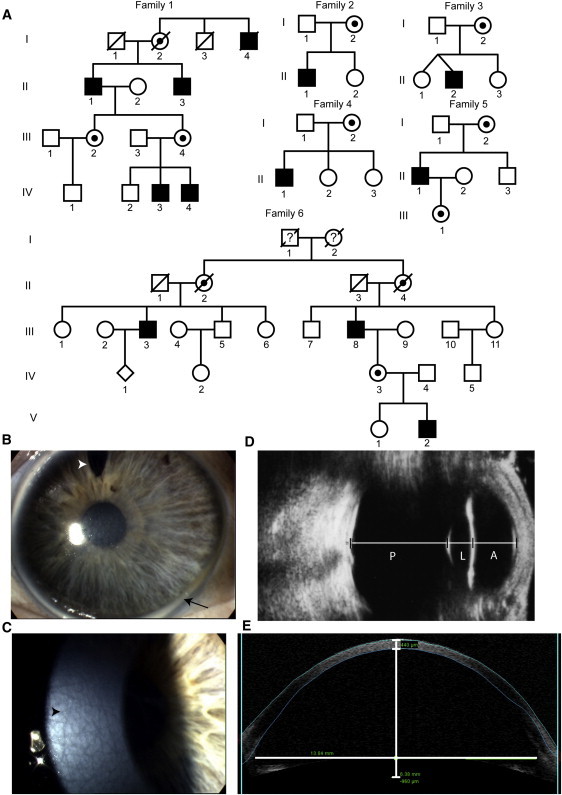
X-Linked Megalocornea Families and Phenotype
(A) Pedigree structure of six families with X-linked megalocornea (MGC1); family 7 has been described elsewhere (pedigree 1 in Mackey et al.2). Black boxes denote affected males; dotted circles, carrier females; clear boxes and circles, unaffected individuals.
(B) Right eye of individual II:3 of family 1 showing a greater than average cornea diameter and a white ring at the limbus (arcus juvenilis, arrow). The superior gap in the iris is from cataract surgery (surgical iridectomy, arrowhead).
(C) Mosaic corneal degeneration of the central corneal stroma (arrowhead) of individual II:1 of family 1.
(D) Axial ultrasound image performed through closed eyelid of the left eye of individual II:2 in family 3 showing a greatly enlarged anterior segment (A), whereas the posterior segment (P) is relatively normal; L is used as an abbreviation of lens.
(E) Optical coherence tomography image of the anterior segment of individual II:1 in family 1 showing thinning of the entire cornea, limbus to limbus, and a deep anterior chamber. Measurements taken for central corneal thickness, anterior chamber depth, and anterior chamber diameter are included.
Table 1.
Detail of Ocular Phenotype in X-Linked Megalocornea Patients
| Individual (Age at last follow up in years) |
Family 1 |
Family 2 |
Family 3 |
Family 4 |
Family 5 |
Family 6 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
II:1 (72) |
II:3 (67) |
II:1 (8) |
II:2 (9) |
II:1 (15) |
II:1 (52) |
III:8 (68) |
V:2 (16) |
|||||||||
| Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | |
| Refractiona | NA | NA | +0.75/−2.00 × 35 | 0.0/−2.50 × 142 | +3.00/−0.5 × 35 | +3.00 | +2.50/−3.00 × 5 | +2.00/−3.00 × 175 | −1.25/−1.00 × 45 | −1.00/−1.25 × 145 | +1.75/−1.25 × 20 | +2.50 | NA | NA | −11.25/−3.50 × 30 | −8.5/−2.75 × 165 |
| Age at cataract surgery (years) | 54 | 58 | 53 | 51 | – | – | – | – | – | – | 48 | 49 | 33 | 41 | – | – |
| −Visual acuity (LogMAR) | 0.3 | 0.0 | −0.1 | −0.1 | 0.4 | 0.25 | 0.2 | 0.1 | 0.0 | 0.0 | −0.1 | 0.6b | 0.0 | 0.2 | 0.18 | 0.1 |
| Iris transillumination | + | + | + | + | NA | NA | + | + | NA | NA | + | + | NA | NA | NA | NA |
| Arcus | + | + | + | + | – | – | – | – | – | – | + | + | + | + | + | + |
| Shagreen | + | + | + | + | - | - | - | - | + | + | + | + | + | + | + | + |
| Anterior chamber depth (mm) | 6.8 | 7.0 | 6.6 | 6.3 | 7.1 | 7.1 | 5.9 | 5.7 | 6.5 | 6.3 | 6.9 | 7.4 | NA | NA | NA | NA |
| WTW (mm) | 16.0 | 16.0 | 15.5 | 15.5 | 15.5 | 15.5 | 15.0 | 15.0 | 15.0 | 15.0 | 16.0 | 16.0 | 14.0 | 14.0 | 14.0 | 14.0 |
| Axial length (mm) | 25.7 | 24.6 | 25.0 | 25.0 | 25.0 | 24.7 | 24.2 | 23.8 | 25.9 | 25.9 | 26.9 | 26.1 | NA | NA | NA | NA |
| Central corneal thickness (μm) | 359 | 365 | 346 | 381 | 377 | 377 | 428 | 446 | 405 | 408 | 439 | 421 | 438 | 437 | 443 | 442 |
The following abbreviations and symbols are used: –, not present; NA, not available; WTW, cornea horizontal diameter, white-to-white.
