

Abstract
The E4 allele of the Apolipoprotein E (APOE) gene is the strongest genetic risk factor for late-onset Alzheimer's disease (AD), and affects clinical outcomes of chronic and acute brain damages. The mechanisms by which apoE affect diverse diseases and disorders may involve modulation of the glial response to various types of brain damages. We examined glial activation in a mouse model where each of the human APOE alleles are expressed under the endogenous mouse APOE promoter, as well as in APOE knock-out mice. APOE4 mice displayed increased glial activation in response to intracerebroventricular lipopolysaccharide (LPS) compared to APOE2 and APOE3 mice by several measures. There were higher levels of microglia/macrophage, astrocytes, and invading T-cells after LPS injection in APOE4 mice. APOE4 mice also displayed greater and more prolonged increases of cytokines (IL-1β, IL-6, TNF-α) than APOE2 and APOE3 mice. We found that APOE4 mice had greater synaptic protein loss after LPS injection, as measured by three different markers: PSD-95, Drebin, and synaptophysin. In all assays, APOE knock-out mice responded similar to APOE4 mice, suggesting that the apoE4 protein may lack anti-inflammatory characteristics of apoE2 and apoE3. Together, these findings demonstrate that APOE4 predisposes to inflammation, which could contribute to its association with Alzheimer's disease and other disorders.
Keywords: Alzheimer's disease, microglia, astrocyte, inflammation, cytokine, synaptic protein loss, lipopolysaccharide
INTRODUCTION
The E4 allele of the Apolipoprotein E (APOE) gene is the strongest genetic risk factor for late-onset Alzheimer's disease (AD) (Genin et al. 2011; Strittmatter et al. 1993). The APOE-ε4 allele reduces the age of onset of AD (Corder et al. 1993), such that the great majority of individuals who develop Alzheimer's disease in their 60's are APOE4 positive (Rebeck et al. 1994). APOE genotype also affects recovery from traumatic brain injury, which is partially characterized by amyloid-beta (Aβ) deposits similar to those seen in AD (Friedman et al. 1999; Smith et al. 2006). APOE4 increases amyloid levels by several types of measures in animal models (Kim et al. 2009) and in humans (Castellano et al. 2011; Rebeck et al. 1993; Schmechel et al. 1993), and is found to associate with Aβ in vivo (Namba et al. 1991) and in vitro (LaDu et al. 1994; Strittmatter et al. 1993). These studies are consistent with models where apoE affects Aβ deposition and clearance (Kim et al. 2009; Wisniewski and Frangione 1992).
The very high risk of AD associated with APOE genotype suggests that apoE may affect more than one step in AD pathogenesis. In a study of AD brains, APOE genotype affected microglial activation, controlling for other disease markers (Egensperger et al. 1998). Genetic studies have also identified several risk factors related to glial activation (Jones et al. 2010), specifically CR1 (a receptor for complement), and CD33 (a receptor on myeloid cells) (Hollingworth et al. 2011; Naj et al. 2011). Gliosis is a common feature in neurodegenerative diseases including AD. It is a response to AD-associated pathological changes and may contribute to cell loss after the accumulation of the plaques and tangles (Mrak and Griffin 2005). Reciprocally, chronic gliosis may also occur prior to AD pathogenesis and actually promote the accumulation of plaques and tangles, and thus the disease progression (Herrup 2010). Suppression of inflammatory responses has been shown to reduce AD risk, especially in APOE4 carriers (McGeer and McGeer 2007; Szekely et al. 2008; Yip et al. 2005).
We hypothesized that APOE4 could augment the risk of AD by increasing chronic state of glial activation or by increasing the glial response of the brain to insults throughout life. We tested this latter hypothesis by injecting LPS in mice expressing each of the three human APOE alleles as well as APOE knock-out mice. The human apoE mice express human APOE alleles under the endogenous mouse APOE promoter (Sullivan et al. 1997), and thus are an excellent mouse model for understanding the effect of APOE genotype on normal brain function. Here we report that the APOE4 genotype was associated with increased glial numbers, increased secretion of cytokines, and decreased synaptic markers.
METHODS
Animals
Homozygous human APOE2, APOE3, and APOE4 knock-in (targeted-replacement) mice were used (Sullivan et al. 1997); in these mice, exons 2-4 of the human APOE2, APOE3 and APOE4 genes replaced the corresponding genomic DNA at the mouse APOE locus. These three mice colonies as well as APOE knock-out mice were maintained at Taconic, Inc. (Hudson, NY). Experiments were performed on age-matched male animals at 4 months of age. Animal experiments were conducted in compliance with the rules and regulations of the Institutional Animal Care and Use Committee at the University of Illinois at Chicago.
Intracerebroventricular (ICV) injection of LPS and animal sacrifice
Mice were anesthetized by intraperitoneal injection of 120 mg/kg ketamine (Abbott Laboratories, Chicago, IL) and then received unilateral ICV injection of LPS (Sigma, St. Louis, MO) or vehicle control. Mice were injected with 2.5 μl of 400ng/μl LPS or 2.5 μl of saline at a rate of 0.5 μl/min, using a syringe pump at the following mouse brain coordinates: anterior/posterior =-0.34 mm, medial/lateral= 1.0 mm, dorsal/ventral=-2.0 mm (n=4-5 per treatment group). After each injection, the syringe was left for an additional 2 min to avoid liquid reflux. These mice were anesthetized with 120mg/kg ketamine and euthanized by transcardial perfusion with ice-cold phosphate-buffered saline (1x PBS) containing 1x protease inhibitor cocktail (Calbiochem, Gibbstown, NJ). For immunohistochemistry, the ipisilateral hemisphere was fixed in 4% paraformaldehyde in 1x PBS, pH7.4, for 48 h and then stored in 30% sucrose, 1x PBS solution for 24 h at 4°C. The contralateral hemisphere was immediately dissected on ice to obtain cerebral cortex, hippocampus, and cerebellum that were snap-frozen in liquid nitrogen and stored at -80°C for biochemical analyses. Experiments were conducted on brains 24 or 72 hours after ICV injection of LPS. These times were chosen to represent early and late responses to inflammation (Maezawa et al. 2006b; Ophir et al. 2003).
