

Abstract
The adjuvant therapy of choice for superficial bladder cancer is the intravesical instillation of live Mycobacterium bovis Bacillus Calmette-Guerin (BCG). In spite of the fact that this therapy is the most effective treatment for superficial bladder cancer, intravesical administration of BCG is associated with high local morbidity and the potential for systemic infection. Therefore, there is a need for the development of safer, less toxic approaches to fight this disease. Since fibronectin attachment protein (FAP) is a key element in BCG retention and targeting to cells, we hypothesize that this protein can be used as targeting agent to deliver cytotoxic cargo for the treatment of bladder tumors. Here we evaluated the ability of bladder tumor cells to bind and endocytose FAP via fibronectin-integrin complexes. We found that microaggregation induced by an anti-FAP polyclonal antibody accelerated FAP uptake by T24 bladder tumor cells. FAP was determined to be internalized via a clathrin-independent, caveolae-dependent mechanism. Further, once within the endosomal compartment, FAP was targeted to the lysosomal compartment with negligible recycling to the plasma membrane. Importantly, we demonstrated that FAP microaggregation and internalization could also be triggered by multivalent Ni2+NTA-bearing liposomes. Overall, our studies validate the use of FAP as a targeting vector and provide the foundation for the design of more effective, less toxic bladder cancer therapeutics.
Keywords: Fibronectin attachment protein, Bacillus calmette-guerin, bladder cancer, caveolae, integrins
INTRODUCTION
Bladder cancer is the fourth most common cancer in men and eleventh most common in women1. Approximately 80% of the cases are classified as superficial cancer and most are treated with surgery alone. Unfortunately, 70% of newly diagnosed patients suffer disease recurrence after surgical treatment2. Therefore, the development of efficient therapeutic countermeasures against this pathology is a high priority.
The bladder displays unique characteristics and challenges as an organ to be targeted for therapy. Specifically, bladder epithelial cells (urothelium) lining the luminal surface, known as umbrella cells, are engaged in tight junctions that prevent access to the lower transitional cell layers3, 4. Furthermore, umbrella cells express characteristic extracellular proteins (uroplakins) that assemble into semi-rigid plaques that effectively shield the apical surface. The urothelium is further isolated from the bladder lumen by a GlycosAminoGlycan (GAG)-rich mucin layer that is produced and assembled on the apical surface of the umbrella cells5. In contrast, malignant bladder cells are usually less differentiated, less polarized, exhibit diminished uroplakin expression and low GAG layer synthesis6. Therefore, as opposed to normal bladder epithelia, neoplastic cells are exposed to the lumen of the bladder. This leads to increased accessibility of tumor lesions to lumen-borne therapeutic agents, compared to the well-protected normal regions of the bladder. However, constant urine influx and periodic voiding of the bladder makes the direct instillation of therapeutic drugs limited in their impact on bladder tumor cells due to poor retention.
At present, intravesical instillation of live Mycobacterium bovis bacillus Calmette-Guerin (BCG) is currently the adjuvant therapy of choice for the treatment of superficial bladder tumors. While the specific mechanism of BCG-mediated antitumor activity is not yet fully understood, direct targeting and binding to bladder tumor cells, followed by cellular uptake and subsequent activation of adaptive immune responses, are required7, 8. Nevertheless, intravesical BCG is associated with high local morbidity and a risk of systemic mycobacterial infection9. Notably, multiple instillations of BCG leads to increased toxicity, thus limiting patient tolerance for the treatment regimen required for effective anti-cancer activity10. Consequently, the development of high-affinity, non-toxic targeting strategies is of high priority in the field.
A breakthrough came from the identification of Fibronectin Attachment Protein (FAP), a bacterial adhesin highly conserved in all mycobacterial species, as the factor that mediates BCG attachment to and internalization by bladder tumor cells11–13. FAP was shown to bind α5β1 Integrins expressed by tumor cells via a fibronectin (FBN) bridge, and to be responsible for the uptake of BCG-FBN-Integrin complexes8, 14–16. Further, FAP interaction did not compete with integrin binding for FBN17. Subsequent in vivo studies, showed that intravesically-instilled, radiolabled recombinant FAP was able to bind bladder implanted tumors. Furthermore, intravesical recombinant FAP was capable of eliciting a T cell-dependent antitumor response in immunized mice indicating recombinant FAP, was internalized, processed and presented in context with MHC class I by bladder tumor cells18. Since FAP by itself was observed to mediate antitumor activity in FAP-immunized mice18, the possibility of using recombinant FAP protein alone as a lower-risk alternative to BCG for therapeutic purposes is currently being explored. In addition, the internalization of FAP by bladder tumor cells provides a novel and potentially powerful approach for the delivery of therapeutics that may greatly enhance the antitumor effect of FAP.