Prior to cataract surgery.
Amblyopia.
Identification of CHRDL1 Mutations Causing MGC1
Using dense X chromosome-specific array comparative genomic hybridization, we identified a novel segmental deletion of approximately 250 kb on Xq23 in an affected male (II:1) from family 1 (Figure 2A). The deletion within the MGC1 critical interval encompassed the 3′ end of CHRDL1 (Chordin-like 1) and identified CHRDL1 as a candidate gene for MGC1 (Figure 2A). To confirm the deletion and define the deletion breakpoint, we amplified and bidirectionally sequenced the breakpoint fragment (Figure 2B). The deletion extends proximally 238 kb from intron 5 of CHRDL1 and segregated with MGC1 in family 1. The junctional sequence showed microhomology between the proximal and distal breakpoints (Figure 2B). The molecular mechanism for this deletion could be either nonhomologous end-joining or a single fork stalling template switching event.16 CHRDL1 mutation screening in other MGC1 families by exon amplification and direct sequencing identified frameshift, nonsense, missense, and splice-site mutations in CHRDL1 in the remaining five families (Figure 2C). The following mutations were identified: a frameshift mutation in exon 9 (c.872delG [p.Cys291LeufsX25]) in family 2, a missense mutation in exon 9 (c.782G>T [p.Cys261Phe]) in family 3, a splice-site mutation in intron 4 (IVS4+2T>G) in family 4, a frameshift mutation in exon 3 (c.101delAG [p.Glu34AspfsX14]) in family 5, and a nonsense mutation in exon 8 (c.652C>T [p.Arg218X]) in family 6 (Figure 2C). Segregation and carrier status were confirmed and none of the sequence changes were detected in 220 control X chromosomes. In addition to the six families described, we also screened a previously reported large MGC1 family of Tasmanian origin (Pedigree 1 in Mackey et al.2). In affected individuals, all CHRDL1 exons failed to amplify, indicating a segmental deletion. The proximal gene RGAG1 was present, and the deletion was found to extend distally to encompass a 5′ alternative noncoding PAK3 (MIM 300142) exon, which is expressed in the fetal brain (dbEST, Figure 2D). Interestingly, mutations in PAK3 cause X-linked mental retardation (MIM 300558).17 Important promoter and enhancer sequences appear to be retained because developmental delay is not a feature of MGC1. We predict that PAK3 protein expression in critical brain regions is unaffected by the deletion. The deletion in the Tasmanian MGC1 family spans a region of 270–600 kb encompassing the entire CHRDL1 gene and does not share breakpoints with family 1 (Figure 2).
Figure 2.
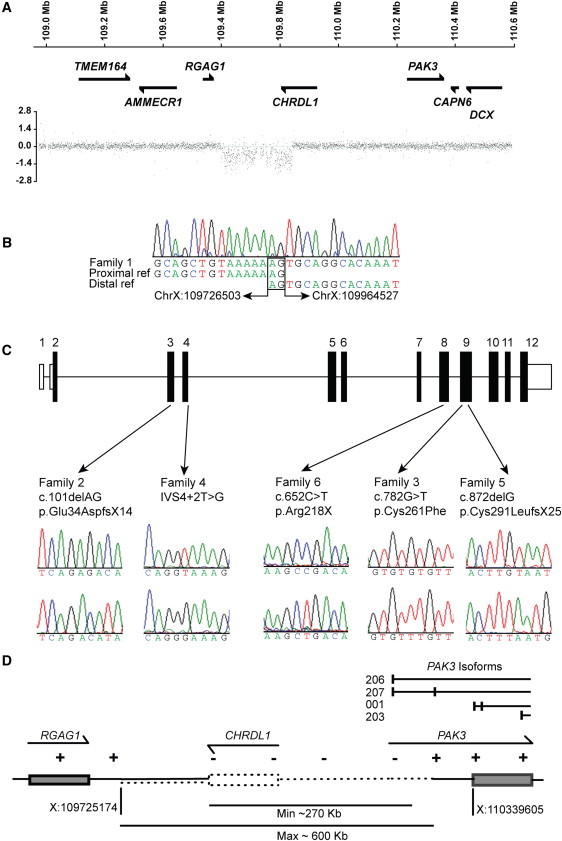
CHRDL1 Mutations Cause X-Linked Megalocornea
(A) X chromosome dense array comparative genomic hybridization for family 1 revealed a deletion within the disease interval of approximately 240 kb that encompassed the 3′ end of CHRDL1. Genes are schematically represented against the position on the X chromosome.
(B) The deletion breakpoint sequence is shown with chromosome X:109726503 joined to sequence chromosome X:109964527 sequence (238,024 bp deleted; hg19 genome build). Microhomology at the deletion breakpoints is highlighted (2 bp; AG).
(C) Schematic of CHRDL1 with patient mutations identified in families 2–6. Nonsense, frameshift, splicing, and missense mutations were identified in exons 3, 4, 8, and 9. Patient sequence electropherograms are shown above control sequence electropherograms.