Immunohistochemistry and stereological analysis
The ipsilateral hemispheres were subsequently cut into 35 μm coronal sections on a Leica SM 2000R microtome, and sections were stored at -20°C in 24-well plates with cryoprotectant (30% glycerol, 30% ethylene glycol, 1x PBS). Every sixth section was immunohistochemically processed for identification of glial cells using a rabbit antibody against Glial Fibrillary Acidic Protein (GFAP) (1:500, Dako, Carpinteria, CA) for astrocytes, and rat anti-F4/80 monoclonal antibody (1:500, Serotec, Raleigh, NC) for microglia/macrophage. Sections were incubated with the primary antibodies at room temperature overnight, washed with TBS-T (25 mM Tris-HCl, 137 mM NaCl, 2.7 mM KCl, pH 7.4, 0.25% Triton X-100), and then incubated at room temperature for 1 h with the corresponding biotinylated goat anti-rabbit and goat anti-rat IgG secondary antibodies. Sections were then incubated in peroxidase-conjugated avidin-biotin complex for 1.5 h. A chromogen solution containing 0.05% 3, 3'-diaminobenzidine and 0.003% H2O2 was used to obtain a brown staining. The total numbers of F4/80-immunoreactive (F4/80-IR) microglia and GFAP-IR astrocytes in the hippocampus were determined using the computerized optical dissector method with Stereo Investigator software (Version 9.03, MBF Bioscience, Williston, VT) with Zeiss Imager A1 microscope. Cells were manually designated by a blinded investigator. The total numbers (N) of IR cells were calculated using the formula N = NV × V, where NV is the numerical density and V is the volume of the hippocampus or frontal cortex. The densities of the F4/80-IR and GFAP-IR cells in the hippocampus ipsilateral to the LPS injection site were determined in three sections per animal, and the average of the counts thus obtained was taken from four to five animals in each group.
Immunofluorescence staining and image analysis
Expression of the T-cell marker CD3 (1:250, Abcam, Cambridge MA), presynaptic marker synaptophysin (1:1000, Chemicon), and postsynaptic markers PSD-95 (1:500, Abcam, Cambridge MA) and drebrin (1:2000, Abcam, Cambridge MA), were evaluated by single immunofluorescence staining. Brain sections (35um) were first blocked by incubation with TBS-T solution containing 5% bovine serum albumin (BSA) for 1 h at room temperature. For PSD-95 staining, the brain sections were pretreated with 100 mg/ml of pepsin (DAKO) at 37°C in a water bath for 5 min prior to blocking (Fukaya and Watanabe 2000). The sections were then incubated with primary antibodies dissolved in TBST solution containing 0.1% Triton X-100 and 2% BSA for 16 h at room temperature. The bound primary antibodies were visualized by incubating the sections for 1 h at room temperature with Alexa 488- or 594-conjugated donkey anti-rabbit IgG (1:1000, Invitrogen). The sections were then mounted on slides, and fluorescence images were captured using a confocal scanning laser microscope (LSM 510; Zeiss, Oberkochen, Germany) with a 40X or 63X oil-immersion lens. Images of CD3, synaptophysin, PSD-95 and drebrin were taken in the CA3 region of the hippocampus or the layer 3-4 of frontal cortex. The numbers of CD3 IR-positive cells in the CA3 region were evaluated by Image J and expressed as numbers per mm2.
Preparation of brain homogenates
The cerebral cortex, hippocampus and cerebellum were homogenized with a polytron homogenizer (Brinkmann Instruments, Rexdale, Ontario, Canada) using 12 rapid pulses in ten volume of ice-cold lysis buffer (150-350 μl, 50mM Tris-HCl, 150mM NaCl, pH7.4, 1% Triton X-100, 1x protease inhibitor cocktail). Homogenates were centrifuged at 14,000 g for 30 min at 4°C and the supernatants were collected for biochemical analyses. Total protein concentration was determined by BCA protein assay kit (Pierce, Rockford, IL).
Determination of cytokine levels
Pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) in the brain homogenates were determined using commercial cytokine ELISA kits following the manufacturer's instructions (R&D, Minneapolis, MN). Briefly, cytokine standards, samples, diluent buffers, and biotinylated anti- IL-1β, IL-6 or TNF-α solutions were pipetted into each well. After 2 h incubation at room temperature, standards and samples were washed and incubated in streptavidin-HRP working solution for 1 h at room temperature. Absorbance was measured at 450 nm using a Molecular Devices microplate reader (Molecular Devices, Sunnydale, CA). The concentration of the IL-1β, IL-6 and TNF-α was determined against a seven-point standard curve. The quantity of IL-1β, IL-6 and TNF-α was expressed as pg/mg total protein.
Immunoblot analysis
For each hippocampal homogenate, 30-50 μg of total protein was separated by 4-12% Bis-Tris gel (Invitrogen). Separated proteins were transferred onto PVDF membranes and analyzed by western blotting. The following primary antibodies from Abcam were used: Rabbit anti-PSD-95 (1:3000), Rabbit anti-synaptophysin (1: 2000), mouse anti-α-tubulin (1:8000), and mouse anti-drebrin antibody (1:1000), respectively. After incubation with the appropriate HRP-conjugated secondary antibody, membranes were developed using ECL-enhanced chemiluminescence (Amersham, Piscataway, NJ). The X-ray film was scanned and the density of bands was quantified using Image J software. The amount of protein was expressed as a relative value to the levels of α-tubulin.
