

Abstract
Methyllysine histone code readers constitute a new promising group of potential drug targets. For instance, L3MBTL1, a Malignant Brain Tumor (MBT) protein, selectively binds mono- and di-methyllysine epigenetic marks (KMe, KMe2) that eventually results in the negative regulation of multiple genes through the E2F/Rb oncogenic pathway. There is a pressing need in potent and selective small-molecule probes that would enable further target validation and might become therapeutic leads. Such an endeavor would require efficient tools to assess the free energy of protein-ligand binding. However, due to an unparalleled function of the MBT binding pocket (i.e. selective binding to KMe/KMe2) and because of its distinctive structure representing a small aromatic “cage”, an accurate assessment of its binding affinity to a ligand appears to be a challenging task. Here, we report a comparative analysis of computationally affordable affinity predictors applied to a set of seven small-molecule ligands interacting with L3MBTL1. The analysis deals with novel ligands and targets, but applies widespread computational approaches and intuitive comparison metrics that makes this study compatible with and incremental to earlier large scale accounts on the efficiency of affinity predictors. Ultimately, this study has revealed three top performers, far ahead of the other techniques, including two scoring functions, PMF04 and PLP, along with a simulation-based method Molecular Mechanics Poisson-Boltzmann/Surface Area (MM-PB/SA). We discuss why some methods may perform better than others on this target class, the limits of their application, as well as how the efficiency of the most CPU-demanding techniques could be optimized.
Introduction
Epigenetic mechanisms are responsible for the cell differentiation and phenotypic changes under the influence of the environment1. These mechanisms are triggered by a set of post-translational modifications (“epigenetic marks”) of histone proteins and DNA2. Among those modifications, histone methylation plays a key role in transcriptional regulation. Abnormal methylation patterns may lead to various pathologies including cancer1. At the molecular level, the spatial arrangements of methylation marks (Kme), i.e. the Kme histone code, act via recruitment of protein complexes (Kme code “readers”) that mediate transcriptional activation or repression3. Malignant Brain Tumor (MBT) repeats that bind to methyllysine marks on histone tails4 form the smallest and the least studied class of “chromatin readers”. Functionally, these proteins localize to chromatin and regulate transcription by a currently unknown molecular mechanism. For instance, L3MBTL1, a protein that features three MBT domains, recognizes mono- and di-methyllysine marks on H1.4K26, H3K4, H3K9, H3K27 and H4K20 in vitro and associates with Heterochromatin Protein 1 (HP1γ), and the Retinoblastoma protein (Rb)5. It has been demonstrated that the Kme recognition by L3MBTL1 occurs in a histone sequence independent manner and is exclusively mediated by the second MBT domain6. These interactions result in a transcriptionally nonpermissive chromatin structure in vitro and the negative regulation of multiple genes through the E2F/Rb oncogenic pathway7, which makes L3MBTL1 and related MBT-containing proteins a group of attractive therapeutic targets. Hence, there is a pressing need in potent and selective small-molecule probes that would enable further target validation and might become leads for therapeutic optimization. Several recent publications have reported small-molecule MBT antagonists8–10. However, their binding affinities are relatively weak and need further improvement.
Computational approaches can provide guidance for the optimization of the current hits and help to identify novel chemical series11. Moreover, recent successes in structure determination for multiple Kme readers open the avenue for the structure-based design and virtual screening12. Such an endeavor will require efficient tools for the assessment of the free energy of protein-ligand binding. However, an accurate assessment of its binding affinity to a novel ligand may appear to be a challenging task. This challenge is dual: first, the binding pocket is quite small (i.e. just enough to accommodate a lysine side chain),, which means that a substantial portion of ligand might be solvent-exposed13; second, the pocket demonstrates a very intriguing ligand selectivity pattern, i.e. recognizes equally well both mono- and di-methyl marks (Kme1/Kme2), but completely ignores unmodified (Kme0) and trimethylated (Kme3) lysine residues. Recently, we have studied how L3MBTL1 discriminates Kme1/Kme2 from Kme0 and Kme3 by means of Molecular Dynamics and Free Energy Perturbation (FEP) which allowed to predict the relative binding free energies for the studied ligands9. This study demonstrated that the binding free energies can in principle be accurately calculated for this class of proteins. However, when it comes to virtual screening and structure-based design, FEP would not be a viable free-energy prediction option. First, its computational cost is very high, especially when scaled to hundreds of ligand-protein complexes. Second, alchemical transformations between ligands as required by FEP are difficult to automate and can only be applied to relatively similar ligands. Therefore, there is a need for computationally efficient predictors of the ligand-protein binding free energy for the histone Kme readers, which constitute a novel class of potential drug targets.