(D) Schematic of segmental deletion identified in family 7. The deletion encompasses the entire CHRDL1 gene and extends distally to PAK3. The first untranslated exon of PAK3 isoforms 206 and 207 (Ensembl nomenclature hg19 genome build) is also deleted.
We have shown that in all seven families, MGC1 is caused by mutation of the CHRDL1 gene (summarized in Table S1, available online). Segmental deletions, frameshift mutations, and a nonsense mutation are effectively null mutations. The splice-site mutation is predicted to result in premature termination following intron 4 missplicing. The predicted functional consequence of the missense mutation identified in one family (p.Cys261Phe) is discussed below.
Developmental networks of key genes and pathways, many of which are poorly understood, control vertebrate eye morphogenesis. Members of the transforming growth factor β (TGFβ) superfamily of secreted proteins, including bone morphogenic proteins (BMPs), are known to be important in ocular development.18 For example, mutations in latent transforming growth factor beta binding protein 2 (LTBP2 [MIM 602091]) cause recessive congenital glaucoma (MIM 613086).19 In heterozygous knockout Tgfβ2 mice, the corneal endothelium fails to differentiate and the anterior chamber never forms.20 Bmp4 heterozygous knockout mice have anterior segment dysgenesis and elevated intraocular pressure, whereas human BMP4 (MIM 112262) mutations cause a range of syndromic anophthalmia/microphthalmia phenotypes (MIM 607932 and 600625).21,22 Binding of BMPs to their receptors on the cell membrane activates the Smad1/5/8 transcription factors.23 BMP gradients are tightly controlled by a network of secreted proteins that determine the concentration of active BMP ligands. These regulators include the chordin family of secreted BMP antagonists. The chordin proteins interact with BMPs via conserved cysteine-rich domains, which are found in extracellular proteins involved in the modulation of BMP signaling (Figure S1, available online).23–25 Chordin-like 1 encodes ventroptin (or neuralin-1, neurogenesin-1), which contains three cysteine-rich domains (von Willebrand factor C domains).26–28 The missense mutation in family 3 (Cys261Phe) disrupts the first cysteine residue of the third cysteine-rich domain and is predicted to abrogate the interaction of ventroptin and BMPs (Figure S1).
Ventroptin was first identified as an antagonist of BMP signaling in developing retina and brain.26–28 Ventroptin acts as an antagonist of Bmp4 and Bmp2 signaling across the anteroposterior and dorsoventral axes in a double-gradient manner to modulate the regional specification of the retina in chick and Xenopus.26,27 In chicks, misexpression studies have shown that ventroptin plays a role in establishing retinal polarity that is important for the topographic expression of axon guidance molecules and the formation of the retinotopic map.26 Our genetic studies establish a previously unidentified requirement for ventroptin in human anterior segment development. We therefore examined the expression pattern of CHRDL1 in the developing human eye to delineate spatiotemporal expression in ocular structures affected by MGC1.
Expression of CHRDL1 in the Developing Human Eye
We detected CHRDL1 expression by RT-PCR in the developing human cornea, lens, and retina as early as week 7 of gestation (Figure 3A). Expression continues into the second trimester and persists into adulthood in the cornea and trabecular meshwork (Figure 3A). In addition to expression within the developing neural retina, RNA in situ hybridization revealed CHRDL1 expression in a distinctive anterior layer of the periocular mesenchyme that contributes to the development of the structure of the anterior segment angle and expression in the corneal stroma and corneal endothelium (Figures 3B–3D, arrowheads). These data suggest that lack of ventroptin function in the anterior neural crest-derived mesenchymal cells of the anterior segment leads to an increase in corneal diameter and deep anterior chamber.
Figure 3.
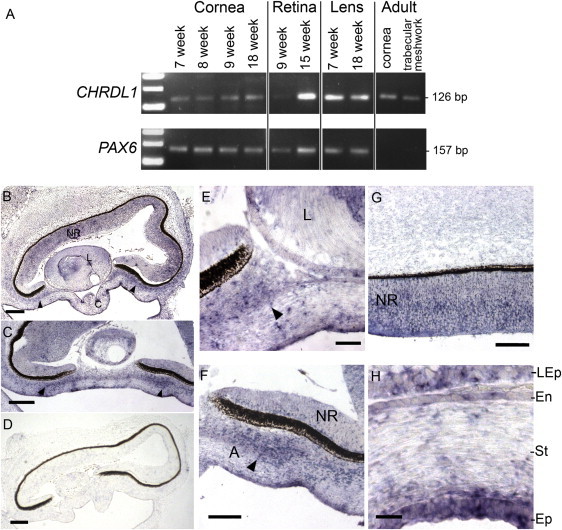
CHRDL1 Is Expressed in the Developing Anterior Segment and Cornea
(A) RT-PCR of human eye tissues showing CHRDL1 expression in the developing cornea from week 7 of gestation continuing into adulthood. CHRDL1 is also expressed in the developing lens and retina and in adult trabecular meshwork. PAX6 (MIM 607108) was used as a control.