Statistical analysis
Data are expressed as mean ± SD. Statistical analysis was performed using the SPSS 10.0 software package (Graphpad, San Diego, CA). Two-way analyses of variance (ANOVA) were used to analyze interaction of APOE genotype, LPS or saline injection and treatment time. Tukey'spost-hoc analyses were used to detect statistical differences among the groups using P< 0.05 as significance values.
RESULTS
Although apoE is predominantly expressed in glial cells, in some instances of neuronal toxicity, apoE expression is observed in injured neurons (Xu et al. 2006). Therefore, in our first set of experiments, we examined which cells expressed apoE in the presence and absence of LPS stimulation in vivo. LPS or vehicle was injected into the ventricles of the different lines of human APOE knock-in and mouse apoE knock-out mice, and brain sections were examined 72 hours later. ApoE is normally expressed by astrocytes and microglia, and indeed, we observed apoE immunoreactivity in GFAP- and F4/80-positive cells (Figure 1A-B, top panels), but not in neurons in our condition (Figure 1C, top panel). Three days after LPS injection, apoE immunoreactivity was increased in both astrocytes and microglia, but was still not observed in neurons (Figure 1A-C, bottom panels).
Figure 1. ApoE protein expression in the hippocampus of apoE-TR mice after treatment.
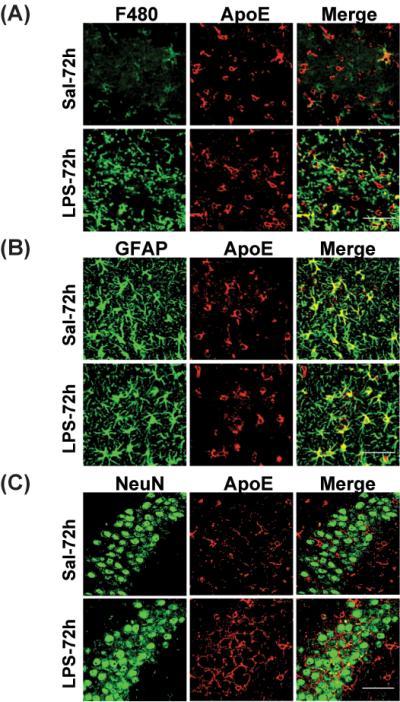
APOE2, APOE3 and APOE4 mice were sacrificed 24 h and 72 h after treatment. ApoE expression in the hippocampus was determined by immunofluorescence staining. Representative images of (A) F4/80 (green) and apoE (red); (B) GFAP (green) and apoE (red); and (C) NeuN (green) and apoE (red) staining in the CA3 region of the hippocampus in saline- and LPS-treated APOE4 mice at 72h. Yellow color in merged image shows apoE-expressing cells. Scale bar is 50 μm.
Chronic inflammation may predispose to Alzheimer's disease (Herrup 2010). We hypothesized that APOE genotype may differentially affect inflammation, thus we examined glial activation in human APOE knock-in mice and mouse APOE knock-out mice. Immunostaining of microglia/macrophages (F4/80) and astrocytes (GFAP) from untreated brains did not demonstrate differences in glial numbers or morphology across genotype (data not shown). Therefore, we hypothesized that APOE genotype may affect glial activation after an inflammatory stimulus. In order to specifically examine the effect of APOE genotype on glial activation, we injected LPS or vehicle into the ventricles of the different lines of APOE knock-in mice and examined brain sections and homogenates 24 or 72 hours later.
We tested the effect of LPS on the number of hippocampal F4/80 positive cells. As expected, in LPS-injected mice, the F4/80 positive cells displayed an “activated” morphology, with larger cell bodies and thicker processes (Figure 2A). After 24 hours, there were significant increases in the numbers of F4/80 positive cells in all mouse lines compared to mice injected with saline. In APOE2 and APOE3 mice, the increases in microglia/macrophage numbers were 70% and 46%, respectively (Figure 2B). However APOE4 mice showed a significantly higher increase, at 133% over control levels (p<0.01 compared to APOE2 or APOE3). The APOE knock-out mice showed an even greater increase (213%, p<0.01) compared to the APOE knock-in mice.
Figure 2. LPS induces F4/80 positive cell activation in an APOE genotype dependent manner in the hippocampus.
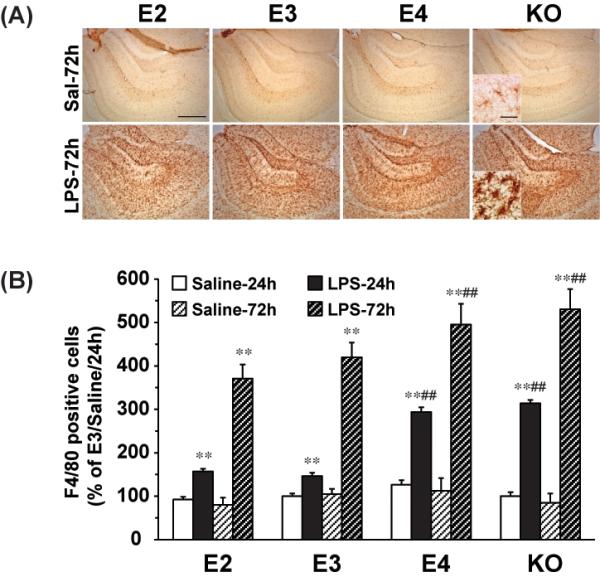
APOE2, APOE3, APOE4 and APOE knock-out mice were sacrificed 24h and 72h after ICV injection of LPS (1000ng) or saline. (A) Representative IHC images of F4/80-positive microglia in the hippocampus under 5X and 63X lens (Inset). The scale bar is 100μm and 10μm, respectively. (B) Quantification of F4/80- positive cells in the hippocampus. The numbers of F4/80-positive microglia are expressed as mean ± SD (n=4-5/group). **P<0.01, compared with saline treatment at the same time point. ##P<0.01, compared with APOE2 or APOE3 at the same LPS treatment time point.