Here, we report a comparative analysis of computationally affordable free energy predictors applied to a set of seven small-molecule ligands interacting with L3MBTL1. A relatively small number of ligands in the test set is because quite few MBT binders have been published so far. However, these compounds cover a broad range of binding affinities (from >−2.32 to −7.23 kcal/mol) that is an absolute prerequisite for such a benchmark analysis. The evaluated predictors were chosen in order to represent various alternative approaches to free energy calculation. We considered the following the method categories. The first category includes dynamics-based Molecular Mechanics-Poisson Boltzmann/Surface Area (MM-PB/SA), Molecular Mechanics-Generalized Born/Surface Area (MM-GB/SA) and Linear Interaction Energy (LIE). These methods are computationally intensive and allow a throughput of about 10 ligand-protein complexes a day. Another category includes single point versions of MM-PB/SA, MM-GB/SA and LIE which do not involve any conformational sampling and make use of a single conformation, most often obtained from a fast docking algorithm. These techniques allow to process hundreds of ligands a day and are generally used as a post-screening rescoring solution. Finally, we have also decided to give a try to a few scoring functions that are often embedded in docking engines and used for ranking docking poses and virtual hits. Here again, we selected a representative subset of popular algorithms. For instance, Dock614 and Autodock415 are illustrative of the force-field-based approach, PMF16, PMF0417 are knowledge-based, while PLP18, LigScore19, Jain20 and GlideSP21 make use of various empirical-based schemas.
We believe that this study is complementary to earlier large scale analyses involving multiple protein targets and small-molecule ligands such as those performed by Wang et al22 and Warren et al23. To this end, we have made use of widespread free energy predictors with standard parameter settings as well as of intuitive comparison metrics such as Pearson’s R2, Kendall’s τb and Predictive Index (PI).
Materials and Methods
System Setup and Molecular Dynamics Simulation
Molecular system was set up in a similar way as described previously in our paper9 and is only briefly summarized here. The starting protein structure is the second MBT domain (residues 366–424) of L3MBTL1 (PDB entry: 2RJF). The protein was treated with Protein Preparation Wizard module of Maestro21. Initial bound structures were obtained by superimposing them to the X-ray structure of the dimethylated lysine residue in complex with L3MBTL1 (PDB entry: 2RJF), followed by a local minimization. The initial bound structure of compound 7 was obtained from its own X-ray structure (PDB entry: 3P8H)10. All simulation systems were solvated in an orthorhombic TIP3P water box with a minimum of 10 Å between the box boundary and any solute atom.
Molecular dynamic simulations were performed using Desmond molecular dynamics program V2.2 24,25. Particle mesh Ewald (PME) summation26,27 was used to treat long-term interactions. SHAKE algorithm28,29 was used to restrain all bonds involving hydrogen atoms. The system was minimized and then gradually heated to 300 K in 0.5 ns. An additional 0.3 ns simulation at 300 K was performed to further equilibrate the system. Two independent 20 ns NPT production runs were performed for each receptor-ligand complexes. System temperature and pressure were regulated by Langevin thermostat and barostat 30,31. The integration time step was 1 fs, 1 fs, 3 fs for, respectively, the bonded term, the near term (short-range electrostatic and van der Waals interactions) and the far term (long range electrostatic interactions). Structure snapshots were saved every 10 ps. Finally, as required for LIE method, each free-state ligand was also simulated in solution with a similar procedure.
MM-PB/SA and MM-GB/SA
In MM-PB/SA and MM-GB/SA schemes, the free energy of binding is estimated as:
| (1) |
Here the molecular mechanics term ΔEMM is the difference between the energy of the complex and the energies of free-state ligand and receptor in gas phase. ΔGsolv and ΔGSA are the electrostatic solvation energy and surface area energy change upon ligand binding. In MM-PB/SA approach, the polar solvation term is computed by iteratively solving Poisson-Boltzmann (PB) equation, while in MM-GB/SA this term is calculated with the linearized Generalized Born (GB) model. The last term TΔSconf is the correction accounting for the conformational entropy loss of the ligand, computed from either normal node or quasiharmonic analysis. This term is often neglected for structurally similar ligands. The symbol 〈 … 〉 denotes the ensemble average over a series of conformations, normally generated by either Molecular Dynamics (MD) or Monte Carlo (MC) simulations. In this study, a “single-trajectory” approach32 was used. MM-PB/SA and MM-GB/SA calculations were performed using mm_pbsa.pl as implemented in AMBER 1033. MM-PB/SA rescoring was conducted using ZAP34. MM-GB/SA rescoring was computed using Prime MM-GB/SA module21.
Linear Interaction Energy (LIE)
LIE method estimates the binding free energy based on a linear electrostatic response approximation and an empirical term accounting for non-polar interactions 35.
| (2) |
Here ‘l-s’ refers to the interaction energy between the ligand and its surroundings. The two energy terms were computed from MD trajectories in explicit solvent. α and β are two adjustable scaling factors, while γ is a constant close to zero. The actual values of α and β vary in different publications and depend on the modeling system36,37. The values recommended by the authors of the approach are α ≈ 0.18 and β in the range from 1/3 to 1/237. In the current study, a “vrun” simulation was performed in Desmond in order to extract the interaction energy between ligand and its surroundings. Similarly to MM-GB/SA, LIE was later extended to a simplified rescoring process by incorporating a continuum solvent model38 and by utilizing a single energy minimization39. Here we used the Surface Generalized Born (SGB)-LIE rescoring model as implemented in Liaison21. In this model, the electrostatic contribution for the net free energy is computed by the sum of Coulomb interaction energy (Ecoul) and the SGB-solvent reaction field energy (Erxn).