(B–H) In situ hybridization of CHRDL1 in the human eye at week 8 of gestation (Carnegie fetal stage F1; sagittal sections) showing expression in the neural retina (NR), and the anterior periocular mesenchyme (arrowheads) of the developing anterior segment angle and cornea. The following abbreviations are used: A, anterior segment angle; C, cornea; L, Lens; En, corneal endothelium; St corneal stroma; Ep, corneal epithelium; LEp, lens epithelium. Sense probe control is shown in (D). The scale bars represent 200 μm (A and D), 100 μm (C), 50 μm (E–G), and 20 μm (H).
Is CHRDL1 a Quantitative Trait Locus for Corneal Diameter?
We speculated that corneal diameter, which is a normally distributed quantitative trait in the general population,13 might be associated with common CHRDL1 variants. We evaluated SNPs within introns of CHRDL1 for association with corneal diameter in two ethnically diverse populations: the Raine cohort (n = 830), which is 88% white, and a Chinese population cohort from Singapore (n = 1345). No significant association with these CHRDL1 polymorphisms was observed (Table S2).
Consequence of Loss of Ventroptin Function on the Retina and Visual System
Given the potential for an associated asymptomatic retinal phenotype, on the basis of retinal expression and the proposed function of ventroptin, we further explored the consequences of the lack of functional ventroptin in MGC1. Detailed clinical examination and retinal and optic nerve head imaging were undertaken in individuals II:1 and II:3 from family 1 and II:1 from family 5. All three MGC1 individuals had normal fundi and optic discs, both clinically and on imaging, and there was no evidence of an increased cup-to-disc ratio, retinal thinning, or coloboma (data not shown). Individual V:2 from family 6 (age 16) had macular drusen (data not shown).
We explored the function of the visual system electrophysiologically. ERGs in individuals II:1 and II:3 from family 1 showed mildly delayed and subnormal cone flicker ERGs in keeping with generalized cone system dysfunction (Figure S2 and Table S3). VEPs were performed to assess the intracranial visual pathway function. VEPs have been widely used to investigate maturational and pathological processes in the visual system. Pattern reversal VEPs for patient II:3 showed increased peak time in the left hemisphere traces from both eyes indicative of interhemispheric asymmetry, and there was a delay in the pattern VEP from the right eye (Figure S2). Pattern reversal VEPs were delayed from both the right and left eyes of II:1, but there was no significant interhemispheric asymmetry in this individual (Figure S2). Analysis of additional patients will be important to determine the potential association of a visual pathway or cone system phenotype with CHRDL1 mutations.
Impact of Loss of Functional Ventroptin on Brain Function and Morphology
In addition to the previously described role in retinotectal patterning, ventroptin has a proposed role as a neurogenic cue in the developing and adult rat brain and in mouse embryonic cortical and neuronal differentiation, which require active BMP signaling for neurogenesis.28,29 Although the presence of an interhemispheric VEP asymmetry does not necessarily indicate a direct relationship with the mutation, given those rodent data, the interhemispheric asymmetry in one MGC1 patient and the observation that mutations in BMP4 result in cognitive impairment, developmental delay, and structural cerebral anomalies,30 we hypothesized that loss of ventroptin function might impact brain function and morphology.
We first examined pre-existing exon expression array data for 14 brain regions in four human fetal specimens (18–23 weeks' gestation) for differential brain expression.9 From this raw dataset, we extracted data for 13 probe sets for CHRDL1 exons.9 CHRDL1 was expressed in all brain regions tested and exhibited regional differential expression; there was high expression in fetal cerebellum, frontal, prefrontal, and occipital neocortex (Figure 4A). Compared to levels of CHRDL1, lower levels of BMP4 were expressed in all regions tested, and there was no evidence of differential expression (Figure S3).
Figure 4.
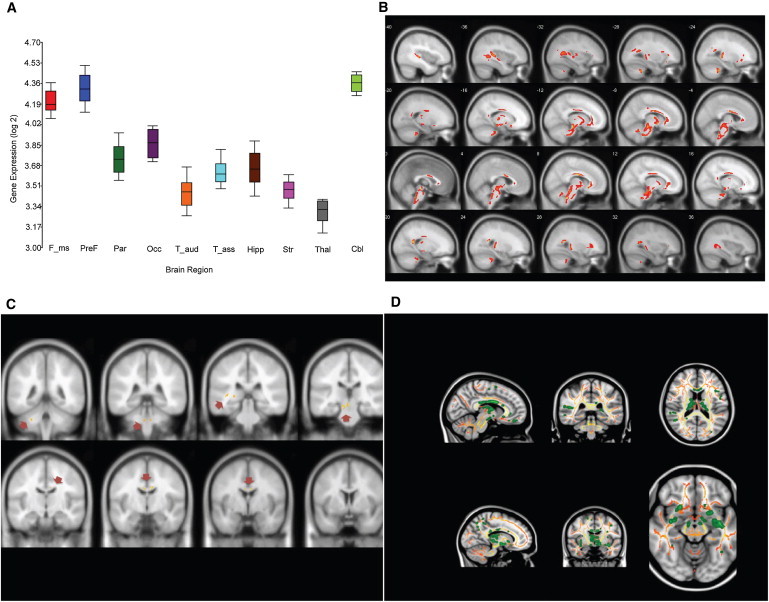
Lack of Ventroptin Function Results in Loss of White Matter or Tract Integrity in Specific Brain Regions
(A) CHRDL1 is differentially expressed in fetal brain regions. Pooled data for four fetal human brains (18, 19, 21, and 23 weeks of gestation) and 13 probes covering CHRDL1 exons are shown for specific brain regions (x axis) on a Log2 scale (y axis). CHRDL1 is expressed in all brain regions tested, with highest expression in cerebellum and neocortex. The following abbreviations are used: F_ms, motor somatosensory neocortex; PreF, prefrontal cortex (orbital dorsolateral, medial and ventrolateral prefrontal neocortex); Par, parietal association neocortex; Occ, occipital visual neocortex; T_aud, temporal auditory neocortex; T_ass, temporal association neocortex; Hipp, hippocampus; Str, striatum; Thal, mediodorsal thalamus; Cbl, cerebellum. Box plots represent median and 25th–75th percentiles. Upper and lower lines show minimum and maximum values, respectively.