When hippocampi were analyzed three days after LPS exposure, there was a further increase in F4/80 positive cell numbers, and the APOE4 and APOE knock-out mice had the highest levels. APOE4 mice averaged 18% more microglia/macrophages, and APOE knock-out mice averaged 26% more than the APOE3 mice (p<0.01) (Figure 2B). There were no significant differences in the numbers of F4/80 positive cells 72 hours after saline injection in any of the strains of mice. These data demonstrate that APOE4 and APOE knock-outs are predisposed to a stronger inflammatory response to LPS in vivo.
We tested whether the effects of APOE genotype on glial activation were limited to the hippocampus, or whether the cortex showed similar effects. ICV injection of LPS induced activation of cortical microglia/macrophage in all APOE lines with significant differences seen after three days (Figure 3A). No significant changes were observed after 24hrs (data not shown). Quantification of these cells showed significantly greater increases in the APOE4 and APOE knock-out mice (Figure 3B), consistent with what we observed in the hippocampi.
Figure 3. LPS induces F4/80 positive cell activation in an APOE genotype dependent manner in the cortex.
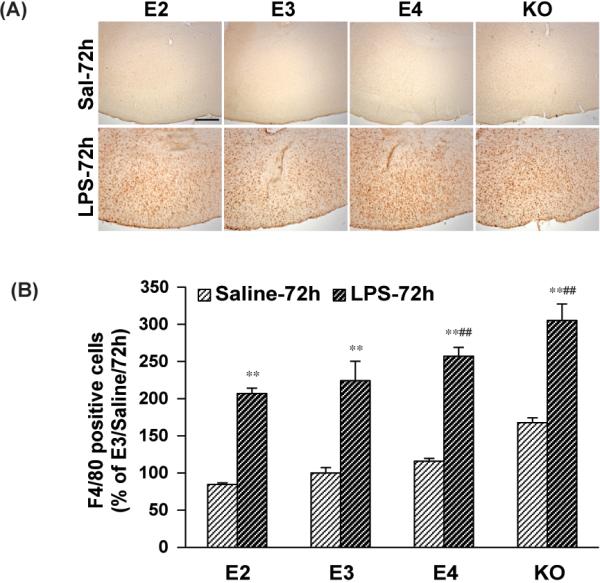
APOE2, APOE3, APOE4 and APOE knock-out mice were sacrificed 24h and 72h after ICV injection of LPS (1000ng) or saline. (A) Representative IHC images of F4/80-positive microglia in the cortex under 5X lens. The scale bar is 100μm. (B) Quantification of F4/80- positive cells in the cortex. The numbers of F4/80-positive microglia are expressed as mean ± SD (n=4-5/group). **P<0.01, compared with saline treatment at the same time point. #P<0.05, ##P<0.01, compared with APOE2 or APOE3 at the same LPS treatment time point.
We further tested whether the effect of APOE genotype on glial activation was limited to F4/80 positive cells, or whether it also affected activation of astrocytes. We analyzed the same brains for astrocyte numbers in the hippocampus by immunostaining for GFAP (Figure 4A). The increase in astrocyte numbers after LPS stimulation was consistently less dramatic than the effects on microglia. None of the mice displayed statistically significant increases in astrocytes 24 hours after LPS. The APOE2 and APOE3 mice also showed no changes in astrocyte numbers at 72 hours, but the APOE4 and APOE knock-out mice displayed significant increases in astrocyte numbers at 72 hours (27% and 33%, respectively, p<0.01) compared to saline-injected controls or to the APOE2 and APOE3 mice (Figure 4B).
Figure 4. LPS effect on hippocampal astrocyte activation.
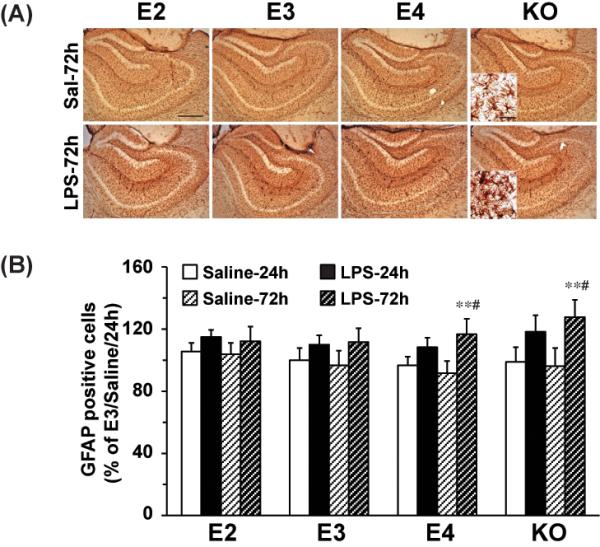
APOE2, APOE3, APOE4 and APOE knock-out were sacrificed 24h and 72h after ICV injection of LPS (1000ng) or saline. (A) Representative IHC images of GFAP-positive astrocyte in the hippocampus under 5X and 63X lens (Inset). The scale bar is 100 μm and 10μm, respectively. (B) Quantification of GFAP-positive astrocyte numbers in the hippocampus. Data are expressed as mean ± SD (n=4-5/group). **P<0.01, compared with saline treatment at the same time point. #P<0.05, compared with APOE2 or APOE3 with LPS treatment at 72 hours.