Statistical criteria
The predictive power of evaluated methods was assessed by three complementary statistical criteria: 1) Pearson’s R2, 2) Kendall’s τb and 3) the Predictive Index (PI). Kendall’s τb is a rank-order correlation coefficient and is defined as:
| (3) |
where nc is the number of concordant pairs of ranks; nd is the number of discordant pairs of ranks. t and u are the number of tied values in experimental ΔG and predicted ΔG, respectively.
Predictive index (PI) is a different indicator of rank ordering first described by Pearlman and Charifson40. It is defined as:
| (4) |
where
| (5) |
Here Ei is the experimental value of ligand i and Pi is the predicted value. The value of PI ranges from −1 to 1, where 1 means a perfect rank prediction and −1 means a completely wrong prediction. It is a more sophisticated ranking measure than the Kendall’s τb because it also includes a weighting term that depends on the differences between the consecutive experimental values.
Results and Discussion
Compound set and experimental data
We selected a total of seven compounds (Table 1) in order to test affordable predictors of binding free energy to the aromatic cage of L3MBTL1, a representative of methyllysine reader proteins. Compounds 1 to 4 were designed to mimic four endogenous methylation states, i.e. N, NMe, NMe2 and NMe3. Compounds 5 (N(Me)Et) and 6 (Pyrrolidine) are two exogenous alkylated lysine analogs, which bind equally well or better than compounds 2 and 3. Further optimization of compound 6 led to compound 7, which contains a restrained linker10. Remarkably, despite quite subtle structural differences, these compounds showed a broad range of binding affinity from >20000 to 5 μM (see Table 1). Five compounds, with exception of compounds 1 (N) and 4 (NMe3), exhibited a dose-dependent interaction with L3MBTL1 as measured by Isothermal Titration Calorimetry (ITC). The binding affinities were in the order of 7 > 6 (Pyrrolidine) > 3 (NMe2) ≈ 5 (NMe(Et)) ≈ 2 (NMe) ≫ 4 (NMe3) ≈ 1 (N).
Table 1.
Structures of seven designed biophysical probes and their experimental binding affinities to L3MBTL1
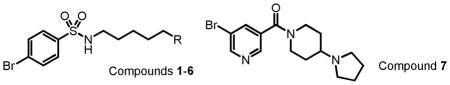 | ||||
|---|---|---|---|---|
| N | Name | R | Kd (μM) | ΔGExp |
| 1 | UNC587 | N | >20000* | >−2.32* |
| 2 | UNC588 | NMe | 104 ± 10 | −5.43 |
| 3 | UNC589 | NMe2 | 73 ± 19 | −5.64 |
| 4 | UNC590 | NMe3 | >10000* | >−2.73* |
| 5 | UNC592 | N(Me)Et | 91 ± 11 | −5.51 |
| 6 | UNC591 | Pyrrolidine | 21 ± 2 | −6.38 |
| 7 | UNC669 | 5 ± 1 | −7.23 | |
Dissociation constants Kd are measured by ITC and are the average of two independent measurements9, 10. The ITC experiments were performed at 25°C. Free energy values (ΔGExp) are expressed in kcal/mol.
: Neither of compounds 1 and 4 showed any binding interaction in ITC experiment. Hence, their Kd values are estimated from the experimental detection limit. The relative binding affinities of 1 and 4 were extrapolated from data by Li et al6.
Free energy predictions
Computational results of our comparative analysis are summarized in Table 2. The results are arranged in three blocks. In the first block, we considered the most computationally intensive, dynamics-based techniques, i.e. PB/SA, GB/SA and LIE. Here, FEP results obtained from our last study are also listed in this block for comparison purposes. The second block includes single-point versions of the first-block methods that do not require dynamics simulations. Finally, the third block comprises 10 popular scoring functions employed by popular virtual screening software. Three popular metrics have been chosen to express the performance of each method: (i) coefficient of determination or Pearson’s R2 is a fraction of variance of experimental data that is explained by predicted data; (ii) Kendall’s τb reflects the similarity of the orderings ranked respectively by experimental and predicted data; and (iii) predictive index (PI) is a hybrid metrics that quantify both similarities in rank orderings and the gaps between consecutive experimental values. Each of the three metrics can be used to rank the predictors from best to worst. As it can be seen from Table 2, all three ranks converge significantly for three affinity prediction methods – PMF04, PLP and MM-PB/SA – that can therefore be regarded as top performers in this study. More generally, we can reasonably assume that all three assessment metrics are important and hence if any of three coefficients rank a method as a poor performer, this method should tend toward the bottom of the overall ranking. Following the above logic, and for the convenience of the data interpretation, Table 2 provides individual ranks for each affinity prediction method according to each of three assessment techniques, as well as a consensual performance measure expressed as an average of the three individual ranks. In the next sections, we propose a detailed comparative analysis of the results obtained and computational protocols used.
Table 2.