(B and C) White matter volume is reduced in patients lacking ventroptin function. (B) voxel-based morphometry showing focal white matter volume reductions (color code red, false discovery rate, p < 0.05, cluster size ≥ 100) and (C) white matter volume reductions (color coded yellow and highlighted with arrows) after family-wise error correction (FWE, p = 0.05, T > 7.4).
(D) Significant fractional anisotropy (FA) decrease in specific brain regions. Tract-based spatial statistics were superimposed on the MNI152 template. Significant FA decrease was observed compared to values in healthy controls. The color scaled red to yellow shows the group skeleton (without difference in FA value); the color green indicates skeletal voxels with a significant FA reduction of at least p = 0.05 (noncorrected).
Neuropsychological tests for intellectual level and executive and memory function were performed for family 1 individuals II:1 (age 71) and II:3 (age 66). No distinctive pattern of cognitive deficit was noted. The verbal IQ was above average with quotients of the 75th (high average ability) and 99th centile (superior ability). MGC1 patients also showed superior verbal memory and executive skills (Table S4). We note that in a clinical MGC1 study published in 1942, the cognitive ability of MGC1 was highlighted: the proband is described as a quick-witted cockney workman and his affected grandson had won a scholarship31. Cognitive ability is therefore preserved in the absence of functional ventroptin.
Next, we explored the impact of loss of ventroptin function on WM anatomy. We compared WM volume of 26 healthy volunteers with the volume of individuals II:1 and II:3 from family 1. VBM showed loss of WM (p = 0.01, false discovery rate correction, cluster = 100 voxels) in the body and genus of the corpus callosum and symmetrically in the internal capsule, inferior fronto-occipital fasciculus, inferior longitudinal fasciculus, dorsal and ventral mesencephalon, and ventral portions of the pons and cerebellum (Figure 4B). After more conservative correction for multiple testing (p < 0.05, familial-wise error correction), clusters in the body of the corpus callosum, the dorsal portion of the mesencephalon, and the left middle cerebellar peduncle remained significant (Figure 4C). MR tractography provides a measure of WM tract integrity. FA is a measure that reflects microstructural properties of the WM, including axon diameter, myelination, and fiber density.32 Group comparison of FA values with tract-based spatial statistics revealed lower FA values in MGC1 patients compared to the values in controls (p < 0.05, noncorrected) in the same WM regions that were found to have significantly reduced WM volume by the VBM analyses (Figure 4D). No regions with higher FA were found in subjects compared to values in controls.
Discussion
Regulation of BMP signaling is important for both patterning and control of organ size during embryogenesis and is tightly controlled by extracellular and intracellular mechanisms. BMPs can form homo- or heterodimers, which are thought to facilitate variable interactions with receptors and activation of different signaling pathways. Both types of BMP receptors (type I and type II) are required to form a functional heteromeric complex and initiate downstream signaling.33 Receptor-BMP ligand binding activates either the canonical BMP signaling pathway (activation of SMAD transcription factors) or the noncanonical BMP signaling pathway (MAPK pathway). BMP signaling is regulated in part by the spatiotemporal expression gradients of BMPs and their receptors but also through the spatiotemporal expression of extracellular BMP antagonists with variable binding affinities for BMPs. BMP signaling can also be modulated by intracellular mechanisms (e.g., inhibitory SMADs).33 The BMP signaling pathway is further complicated by crosstalk with Wnt and Sonic hedgehog signaling during development.33
Reduced BMP signaling caused by mutations in BMP4 is the mechanism described as underlying human syndromic anophthalmia/microphthalmia phenotypes.22,30 In contrast to the reduced ocular organ size observed in these conditions, lack of functional extracellular BMP antagonist ventroptin described here causes an increase in anterior segment size. Although the precise mechanism remains to be determined, we hypothesize that in the absence of functional ventroptin, the tightly regulated gradient of BMPs is perturbed, leading to dysregulation of BMP signaling (via BMP4 and/or other BMPs) and subsequent misexpression of downstream genes. This could result from either BMP overactivity caused by loss of the inhibitory function of ventroptin or the loss of the restricted BMP expression gradient in specific ocular tissues during development.