We next tested whether, in addition to affected glial numbers, APOE genotype also affected release of pro-inflammatory cytokines. We measured three important pro-inflammatory cytokines, IL-1β, IL-6, and TNF-α, at 24 and 72 hours after LPS injection, using ELISAs on hippocampal brain extracts. Consistent with their roles as transient mediators of damage, levels of each of these cytokines were increased dramatically 24 hours after LPS, and then returned toward baseline levels by 72 hours. At 24 hours, levels of IL-1β were increased from approximately 1.3 pg/mg protein in all lines to 17.0 pg/mg protein in APOE2 mice, 18.4 pg/mg protein in APOE3 mice, 24.5 pg/mg protein in APOE4 mice and 34.6 pg/mg protein in APOE knock-out mice (Figure 5A). The increases observed in both APOE4 and APOE knock-out mice were significantly greater than those observed in APOE2 and APOE3 mice (P<0.01). After 72 hours, the levels of IL-1β returned to baseline levels in the APOE2 and APOE3 mice, but were still significantly elevated in the APOE4 (7.9 pg/mg protein, p<0.01) and the APOE knock-out (8.4 pg/mg protein, p<0.01) mice (Figure 5A).
Figure 5. LPS induces pro-inflammatory cytokine release in an APOE genotype dependent manner in the hippocampus.
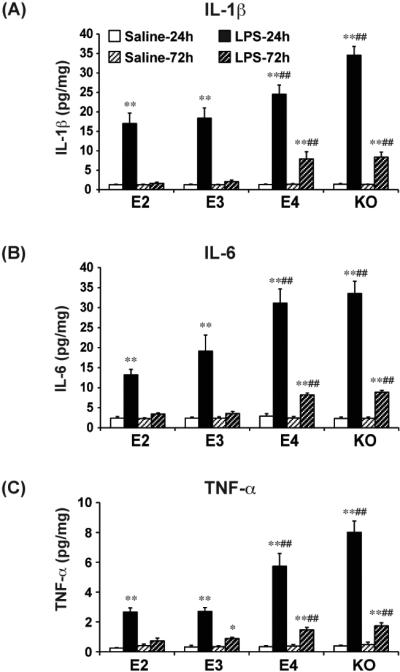
APOE2, APOE3, APOE4 and APOE knock-out were sacrificed 24h and 72h after treatment. The hippocampus was dissected out and homogenates were prepared for TNF-α (A), IL-1β (B), and IL-6 (C) ELISA. Cytokine levels are expressed as mean ± SD (n=4-5/group). *P<0.05, **P<0.01, compared with saline treatment at the same time point. ##P<0.01, compared with APOE2 or APOE3 at the same LPS treatment condition.
Similar patterns were also seen for both IL-6 and TNF-α. For IL-6, at 24 hours after LPS, levels increased from a baseline of approximately 2.4 pg/mg protein to 13.2 pg/mg protein (APOE2), 19.1 pg/mg protein (APOE3), 31.1 pg/mg protein (APOE4) and 33.5 pg/mg protein (APOE knock-out) (Figure 5B). The increases in APOE4 knock-in and APOE knock-out mice were significantly greater than those in APOE2 and APOE3 mice (p<0.01). At 72 hours, levels of IL-6 in APOE2 and APOE3 mice were similar to untreated mice, but remained significantly elevated for APOE4 and APOE knock-out mice (8.2 and 8.9 pg/mg protein, respectively) (Figure 5B).
At 24 hours after LPS treatment, TNF-α levels increased from a baseline of approximately 0.3 pg/mg protein to 2.7 pg/mg protein for APOE2 and APOE3 mice, and to significantly greater levels in APOE4 (5.7 pg/mg protein) and APOE knock-out (8.0 pg/mg protein) mice (Figure 5C). The increases observed in APOE4 and APOE knock-out mice again were significantly greater than those observed in APOE2 and APOE3 mice (p<0.01). At 72 hours after LPS, levels of TNF-α in APOE2 and APOE3 mice were not significantly different than untreated mice, but were significantly elevated for APOE4 and APOE knock-out mice (1.5 and 1.7 pg/mg protein, respectively, p<0.01) (Figure 5C). Thus, for all three cytokines, APOE4 and APOE knock-out mice were associated with a greater induction and a longer increase in cytokine levels.
These data indicate that APOE4 compared to the other alleles increases the endogenous inflammatory response of the brain. We next tested whether it also increased the influx of T-cells from the periphery by measuring CD3-positive cells in the brain 72 hours after LPS injection. Very few CD-3 positive cells were observed in the brains of animals injected with saline. However, after LPS, numbers of CD3-positive cells increased significantly in all lines of mice (Figure 6), demonstrating the large influx of T-cells after brain LPS injection. Again, APOE2 and APOE3 mice showed the lowest levels of CD3 cells (28 and 42 times the levels in the control APOE3 brains, respectively). APOE4 mice (64 times) and APOE knock-out mice (68 times) showed significantly higher levels than the APOE2 and APOE3 mice (p<0.01) (Figure 6).
Figure 6. LPS leads to peripheral T cell migration into the brain.
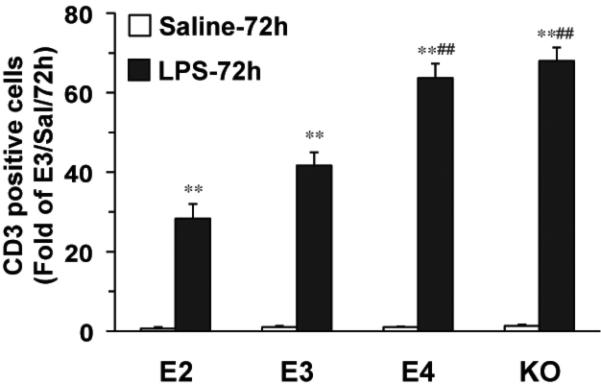
APOE2, APOE3, APOE4 and APOE knock-out were sacrificed 72h after treatment and CD3-positive T cells in the CA3 area of the hippocampi were quantified. Data are expressed as mean ± SD (n=4-5/group). **P<0.01, compared with saline treatment. ##P<0.01, compared with APOE2 or APOE3 mice with LPS treatment at 72 hours.