The summary of computational results for all tested free energy predictors.
| Individual ΔGbind predictions for compounds 1–7 | Overall method performance | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method category | Method | 1 | 2 | 3 | 4 | 5 | 6 | 7 | R2 (rank) | τb (rank) | PI (rank) | 〈Rank〉 |
| Experiment (kcal/mol) | ITC | >−2.32 | −5.43 | −5.64 | >−2.73 | −5.51 | −6.38 | −7.23 | 1.00 | 1.00 | 1.00 | |
| Simulation-based Methods (kcal/mol) | FEP* | 4.73 | 0.00 | −0.30 | 1.81 | −0.65 | −1.82 | N/A | 0.88 | 0.87 | 0.99 | |
| PB/SA | −21.69 | −27.50 | −27.40 | −23.78 | −26.79 | −31.98 | −30.60 | 0.88 (2) | 0.68 (5) | 0.95 (3) | 3.3 | |
| GB/SA | −25.62 | −31.77 | −28.77 | −27.97 | −29.71 | −32.69 | −31.26 | 0.71 (5) | 0.49 (11) | 0.86 (6) | 7.3 | |
| LIE | −6.54 | −1.85 | −3.57 | −3.85 | −7.74 | −8.38 | −9.78 | 0.16 (18) | 0.43 (16) | 0.42 (18) | 17.3 | |
| LIE fitted | −4.16 | −4.46 | −3.83 | −5.03 | −4.79 | −5.96 | −6.52 | 0.30 (16) | 0.43 (15) | 0.45 (17) | 16 | |
| Single-point Rescoring (kcal/mol) | PB/SA | −35.39 | −41.67 | −43.85 | −38.94 | −46.64 | −43.29 | −43.83 | 0.69 (6) | 0.49 (12) | 0.77 (10) | 9.3 |
| GB/SA | −37.14 | −44.42 | −42.17 | −43.06 | −41.69 | −51.72 | −43.45 | 0.34 (14) | 0.39 (17) | 0.59 (13) | 14.7 | |
| LIE | −12.21 | −14.89 | −18.39 | −16.59 | −17.35 | −18.32 | −16.12 | 0.34 (13) | 0.43 (13) | 0.47 (15) | 13.7 | |
| LIE fitted | −3.17 | −4.58 | −5.64 | −4.14 | −5.21 | −7.24 | −4.78 | 0.49 (9) | 0.71 (4) | 0.83 (8) | 7 | |
| Scoring functions | GlideSP | −5.07 | −5.20 | −5.28 | −4.46 | −5.42 | −6.62 | −5.34 | 0.39 (12) | 0.62 (8) | 0.87 (4) | 8 |
| GlideXP | −7.63 | −8.19 | −7.56 | −6.38 | −8.72 | −8.24 | −8.05 | 0.42 (11) | 0.24 (18) | 0.59 (14) | 14.3 | |
| LigScore | −2.15 | −1.96 | −2.73 | −1.33 | −3.04 | −3.26 | −2.17 | 0.31 (15) | 0.43 (14) | 0.66 (12) | 13.7 | |
| PLP | −51.89 | −55.31 | −65.23 | −45.94 | −66.66 | −67.76 | −69.71 | 0.80 (3) | 0.81 (2) | 0.98 (2) | 2.3 | |
| Jain | −2.22 | −1.46 | −3.52 | −2.56 | −5.64 | −5.03 | −6.62 | 0.49 (10) | 0.62 (7) | 0.69 (11) | 9.3 | |
| PMF | −67.68 | −72.10 | −83.17 | −74.39 | −86.01 | −77.01 | −96.27 | 0.56 (8) | 0.62 (6) | 0.80 (9) | 7.7 | |
| PMF04 | −46.72 | −56.45 | −59.32 | −49.27 | −57.27 | −59.97 | −67.43 | 0.95 (1) | 1.00 (1) | 1.00 (1) | 1 | |
| Autodock4 | −3.38 | −3.95 | −4.56 | −4.00 | −4.99 | −4.76 | −5.22 | 0.74 (4) | 0.71 (3) | 0.83 (7) | 4.7 | |
| Dock6 | −33.38 | −36.35 | −36.34 | −30.04 | −40.06 | −42.20 | −37.50 | 0.62 (7) | 0.52 (10) | 0.86 (5) | 7.3 | |
| Contact | −115 | −123 | −143 | −147 | −132 | −145 | −156 | 0.28 (17) | 0.62 (9) | 0.45 (16) | 14 | |
: For FEP method, computed binding free energies are given relative to the binding free energy of compound 2, which were obtained in our previous study9. The performance of FEP was evaluated using compounds 1–6 only.
MM-PB/SA outperforms MM-GB/SA
All dynamics-based predictions including MM-PB/SA and MM-GB/SA were obtained by processing snapshots from two independent 20 ns MD simulations. The gas phase interaction term ΔEMM, polar solvation term ΔGsolv approximated by PB and GB equations, as well as the non-polar solvation term ΔGSA are summarized in Supplemental Table 1. The binding energies predicted by MM-PB/SA are in a good agreement with experimental data with an R2 value of 0.88 (Table 2 and Figure 1). It also effectively discriminates among the three apparent groups of weak (1, 4), moderate (2, 3, 5) and stronger (6, 7) binders (as can be seen from Figure 1). The only minor inaccuracy is an inversion of the predicted rank orders for compounds 6 and 7. However, on the energetic scale, MM-PB/SA has predicted quite unrealistic figures. For example, the predicted affinities of the compounds 2, 3, 5, 6 and 7 lie in the range of ~5.2 kcal/mol (from −31.98 to −26.79 kcal/mol), which is nearly 3.5-fold as compared to the experimentally determined range of ~1.5 kcal/mol (from −5.59 to −7.06 kcal/mol). This large energetic range was also observed when MM-PB/SA is applied in several other protein systems41,42. Hence, unlike FEP, MM-PB/SA can only be considered as a ranking tool rather than a true free energy predictor in the case of the KMe reader L3MBTL1. MM-PB/SA is ranked 2nd according to R2 (0.88), 5th according to τb (0.68) and 3rd according to PI (0.95) that, with an average rank of 3.3, places it third in the consensus ranking.