This hypothesis is supported by a study describing a chordin knockout mouse.34 Chordin is a related BMP antagonist expressed in many tissues throughout development. Interestingly, surviving chordin-null mice were found to have a significant increase in presynaptic transmitter release from hippocampal neurons and altered cognitive behavior including a significantly enhanced learning rate in Morris water maze tests. These data support the hypothesis that BMP signaling regulates synaptic function in the presynaptic terminal. Perfusion of BMP ligands onto hippocampal slices replicated the presynaptic phenotype of the chordin-null mouse and the authors suggest that enhanced BMP signaling in the absence of chordin is the mechanism that results in enhanced short-term plasticity and spatiotemporal processing.34
Intriguingly, these data in chordin knockout mice appear to correlate with the observed high performance in tests of memory in our two MGC1 subjects, despite a striking focal loss of WM. This suggests that chordin and ventroptin have functionally similar effects on BMP signaling; chordin primarily exerts its' inhibitory effects on the hippocampus, and ventroptin on the hippocampus and prefrontal brain regions. None of our MGC1 cases has developmental or cognitive problems, which also suggests that loss of WM and tract integrity does not compromise brain function. Studies of cerebral WM development in human neonates and infants in vivo generally show an increase in the degree of anisotropy with maturation.32 These diffusion changes are linked to a concomitant increase in myelination, reduction in brain water, greater cohesiveness and compactness of fiber tracts, and reduced extra-axonal space (i.e., greater packing) as WM matures over time.35 Changes in diffusion anisotropy in MGC1 might be caused by loss of BMP inhibition by ventroptin, and therefore loss of oligodendrocytic differentiation and myelination; disorganization or maldevelopment of the fiber tracts could be another explanation for the observed lower FA in our MGC1 patients. Several studies have shown that overexpression of BMPs inhibits oligodendrocyte development and myelin protein expression, which suggests an inhibitory effect of enhanced BMP signaling.36,37,38 This supports the idea that the changes in diffusion anisotropy in MGC1 patients are caused by loss of oligodendrocytic differentiation and myelination as a result of enhanced BMP signaling in the absence of functional ventroptin.
Overexpression of Chrdl1 results in altered projection of retinal axons to the tectum along the anteroposterior and dorsoventral axes in chick.25 Our findings in humans also suggest that anatomical connectivity of the visual system might be altered, especially in the mesencephalon. Although CHRDL1 appears widely expressed in the developing human brain, the consequences of mutation for the brain are both spatially and functionally limited. Redundancy and compensatory changes might underlie this resilience, but the full phenotype of CHRDL1 mutation likely extends beyond the eye. Our current extraocular data are limited to two related MGC1 patients, such that the complex anatomical and neurosychological features could be associated with other common alleles in essential genes expressed in the brain. It will be important to recruit new patients with this rare condition for similar studies to definitively link these observed phenotypes with mutations in the CHRDL1 gene.
We show that MGC1 is genetically homogeneous, as all cases studied are caused by mutation of the CHRDL1 gene. We also demonstrate that CHRDL1 is expressed in the developing human periocular mesenchyme, corneal stroma, and corneal endothelium, delineating MGC1 as a neural crest-derived anterior segment disorder. Because CHRDL1 is expressed in the developing human retina and ventroptin has a proposed role in retinotectal patterning, we explored the potential for an asymptomatic retinal or visual pathway phenotype in MGC1 patients. Intriguingly, mild cone photoreceptor system dysfunction was observed in two related subjects and suggestive occipital interhemispheric asymmetry in one subject. Although other affected individuals need to be tested before conclusions can be drawn, these results might represent asymptomatic MGC1 phenotypes associated with lack of ventroptin function caused by a similar mechanism of enhanced and aberrant spatiotemporal BMP signaling interfering with tightly regulated neurodevelopment or synaptic functional maturation.