Finally, we tested whether these increases in glial activation correlated with neuronal changes. We did not observe any difference in the density of NeuN-positive cells between saline and LPS-treated brains (data not shown). We analyzed changes in synaptic proteins as markers of neurotoxicity. Three synaptic proteins, PSD-95, drebin, and synaptophysin, were analyzed 72 hours after LPS and vehicle exposure by both immunohistochemistry and immunoblotting. Hippocampal staining for each of these synaptic markers showed the expected distributions on cell bodies and processes (Figure 7A-C). From the immunostainings, each synaptic marker seemed decreased in the CA3 region of mice treated with LPS, when compared with saline treatment (Figure 7A-C). To accurately quantify levels of each synaptic marker, we used total hippocampal lysates for immunoblotting. In the vehicle treatments, there were no significant differences of synaptophysin levels across groups. However, levels of PSD95 and Drebin were significantly less in the APOE4 (15% and16%, respectively) and APOE knock-out (24% and 13%, respectively) mice compared to APOE3 mice, suggesting that there may be underlying basal differences in post synaptic densities among the genotypes. When comparing LPS to vehicle, for PSD-95 (Fig 7A), we observed a 33% decrease in the APOE4 mice and a 39% decrease in the APOE knock-out mice, significantly greater reductions than seen in APOE2 (10%) and APOE3 (16%) mice at 72 hours. Similarly, for drebin (Fig 7B), we found that LPS compared to vehicle resulted in a 26% decrease in the APOE4 mice and a 35% decrease in the APOE knock-out mice, greater reductions than seen in APOE2 (25%) and APOE3 (22%) mice. Finally, for synaptophysin (Fig 7C), we found that LPS resulted in an 11% decrease in the APOE4 mice and a 15% decrease in the APOE knock-out mice, whereas APOE2 mice showed no decrease and APOE3 mice showed a 9% decrease. The levels of each of these synaptic markers in APOE4 and APOE knock-out mice were significantly less after LPS treatment than the levels seen in APOE2 or APOE3 mice (PSD-95, p<0.01; Drebin, p<0.01; synaptophysin, p<0.05). Thus, LPS caused a greater loss of synaptic proteins in APOE4 and APOE-knock-out mice.
Figure 7. APOE genotype dependent effects on synaptic proteins.

APOE2, APOE3, APOE4 and APOE knock-out were sacrificed 72h after treatment. Shown on the left are representative immunohistochemistry images of the CA3 region of the hippocampus. Scale bar is 25μm. Levels of PSD-95 (A), Drebrin (B), and synaptophysin (C) were determined by western blot. α-tubulin was used as a protein loading control. Western blot data are expressed as mean ± SD (n=3-4/group). *P<0.05, **P<0.01, compared with saline treatment at 72 hours. #P<0.05, ##P<0.01, compared with APOE2 or APOE3 with LPS treatment at 72 hours.
DISCUSSION
We found that APOE genotype affected glial activation in mice such that those expressing the APOE4 allele responded to an acute, brain inflammatory challenge (ICV injection of LPS) more strongly than those expressing APOE2 and APOE3, and that APOE2 mice responded similarly as APOE3 mice. The heightened levels of glial activation in APOE4 mice were also evidenced by increased levels of three cytokines (IL-1β, IL-6, and TNF-α), and increased numbers of F4/80 positive cells and infiltrating T-cells. The APOE4 mice also showed the greatest reductions in synaptic markers after the inflammatory challenge, which was not great enough to result in observable neuron loss. Acute injection of LPS does not model the chronic effects of discrete amyloid deposits, neurofibillary tangles, and cell death seen in AD. However, our data importantly show that the effects of APOE4 on inflammation may occur in conditions beyond AD. We are currently using mouse models of amyloid deposition to test whether APOE genotype affects glial responses to Aβ deposits.
These data provide in vivo evidence consistent with other findings that define apoE as an anti-inflammatory agent, with the apoE4 protein being less efficacious than apoE2 and apoE3. In vitro, LPS-induced inflammation was greatest in the presence of apoE4-expressing glia, compared to apoE2 or apoE3-expressing glia (Maezawa et al. 2006b). In vivo, APOE4 genotype also increased susceptibility to central and peripheral inflammation, and depended on gene dosage (Vitek et al. 2009). APOE4 knock-in mice showed increased LPS-induced inflammation compared to APOE3 knock-in mice (Lynch et al. 2003) and decreased dendritic lengths (Maezawa et al. 2006a). APOE knockout mice and cells are more susceptible to induction of inflammatory cytokines after LPS treatment (Laskowitz et al. 1998; Lynch et al. 2001). Similar to the APOE knock-out mice, APOE4 knock-in mice showed increased brain inflammation in response to LPS, suggesting that apoE4 is impaired at blocking inflammation.
The mechanism of the anti-inflammatory effect of apoE is being elucidated. ApoE interacts with its receptor LRP1 and inhibits downstream JNK activation (Pocivavsek et al. 2009a; Pocivavsek et al. 2009b; Pocivavsek and Rebeck 2009). Gene expression studies found that LPS induced stronger inflammatory responses in APOE4 mice through greater activation of the NF-kB signaling cascade (Ophir et al. 2005). ApoE also reduces activation of macrophage, again through interactions with apoE receptors (Baitsch et al. 2011). Some of the observed effects in APOE4 mice may be due to lower levels of apoE protein present in the brains of these mice. Total brain apoE levels were found to be reduced by various assays, including immunoblots (Vitek et al. 2009), ELISA (Riddell et al. 2008) and mass spectrometry (Sullivan et al. 2011).