Figure 1.
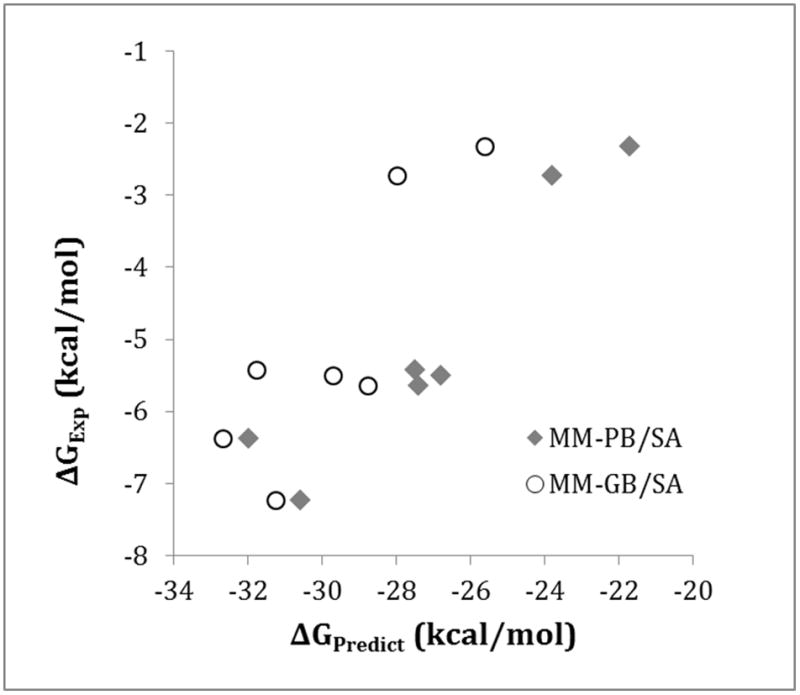
Scatter plots of predicted MM-PB/SA (diamonds) and MM-GB/SA (circles) binding free energies versus experimental data.
The dynamics-based MM-GB/SA method has shown a significantly lower performance than MM-PB/SA as instantiated by all three correlation coefficients. Moreover, MM-GB/SA was not able to discriminate between the weak, moderate and strong binders. For example, despite a large predicted energetic scale, the computed free energy difference between a moderate binder 3 (−28.7 kcal/mol) and a non-binder 4 (−28.0 kcal/mol) was just 0.7 kcal/mol. The only bright spot in the poor outcome of MM-GB/SA predictions was a robust discrimination between the weakest and the strongest binders. It should be noted that the mediocre performance of MM-GB/SA is, to some extent, in contrast with the results of a recent study by Hou et al43. In their comparative analysis performed on a set of six proteins (structurally different from L3MBTL1), MM-GB/SA was a slightly better predictor than MM-PB/SA on most protein targets.
MM-PB/SA tolerates short simulation times
We then investigated on how to optimize the computational efficiency of the MM-PB/SA method. First, we made predictions by means of a single-point version of MM-PB/SA, in which only a local energy minimization on docking poses was conducted instead of the MD conformational sampling. In recent years, this method has become a popular rescoring scheme for the docking-based virtual screening and, according to some reports, demonstrated a better predictive power as compared to scoring functions embedded in the docking software44–47. In the current study, the single-point MM-PB/SA was significantly less predictive than its dynamics-based counterpart (Table 2). Therefore, the next step was to determine the optimal simulation time. We made use of our 20 ns simulations in order to calculate MM-PB/SA free energy values at progressively increasing simulation times starting from 0.1 ns. The plot in Figure 2 demonstrates that even relatively short simulations lead to high quality predictions. For instance, the shortest tested simulation results in a best possible compound ranking (PI = 0.95). Additional 3 ns are then necessary to obtain the highest possible R2.
Figure 2.
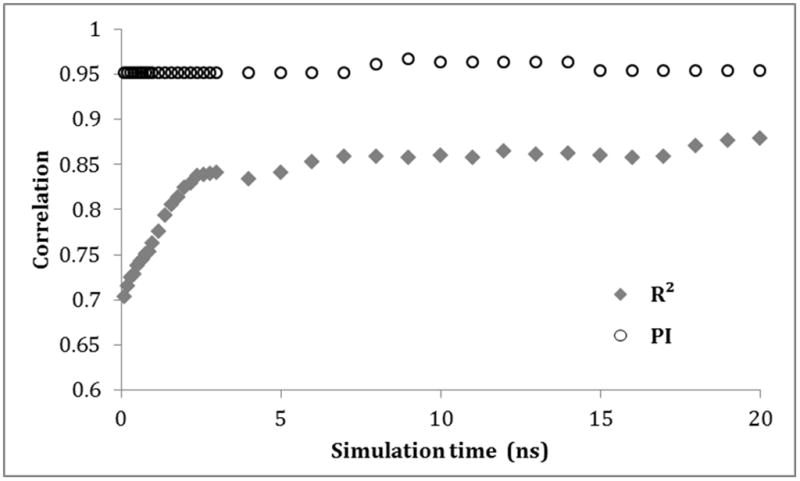
Impact of simulation time on the predictive power of MM-PB/SA represented by R2 (circles) and PI (diamonds).