Acknowledgments
This research was funded by the Department of Health through the National Institute for Health Research (NIHR) award to Moorfields Eye Hospital National Health Service Foundation Trust and UCL Institute of Ophthalmology for a Specialist Biomedical Research Centre for Ophthalmology (BMRC) and by Moorfields Special Trustees. We were supported in part by Fight for Sight and Retintis Pigmentosa Fighting Blindness. We are grateful to the Big Lottery Fund, Wolfson Trust, and the National Society for Epilepsy for supporting the National Society for Epilepsy Magnetic Resonance Imaging unit scanner. This research was also supported by funding from the NIHR BMRC to Great Ormond Street Hospital for Children/UCL Institute of Child Health and University College London Hospitals/UCL. We are grateful to all families for their participation in this study, and we thank Beverly Scott for technical assistance. J.C.S. and D.K. are funded by Great Ormond Street Hospital Children's Charity and the Ulverscroft Foundation. The authors are grateful to the Raine Study participants and the research staff. Funds for the Raine Eye Health Study were provided by the University of Western Australia, Australian Foundation for the Prevention of Blindness and National Health and Medical Research Council. Funding for Core Management of the Raine Study was provided by The University of Western Australia (UWA), Raine Medical Research Foundation, UWA Faculty of Medicine, Dentistry and Health Sciences, The Telethon Institute for Child Health Research and Women and Infants Research Foundation. The authors gratefully acknowledge the assistance of the Western Australian DNA Bank and the support of the National Health and Medical Research Council of Australia and the Canadian Institutes of Health Research.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FMRIB software, http://www.fmrib.ox.ac.uk/fsl/tbss
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Statistical Parametric Mapping, http://www.fil.ion.ucl.ac.uk/spm
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Idrees F., Vaideanu D., Fraser S.G., Sowden J.C., Khaw P.T. A review of anterior segment dysgeneses. Surv. Ophthalmol. 2006;51:213–231. doi: 10.1016/j.survophthal.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Mackey D.A., Buttery R.G., Wise G.M., Denton M.J. Description of X-linked megalocornea with identification of the gene locus. Arch. Ophthalmol. 1991;109:829–833. doi: 10.1001/archopht.1991.01080060093033. [DOI] [PubMed] [Google Scholar]
- 3.Meire F.M., Delleman J.W. Biometry in X linked megalocornea: Pathognomonic findings. Br. J. Ophthalmol. 1994;78:781–785. doi: 10.1136/bjo.78.10.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skuta G.L., Sugar J., Ericson E.S. Corneal endothelial cell measurements in megalocornea. Arch. Ophthalmol. 1983;101:51–53. doi: 10.1001/archopht.1983.01040010053007. [DOI] [PubMed] [Google Scholar]
- 5.Chen J.D., Mackey D., Fuller H., Serravalle S., Olsson J., Denton M.J. X-linked megalocornea: Close linkage to DXS87 and DXS94. Hum. Genet. 1989;83:292–294. doi: 10.1007/BF00285176. [DOI] [PubMed] [Google Scholar]
- 6.Meire F.M., Bleeker-Wagemakers E.M., Oehler M., Gal A., Delleman J.W. X-linked megalocornea. Ocular findings and linkage analysis. Ophthalmic Paediatr. Genet. 1991;12:153–157. doi: 10.3109/13816819109029398. [DOI] [PubMed] [Google Scholar]
- 7.Marmor M.F., Fulton A.B., Holder G.E., Miyake Y., Brigell M., Bach M., International Society for Clinical Electrophysiology of Vision ISCEV Standard for full-field clinical electroretinography (2008 update) Doc. Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 8.Odom J.V., Bach M., Brigell M., Holder G.E., McCulloch D.L., Tormene A.P., Vaegan ISCEV standard for clinical visual evoked potentials (2009 update) Doc. Ophthalmol. 2010;120:111–119. doi: 10.1007/s10633-009-9195-4. [DOI] [PubMed] [Google Scholar]
- 9.Johnson M.B., Kawasawa Y.I., Mason C.E., Krsnik Z., Coppola G., Bogdanović D., Geschwind D.H., Mane S.M., State M.W., Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sisodiya S.M., Thompson P.J., Need A., Harris S.E., Weale M.E., Wilkie S.E., Michaelides M., Free S.L., Walley N., Gumbs C. Genetic enhancement of cognition in a kindred with cone-rod dystrophy due to RIMS1 mutation. J. Med. Genet. 2007;44:373–380. doi: 10.1136/jmg.2006.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook P.A., Symms M., Boulby P.A., Alexander D.C. Optimal acquisition orders of diffusion-weighted MRI measurements. J. Magn. Reson. Imaging. 2007;25:1051–1058. doi: 10.1002/jmri.20905. [DOI] [PubMed] [Google Scholar]
- 12.Smith S.M., Johansen-Berg H., Jenkinson M., Rueckert D., Nichols T.E., Miller K.L., Robson M.D., Jones D.K., Klein J.C., Bartsch A.J., Behrens T.E. Acquisition and voxelwise analysis of multi-subject diffusion data with tract-based spatial statistics. Nat. Protoc. 2007;2:499–503. doi: 10.1038/nprot.2007.45. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann P.C., Hütz W.W. Analysis of biometry and prevalence data for corneal astigmatism in 23,239 eyes. J. Cataract Refract. Surg. 2010;36:1479–1485. doi: 10.1016/j.jcrs.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 14.Foster P.J., Broadway D.C., Hayat S., Luben R., Dalzell N., Bingham S., Wareham N.J., Khaw K.T. Refractive error, axial length and anterior chamber depth of the eye in British adults: the EPIC-Norfolk Eye Study. Br. J. Ophthalmol. 2010;94:827–830. doi: 10.1136/bjo.2009.163899. [DOI] [PubMed] [Google Scholar]