The anti-inflammatory nature of apoE is supported by the observations that APOE knockout mice and APOE4 mice have worse responses to brain injuries accompanied by strong components of glial activation, such as ischemia (Sheng et al. 1999), experimental autoimmune encephalomyelitis (Li et al. 2006), traumatic brain injury (Lynch et al. 2002), and induced neuroinflammation (Laskowitz et al. 1998; Lynch et al. 2001; Lynch et al. 2003). Treatments with peptides that activate apoE receptors have proven therapeutic for mouse models of brain damages. For example, peptides based on the apoE receptor-binding domain decrease glial activation and reduce damage in traumatic brain injury (Laskowitz et al. 2010), promote axon regeneration after peripheral nerve injury (Li et al. 2010), and decrease lesion volume after focal brain ischemia (Tukhovskaya et al. 2009). These protective effects of apoE-based therapeutics may depend on their roles in reducing inflammation.
Even in the absence of induced inflammation, APOE4 mice have neuronal and cognitive deficits. APOE4 knock-in mice have reduced neuronal complexity in the amygdala and the cortex (Dumanis et al. 2009; Wang et al. 2005), as well as a reduced density of cortical dendritic spines (Dumanis et al. 2009). Our findings showing reduced PSD95 and drebin staining under control conditions (Fig 7A,B) are consistent with these results. Moreover, APOE4 mice have behavioral differences in retention of spatial memory (Bour et al. 2008) and avoidance behavior (Grootendorst et al. 2005). APOE4 mice also show reduced LTP in the dentate gyrus of the hippocampus (Trommer et al. 2004), although enhanced LTP in the CA1 region has been reported (Korwek et al. 2009). Thus, normal expression of APOE4 is associated with structural and functional alterations in various regions of the brain. We hypothesize that these changes may result from chronic induction of low levels of inflammation throughout life, and may predispose to brain damages that accumulate with aging.
The importance of inflammation in AD is emphasized by epidemiological and clinical studies. The use of non-steroidal anti-inflammatory drugs (NSAIDs) decreases the risk of AD (McGeer and McGeer 2007). Several studies have specifically addressed whether this risk depended on APOE genotype and found the reduced risk was largely attributable to the APOE4-positive individuals (Szekely et al. 2008; Yip et al. 2005). If NSAIDs can act by inhibiting inflammatory processes in the CNS, they may be particularly effective in individuals with the apoE4 proinflammatory phenotype. Determining the roles of apoE isoforms in modulating the effects of NSAIDs on measures of neurotoxicity may help elucidate how NSAIDs could be protective (Varvel et al. 2009). Clinical trials of Aβ immunotherapies have found that some participants have adverse neuroinflammatory side effects, for example, vasogenic edema (Salloway et al. 2009). Interestingly, almost all the adverse side effects reported were in APOE4 carriers, prompting segregation of E4 positive and E4 negative carriers in future clinical studies. These findings are consistent with our reports here of APOE4 carriers having an enhanced neuroinflammatory response compared to the other APOE genotypes.
Our work demonstrates that APOE genotype affects several aspects of the inflammatory response in mouse brains, leading to increased neuronal damages in APOE4 mice. These studies support the model where apoE4 increases susceptibility to inflammation brought about by various types of brain damages, such as stroke, AD and TBI. The model of LPS injection was chosen for these studies to avoid effects of APOE genotype on other aspects of AD pathology, such as plaques and tangles. It would be interesting to investigate whether the susceptibility of the APOE4 brain to inflammation was observed in areas not vulnerable to AD pathogenesis, such as the cerebellum or striatum. Together, these data suggest that strategies to reduce inflammatory processes may be particularly effective in APOE4 individuals.
ACKNOWLEDGEMENTS
This work was supported by NIH P01 AG030128 (MJL and GWR) and P01 AG030128-03S1 (ENH).
REFERENCES
- Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011;31(5):1160–8. doi: 10.1161/ATVBAHA.111.222745. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour A, Grootendorst J, Vogel E, Kelche C, Dodart JC, Bales K, Moreau PH, Sullivan PM, Mathis C. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav Brain Res. 2008;193(2):174–82. doi: 10.1016/j.bbr.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3(89):89ra57. doi: 10.1126/scitranslmed.3002156. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29(48):15317–22. doi: 10.1523/JNEUROSCI.4026-09.2009. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egensperger R, Kosel S, von Eitzen U, Graeber MB. Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol. 1998;8(3):439–47. doi: 10.1111/j.1750-3639.1998.tb00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, Babaey S, Yehuda B, Groswasser Z. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52(2):244–8. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Watanabe M. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: a study in the adult mouse brain. J Comp Neurol. 2000;426(4):572–86. [PubMed] [Google Scholar]
- Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, Bullido MJ, Engelborghs S, De Deyn P, Berr C. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.52. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootendorst J, Bour A, Vogel E, Kelche C, Sullivan PM, Dodart JC, Bales K, Mathis C. Human apoE targeted replacement mouse lines: h-apoE4 and h-apoE3 mice differ on spatial memory performance and avoidance behavior. Behav Brain Res. 2005;159(1):1–14. doi: 10.1016/j.bbr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Herrup K. Reimagining Alzheimer's disease--an age-based hypothesis. J Neurosci. 2010;30(50):16755–62. doi: 10.1523/JNEUROSCI.4521-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43(5):429–435. doi: 10.1038/ng.803. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One. 2010;5(11):e13950. doi: 10.1371/journal.pone.0013950. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63(3):287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korwek KM, Trotter JH, Ladu MJ, Sullivan PM, Weeber EJ. ApoE isoformdependent changes in hippocampal synaptic function. Mol Neurodegener. 2009;4:21. doi: 10.1186/1750-1326-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269(38):23403–6. [PubMed] [Google Scholar]