It is worth to note that a too short simulation may suffer from statistical uncertainty. This is especially important for MM-PB/SA and MM-GB/SA as these calculations sometimes yield a large standard deviation of the estimated free energy48. As pointed out by a recent article48, 1.5 to 15 ns simulation is needed to obtain a statistical precision of 1 kJ/mol (0.24 kcal/mol) for seven complexes of avidin-biotin analogues. This is nearly an order of magnitude longer than the length of most reported MM-PB/SA simulations. In our study, the standard deviations of the binding free energies obtained from alternative snapshot samplings varied from 3 to 5 kcal/mol (Supplemental Table 1), which is quite low compared to a typical reported values of 5 to 35 kcal/mol48. Hence, at the sampling frequency of 10 ps, a 3 ns simulation was sufficient to obtain a standard error of mean (SEM) around 0.25 kcal/mol. Finally, simulation times longer than 3 ns did not result in any increase of the prediction quality in this study.
In all simulations the actual data have been collected after a short equilibration time. For obvious reasons, this equilibration step is especially important for short simulations in order to “smoothen” unfavorable interactions mainly due to soaking the protein-ligand system in the explicit solvent medium. In our setting, a 0.3 ns equilibration was sufficient for a convergent “production” run. However, certain compounds required a longer time. For example, compound 4 needed at least a 1 ns equilibration, mainly because the bulky tri-methyl amino group did not fit the initial receptor structure. However, given that compound 4 is a very weak binder, even a partially equilibrated structure did not dramatically affect the prediction quality.
Deficient performance of LIE
The LIE method was applied in four different settings including the dynamics-based and single-point protocol, each with either predetermined α and β parameters or fitted to our data in order to reach a better correlation. None of these protocols has demonstrated an adequate performance as reflected by all three statistical criteria (Table 2). When using the recommended parameter set (α = 0.18 and β = 0.50), predicted binding affinities correlated very weakly with experimental data (R2 = 0.16, τb = 0.43, PI = 0.42). We have also tried to fit the scaling factors for a better match with experimental binding energies. However, the fitted parameter set (with α = 0.71 and β = 0.17) only led to an insignificant gain in R2 to 0.29. In terms of compound ranking, LIE method can clearly distinguish two strong binders 6 and 7 from the other five compounds. On the other hand, the binding affinity of compounds 1 and 4 was overestimated, partially due to an unexpected large electrostatic term. In addition, LIE incorrectly predicted that there is a large free energy difference among the three weak binders 2, 3 and 5.
Here we used our previous free energy decomposition results9 to analyze the possible deficiencies of LIE terms in this test. Although there is no direct one-to-one correspondence, a qualitative relationship can be anticipated between the polar and non-polar FEP terms and the two energy terms in LIE. For instance, our previous decomposition suggested that compound 2 benefits from more efficient vdW interactions and higher bound-state mobility and that compounds 3, 5 and 6 have a higher electrostatic contribution due to their highly localized positive charge on the hydrogen pointing to Asp355. In LIE, the relative values of the vdW term are clearly correlated with our FEP non-polar contributions (Supplemental Table 2). On the other hand, the electrostatic term is practically uncorrelated to the electrostatic contributions from our FEP study. Moreover, it has already been reported that LIE may have issues in treating charged ligands, particularly their electrostatic terms37,49. A possible solution might involve constant pH simulation or a polarizable force field. Another LIE problem in this study was an excessively high variation of both energy terms of Equation 2 across different MD snapshots. The latter fact is in contrast with a relative stability of the MM-PB/SA energy terms, as well as with a good convergence of our FEP simulations on the same molecular systems9.
In addition to the original LIE scheme, we also examined the single-point LIE method. This scheme was performed on a single minimized structure with implicit SGB solvation. Using the default scaling factors, a very pour correlation is found (R2 = 0.34). After fitting the scaling factors to experimental data, R2 increased to a still low 0.49.
Scoring functions
A total of 10 scoring functions were evaluated in this test, including GlideSP, GlideXP from Schrodinger21; Ligscore19, PLP18, Jain20, PMF16 and PMF0417, as implemented in Discovery Studio; Dock6 grid score and contact score in Dock614; as well as AutoDock4 score15. These 10 scoring functions represent all three major types of energy functions, i.e. force-field-based, knowledge-based and empirical-based. Before scoring each ligand, we performed a local minimization to optimize the initial binding pose. In Discovery Studio, a minimization step by means of the CHARMM force field was performed prior to the scoring step. In Maestro, Dock6 and AutoDock4, we relied upon native energy minimization methods intrinsic to the scoring algorithms.