- 15.Doughty M.J., Zaman M.L. Human corneal thickness and its impact on intraocular pressure measures: A review and meta-analysis approach. Surv. Ophthalmol. 2000;44:367–408. doi: 10.1016/s0039-6257(00)00110-7. [DOI] [PubMed] [Google Scholar]
- 16.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 17.Allen K.M., Gleeson J.G., Bagrodia S., Partington M.W., MacMillan J.C., Cerione R.A., Mulley J.C., Walsh C.A. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 1998;20:25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- 18.Wordinger R.J., Clark A.F. Bone morphogenetic proteins and their receptors in the eye. Exp. Biol. Med. (Maywood) 2007;232:979–992. doi: 10.3181/0510-MR-345. [DOI] [PubMed] [Google Scholar]
- 19.Ali M., McKibbin M., Booth A., Parry D.A., Jain P., Riazuddin S.A., Hejtmancik J.F., Khan S.N., Firasat S., Shires M. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 2009;84:664–671. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saika S., Saika S., Liu C.Y., Azhar M., Sanford L.P., Doetschman T., Gendron R.L., Kao C.W., Kao W.W. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev. Biol. 2001;240:419–432. doi: 10.1006/dbio.2001.0480. [DOI] [PubMed] [Google Scholar]
- 21.Chang B., Smith R.S., Peters M., Savinova O.V., Hawes N.L., Zabaleta A., Nusinowitz S., Martin J.E., Davisson M.L., Cepko C.L. Haploinsufficient Bmp4 ocular phenotypes include anterior segment dysgenesis with elevated intraocular pressure. BMC Genet. 2001;2:18. doi: 10.1186/1471-2156-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reis L.M., Tyler R.C., Schilter K.F., Abdul-Rahman O., Innis J.W., Kozel B.A., Schneider A.S., Bardakjian T.M., Lose E.J., Martin D.M. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 2011;130:495–504. doi: 10.1007/s00439-011-0968-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rider C.C., Mulloy B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 2010;429:1–12. doi: 10.1042/BJ20100305. [DOI] [PubMed] [Google Scholar]
- 24.Larraín J., Bachiller D., Lu B., Agius E., Piccolo S., De Robertis E.M. BMP-binding modules in chordin: A model for signalling regulation in the extracellular space. Development. 2000;127:821–830. doi: 10.1242/dev.127.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abreu J.G., Ketpura N.I., Reversade B., De Robertis E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakuta H., Suzuki R., Takahashi H., Kato A., Shintani T., Iemura Si, Yamamoto T.S., Ueno N., Noda M. Ventroptin: A BMP-4 antagonist expressed in a double-gradient pattern in the retina. Science. 2001;293:111–115. doi: 10.1126/science.1058379. [DOI] [PubMed] [Google Scholar]
- 27.Coffinier C., Tran U., Larraín J., De Robertis E.M. Neuralin-1 is a novel Chordin-related molecule expressed in the mouse neural plate. Mech. Dev. 2001;100:119–122. doi: 10.1016/s0925-4773(00)00507-4. [DOI] [PubMed] [Google Scholar]
- 28.Ueki T., Tanaka M., Yamashita K., Mikawa S., Qiu Z., Maragakis N.J., Hevner R.F., Miura N., Sugimura H., Sato K. A novel secretory factor, Neurogenesin-1, provides neurogenic environmental cues for neural stem cells in the adult hippocampus. J. Neurosci. 2003;23:11732–11740. doi: 10.1523/JNEUROSCI.23-37-11732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaughwin P., Ciesla M., Yang H., Lim B., Brundin P. Stage-specific modulation of cortical neuronal development by Mmu-miR-134. Cereb. Cortex. 2011;21:1857–1869. doi: 10.1093/cercor/bhq262. [DOI] [PubMed] [Google Scholar]
- 30.Bakrania P., Efthymiou M., Klein J.C., Salt A., Bunyan D.J., Wyatt A., Ponting C.P., Martin A., Williams S., Lindley V. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: Overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet. 2008;82:304–319. doi: 10.1016/j.ajhg.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savin L.H. A Note on Two Cases of Megalocornea. Br. J. Ophthalmol. 1942;26:265–271. doi: 10.1136/bjo.26.6.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beaulieu C. The basis of anisotropic water diffusion in the nervous system - a technical review. NMR Biomed. 2002;15:435–455. doi: 10.1002/nbm.782. [DOI] [PubMed] [Google Scholar]
- 33.Walsh D.W., Godson C., Brazil D.P., Martin F. Extracellular BMP-antagonist regulation in development and disease: Tied up in knots. Trends Cell Biol. 2010;20:244–256. doi: 10.1016/j.tcb.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Sun M., Thomas M.J., Herder R., Bofenkamp M.L., Selleck S.B., O'Connor M.B. Presynaptic contributions of chordin to hippocampal plasticity and spatial learning. J. Neurosci. 2007;27:7740–7750. doi: 10.1523/JNEUROSCI.1604-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinstry R.C., Mathur A., Miller J.H., Ozcan A., Snyder A.Z., Schefft G.L., Almli C.R., Shiran S.I., Conturo T.E., Neil J.J. Radial organization of developing preterm human cerebral cortex revealed by non-invasive water diffusion anisotropy MRI. Cereb. Cortex. 2002;12:1237–1243. doi: 10.1093/cercor/12.12.1237. [DOI] [PubMed] [Google Scholar]
- 36.Mekki-Dauriac S., Agius E., Kan P., Cochard P. Bone morphogenetic proteins negatively control oligodendrocyte precursor specification in the chick spinal cord. Development. 2002;129:5117–5130. doi: 10.1242/dev.129.22.5117. [DOI] [PubMed] [Google Scholar]
- 37.Gomes W.A., Mehler M.F., Kessler J.A. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev. Biol. 2003;255:164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- 38.See J.M., Grinspan J.B. Sending mixed signals: bone morphogenetic protein in myelination and demyelination. J. Neuropathol. Exp. Neurol. 2009;68:595–604. doi: 10.1097/NEN.0b013e3181a66ad9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.