- Laskowitz DT, Matthew WD, Bennett ER, Schmechel D, Herbstreith MH, Goel S, McMillian MK. Endogenous apolipoprotein E suppresses LPS-stimulated microglial nitric oxide production. Neuroreport. 1998;9(4):615–8. doi: 10.1097/00001756-199803090-00010. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP, Dawson HN. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma. 2010;27(11):1983–95. doi: 10.1089/neu.2010.1396. [DOI] [PubMed] [Google Scholar]
- Li FQ, Fowler KA, Neil JE, Colton CA, Vitek MP. An apolipoprotein E-mimetic stimulates axonal regeneration and remyelination after peripheral nerve injury. J Pharmacol Exp Ther. 2010;334(1):106–15. doi: 10.1124/jpet.110.167882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Sempowski GD, McKenna SE, Laskowitz DT, Colton CA, Vitek MP. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J Pharmacol Exp Ther. 2006;318(3):956–65. doi: 10.1124/jpet.106.103671. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT. Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol. 2001;114(1-2):107–13. doi: 10.1016/s0165-5728(00)00459-8. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Pineda JA, Morgan D, Zhang L, Warner DS, Benveniste H, Laskowitz DT. Apolipoprotein E affects the central nervous system response to injury and the development of cerebral edema. Ann Neurol. 2002;51(1):113–7. doi: 10.1002/ana.10098. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, Warner DS, Laskowitz DT. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278(49):48529–33. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Maeda N, Montine TJ, Montine KS. Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation. 2006a;3:10. doi: 10.1186/1742-2094-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Nivison M, Montine KS, Maeda N, Montine TJ. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. Faseb J. 2006b;20(6):797–9. doi: 10.1096/fj.05-5423fje. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28(5):639–47. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26(3):349–54. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43(5):436–41. doi: 10.1038/ng.801. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541(1):163–6. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- Ophir G, Amariglio N, Jacob-Hirsch J, Elkon R, Rechavi G, Michaelson DM. Apolipoprotein E4 enhances brain inflammation by modulation of the NFkappaB signaling cascade. Neurobiol Dis. 2005;20(3):709–18. doi: 10.1016/j.nbd.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Ophir G, Meilin S, Efrati M, Chapman J, Karussis D, Roses A, Michaelson DM. Human apoE3 but not apoE4 rescues impaired astrocyte activation in apoE null mice. Neurobiol Dis. 2003;12(1):56–64. doi: 10.1016/s0969-9961(02)00005-0. [DOI] [PubMed] [Google Scholar]
- Pocivavsek A, Burns MP, Rebeck GW. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia. 2009a;57(4):444–53. doi: 10.1002/glia.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Mikhailenko I, Strickland DK, Rebeck GW. Microglial low-density lipoprotein receptor-related protein 1 modulates c-Jun N-terminal kinase activation. J Neuroimmunol. 2009b;214(1-2):25–32. doi: 10.1016/j.jneuroim.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Rebeck GW. Inhibition of c-Jun N-terminal kinase increases apoE expression in vitro and in vivo. Biochem Biophys Res Commun. 2009;387(3):516–20. doi: 10.1016/j.bbrc.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW, Perls TT, West HL, Sodhi P, Lipsitz LA, Hyman BT. Reduced apolipoprotein epsilon 4 allele frequency in the oldest old Alzheimer's patients and cognitively normal individuals. Neurology. 1994;44(8):1513–6. doi: 10.1212/wnl.44.8.1513. [DOI] [PubMed] [Google Scholar]
- Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993;11(4):575–80. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28(45):11445–53. doi: 10.1523/JNEUROSCI.1972-08.2008. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–70. doi: 10.1212/WNL.0b013e3181c67808. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, Laskowitz DT, Mackensen GB, Kudo M, Pearlstein RD, Warner DS. Apolipoprotein E deficiency worsens outcome from global cerebral ischemia in the mouse. Stroke. 1999;30(5):1118–24. doi: 10.1161/01.str.30.5.1118. [DOI] [PubMed] [Google Scholar]
- Smith C, Graham DI, Murray LS, Stewart J, Nicoll JA. Association of APOE e4 and cerebrovascular pathology in traumatic brain injury. J Neurol Neurosurg Psychiatry. 2006;77(3):363–6. doi: 10.1136/jnnp.2005.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Han B, Liu F, Mace BE, Ervin JF, Wu S, Koger D, Paul S, Bales KR. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272(29):17972–80. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Breitner JC, Fitzpatrick AL, Rea TD, Psaty BM, Kuller LH, Zandi PP. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70(1):17–24. doi: 10.1212/01.wnl.0000284596.95156.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Ye GL, Sotak M, Sullivan PM, Pasternak JF, LaDu MJ. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 2004;15(17):2655–8. doi: 10.1097/00001756-200412030-00020. [DOI] [PubMed] [Google Scholar]
- Tukhovskaya EA, Yukin AY, Khokhlova ON, Murashev AN, Vitek MP. COG1410, a novel apolipoprotein-E mimetic, improves functional and morphological recovery in a rat model of focal brain ischemia. J Neurosci Res. 2009;87(3):677–82. doi: 10.1002/jnr.21874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Bhaskar K, Kounnas MZ, Wagner SL, Yang Y, Lamb BT, Herrup K. NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Invest. 2009;119(12):3692–702. doi: 10.1172/JCI39716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30(9):1350–60. doi: 10.1016/j.neurobiolaging.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, Sullivan PM. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18(2):390–8. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135(2):235–8. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26(19):4985–94. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip AG, Green RC, Huyck M, Cupples LA, Farrer LA. Nonsteroidal antiinflammatory drug use and Alzheimer's disease risk: the MIRAGE Study. BMC Geriatr. 2005;5:2. doi: 10.1186/1471-2318-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]