As can be seen from Table 1, the 10 scoring methods performed quite differently in this study, with R2 ranging from 0.28 to 0.95. Three scoring functions (PLP, PMF04 and Autodock4) demonstrated a very high R2 value (>0.70). There are three scoring functions (GlideSP, LigScore and Contact) where the correlation is almost negligible (R2 < 0.40). In general, the rank-order correlation coefficients τb and PI followed the same trend as that of R2, which ranks both PLP and PMF04 as the top two performers. However, there are a few exceptions: for example, GlideSP has a poor R2 value of 0.39, mainly because of a single outlier (compound 7), while it has correctly ranked the other six molecules that resulted in a PI value of 0.87. Remarkably, the best scorer, PMF04, is also the best overall performer in the current study, ahead of computationally demanding MM-PB/SA. However, a larger number of compounds would be necessary to confirm this finding.
Interestingly, the top scorers represent all three major categories of scoring functions. Piecewise Linear Potential (PLP) is an empirical function that accounts for all atom-atom interactions intermolecular interactions18. Potential of Mean Force 04 (PMF04) is a knowledge-based function derived from statistical analysis of protein ligand complexes16,17. It outperformed its ancestor, PMF, probably due to a larger parameterization data set. Autodock4 is a semi-empirical energy function, which combines force field empirical weights and/or empirical functional forms15. Finally, a force-field-based function, Dock6, performed moderately well in this test (R2 = 0.62). Dock6 score is a grid-based score derived from the non-bonded terms of a molecular mechanics force field14.
Six of the ten scoring functions have correctly predicted compound 7 to have the strongest binding affinity in this set. Among the six functions, only PLP and PMF04 have correctly ranked compound 6 to be the 2nd strongest binder. Another challenge was to correctly rank the binding affinities of moderate binders 2, 3 and 5, whose experimental binding free energies differ only by ~0.2 kcal/mol. Of all scoring functions, only PLP, PMF04 and GildeSP have correctly predicted these three compounds to have comparable bind affinities. On the other hand, most scoring functions were correct in ranking compounds 1 and 4 as the weakest binders. This is likely due to the ability to recognize a weaker electrostatic interaction with Asp355 that was experimentally demonstrated to be critical for ligand binding6.
Interestingly, the average R2 over 10 scoring functions amounts to 0.56 that, despite the novelty of the protein target, is overall superior than what was observed in earlier benchmark studies on more familiar therapeutic targets22,23. For instance, in a test against 800 diverse protein-ligand complexes, the R2 values demonstrated by 14 energy functions ranged from 0.02 to 0.3222. The authors have also evaluated the performance of those scoring functions on three individual targets: HIV-1 protease, trypsin and carbonic anhydrase II and found that most scoring functions failed to correctly rank ligands. The R2 values ranged from 0.04 to 0.41, with an average of 0.2022. Another study that involved eight receptors and ten docking programs resulted in R2 values ranging between 0.01 and 0.1623. The authors concluded that “no single program performed well for all of the targets and none of the docking programs made a useful prediction of the ligand binding affinity”. In our study, the surprisingly good performance of the standard scoring functions may have two complementary explanations: (i) scoring functions are subject to continuous improvements (e.g., superior performance of PMF04 as compared to the original PMF), (ii) the MBT binding pocket is relatively small and forms fewer interactions with the ligand as compared to other proteins that leads to a smaller cumulative error.
Conclusions
In this study, we evaluated how various computational predictors of the ligand-protein binding affinity perform on a series of seven novel small-molecule binders to a methyllysine histone code “reader”, L3MBTL1. As a consensus of the comparison metrics used, the study has revealed three top performers, far ahead of the other techniques. They include two scoring functions, PMF04 (R2 = 0.95; τb = 1.0; PI = 1.0) and PLP (R2 = 0.80; τb = 0.81; PI = 0.98); and a simulation-based method MM-PB/SA (R2 = 0.88; τb = 0.68; PI = 0.95). It should be noted that while PMF04 and PLP show excellent results on L3MBTL1, their performance varies greatly from one protein target to another22,23. Therefore, their general utility for the KMe histone code readers is still to be confirmed when more experimental data become available. Alternatively, MM-PB/SA is a less empirical and more CPU-intensive approach (compared to PMF04 and PLP) with a thorough handling of solvation effects. Therefore, despite being ranked only third on the current data set, it is more likely to demonstrate a comparable predictivity on other ligands and proteins similar to those used in the current analysis. Here, we report a computational protocol that enables an optimal speed-predictivity ratio when applying MM-PB/SA to histone KMe readers.
Supplementary Material
Acknowledgments
This research was supported by the grants RC1-GM090732-01 from the National Institutes of Health and the Innovation Award from the University Cancer Research Fund.
References
- 1.Rodriguez-Paredes M, Esteller M. Nat Med. 2011:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 2.Turner BM. Nature Cell Biol. 2007;9:2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 3.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Cell. 2010;143(3):470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonasio R, Lecona E, Reinberg D. Semin Cell Dev Biol. 2009;21(2):221–230. doi: 10.1016/j.semcdb.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trojer P, Reinberg D. Cell Cycle. 2008;7(5):578–585. doi: 10.4161/cc.7.5.5544. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ. Mol Cell. 2007;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalakonda N, Fischle W, Boccuni P, Gurvich N, Hoya-Arias R, Zhao X, Miyata Y, Macgrogan D, Zhang J, Sims JK, Rice JC, Nimer SD. Oncogene. 2008;27(31):4293–4304. doi: 10.1038/onc.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. J Med Chem. 2010;53(21):7625–7631. doi: 10.1021/jm1007374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao C, Herold JM, Kireev DB, Wigle TJ, Norris JL, Frye SV. J Am Chem Soc. 2011;133(14):5357–5362. doi: 10.1021/ja110432e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, Ingerman LA, Kireev DB, Senisterra G, Vedadi M, Tripathy A, Brown PJ, Arrowsmith CH, Jin J, Janzen WP, Frye SV. J Med Chem. 2011;54(7):2504–2511. doi: 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jhoti H, Leach A. Structure-based Drug Discovery. Springer; 2007. [Google Scholar]
- 12.Adams-Cioaba MA, Min J. Biochemistry and Cell Biology. 2009;87(1):93–105. doi: 10.1139/O08-129. [DOI] [PubMed] [Google Scholar]
- 13.Min J, Allali-Hassani A, Nady N, Qi C, Ouyang H, Liu Y, MacKenzie F, Vedadi M, Arrowsmith CH. Nat Struct Mol Biol. 2007;14(12):1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 14.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. RNA. 2009;15(6):1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huey R, Morris GM, Olson AJ, Goodsell DS. J Comput Chem. 2007;28(6):1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 16.Muegge I, Martin YC. J Med Chem. 1999;42(5):791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 17.Muegge I. J Med Chem. 2005;49(20):5895–5902. doi: 10.1021/jm050038s. [DOI] [PubMed] [Google Scholar]
- 18.Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DR, Fogel LJ, Freer ST. Chem Biol. 1995;2(5):317–324. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 19.Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. J Mol Graphics Model. 2005;23(5):395–407. doi: 10.1016/j.jmgm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Jain AN. J Comput-Aided Mol Des. 1996;10(5):427–440. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 21.Schrödinger LLC. 2010 [Google Scholar]
- 22.Wang R, Lu Y, Fang X, Wang S. J Chem Inf Comp Sci. 2004;44(6):2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 23.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. Journal of Medicinal Chemistry. 2006;49(20):5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 24.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida. 2006. [Google Scholar]
- 25.Shaw DE. Research. New York, NY: 2009. [Google Scholar]
- 26.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577. [Google Scholar]
- 27.Shan Y, Klepeis JL, Eastwood MP, Dror RO, Shaw DE. J Chem Phys. 2005;122:54101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 28.Ryckaert J, Ciccotti G, Berendsen H. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 29.Andersen H. Journal of Computational Physics. 1983;52:24–34. [Google Scholar]
- 30.Pastor R, Brooks B, Szabo A. Mol Phys. 1988;65:1409–1419. [Google Scholar]
- 31.Feller SE, Zhang Y, Pastor RW, Brooks BR. J Chem Phys. 1995;103:4613. [Google Scholar]
- 32.Gohlke H, Case DA. J Comput Chem. 2004;25(2):238–250. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 33.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. University of California; San Francisco: 2008. [Google Scholar]
- 34.Grant JA, Pickup BT, Nicholls A. J Comput Chem. 2001;22(6):608–640. [Google Scholar]
- 35.Åqvist J, Medina C, Samuelsson JE. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 36.Wang W, Wang J, Kollman PA. Proteins. 1999;34:395–402. [PubMed] [Google Scholar]
- 37.Åqvist J, Marelius J. Combinatorial Chem High Throughput Screening. 2001;4(8):613. doi: 10.2174/1386207013330661. [DOI] [PubMed] [Google Scholar]
- 38.Carlsson J, Andér M, Nervall M, Åqvist J. J Phys Chem B. 2006;110:12034–12041. doi: 10.1021/jp056929t. [DOI] [PubMed] [Google Scholar]
- 39.Huang D, Caflisch A. J Med Chem. 2004;47:5791–5797. doi: 10.1021/jm049726m. [DOI] [PubMed] [Google Scholar]
- 40.Charifson PS. J Med Chem. 2001;44(21):3417–3423. doi: 10.1021/jm0100279. [DOI] [PubMed] [Google Scholar]
- 41.Brown SP, Muchmore SW. J Chem Inf Model. 2006;46(3):999–1005. doi: 10.1021/ci050488t. [DOI] [PubMed] [Google Scholar]
- 42.Guimarães CRW, Mathiowetz AM. J Chem Inf Model. 2010;50(4):547–559. doi: 10.1021/ci900497d. [DOI] [PubMed] [Google Scholar]
- 43.Hou T, Wang J, Li Y, Wang W. J Chem Inf Model. 2011;51(1):69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyne PD, Lamb ML, Saeh JC. Journal of Medicinal Chemistry. 2006;49(16):4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- 45.Guimarães CRW, Cardozo M. J Chem Inf Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- 46.Thompson DC, Humblet C, Joseph-McCarthy D. J Chem Inf Model. 2008;48:1081–1091. doi: 10.1021/ci700470c. [DOI] [PubMed] [Google Scholar]
- 47.Kuhn B, Gerber P, Schulz-Gasch T, Stahl M. J Med Chem. 2005;48:4040–4048. doi: 10.1021/jm049081q. [DOI] [PubMed] [Google Scholar]
- 48.Genheden S, Ryde U. J Comput Chem. 2010;31(4):837–846. doi: 10.1002/jcc.21366. [DOI] [PubMed] [Google Scholar]
- 49.Åqvist J. J Comput Chem. 1996;17(14):1587–1597. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.