As a first step towards the design of a FAP-targeted delivery strategy, we investigated the mechanism and kinetics of FAP uptake by T24 bladder tumor cells. We determined that in a serum-free environment such as the lumen of the bladder, internalization of FAP-fibronectin (FBN)-Integrin complexes occurred with a very slow kinetics. This internalization process could be greatly accelerated by microaggregation of FAP-FBN-integrin complexes. Mechanistically, we established that purified FAP was internalized by a clathrin-independent mechanism and routed to the lysosome in T24 cells. We also provide a proof-of-principle for the utilization of FAP in promoting liposome internalization. We envision that the results obtained in this study will constitute the foundations for the design of FAP-targeted carriers for the efficient delivery of cytotoxic agents.
MATERIALS AND METHODS
Materials
Materials were purchased from Fisher Scientific (Fairlawn, NJ) or Sigma (St. Louis, MO) unless stated otherwise.
Plasmids, siRNAs and antibodies used in this study are listed in Supplemental Table I. Ni2+:trisNTA-AlexaFluor647 and DSPE- CF633 fluorescent lipid were synthesized as described in Supplemental Materials.
Cell culture and transfection
T24 bladder carcinoma cells were obtained from ATCC and cultured on polylysine-coated surfaces with DMEM, streptomycin/penicillin, 2mM L-glutamine and 10% fetal bovine serum. Normal human dermal fibroblasts were obtained from the Coriell cell repository and cultured as previously described19.
Transfections of plasmid DNA and siRNAs were performed using FugeneHD (Roche) and TransIT-TKO (Mirus) reagents, respectively, according to the manufacturer’s instructions. Cells were used for experiments after 18–30h following plasmid transfection. siRNA transfections were performed twice at 48 and 72h before using knock-down cells.
Protein purification
Bacterially-produced recombinant His6-FAP was generated in Rosetta cells (Novagen) by inducing expression of pTrc-his plasmids with 0.05mM IPTG for 5h at 30°C. Proteins were purified in PBS, 0.1%Tween, and 15% glycerol using Ni2+NTA resin (Novagen) according to standard protocols and eluted with PBS, 250mM imidazole for 2–8h at 4°C. The buffer from the protein preparation was exchanged by PBS using Zeba Spin columns (Pierce). The concentration of purified His6-FAP was measured using Precision Red protein assay reagent (Cytoskeleton) and diluted to a final concentration of 10μM in 1) serum-free DMEM, 2) PBS or 3) Buffer 9B (Modified PBS).
FAP binding
Cells were seeded on coverslips for approximately 24h before use to allow secretion and organization of extracellular fibronectin structures. 10μM His6-FAP in the indicated media was added and incubated with cells for 3h at 37°C to enable optimal cell binding. Cells were then fixed in 3% formaldehyde for 10min, processed for immunofluorescence and imaged as previously described19.
Internalization experiments
Transferrin-AlexaFluor488 internalization experiments were performed as previously described19.
To assay FAP endocytosis, T24 cells were seeded for 8–24h and then starved in DMEM+0.1%FBS for 4h. Cells were allowed to bind 10μM His6-FAP in serum-free DMEM for 1h at 37°C. Coverslips were rinsed and incubated with 25μM Ni2+:trisNTA-AlexaFluor647 and indicated antibodies (or liposome suspension) in serum-free DMEM at 10°C for 45min. After washing with PBS, cells were allowed to internalize at 37°C for the indicated times.
Since FAP binding is pH-resistant we devised two alternative approaches to detect internalized FAP:
i) Inclusion of internalized FAP in GFP-Rab5Q79L-endosomes
Following endocytosis, GFP-Rab5Q79L-expressing cells were fixed in 3% formaldehyde for 10min at 20°C before mounting and imaging. To quantify internalization, 3-slice z-stack images were acquired with a Zeiss Axiovert 200M microscope equipped with an Axiocam MRm camera and standard Zeiss filter cubes including filter set 50 (ex: BP640/30, bs: FT660, em: BP690/50) for AlexaFluor647 and CF633 imaging. The images were processed with ImageJ as described before19. The signal intensity of Ni2+:trisNTA-AlexaFluor647-labeled His6-FAP was then measured only within the Rab5Q79L-GFP structures.
ii) Stripping of anti-FAP or Ni2+:trisNTA-AlexaFluor647 probe from leupeptin-treated cells
Cells were treated with 100μg/mL leupeptin to prevent lysosomal degradation. Following uptake, internalized FAP:Ni2+:trisNTA-AlexaFluor647 was quantified either fluorescence microscopy or flow cytometry.
In the fluorescence microscopy experiments, non-internalized surface probes (Ni2+:trisNTA-AlexaFluor647 or anti-FAP antibody) were removed by incubating cells with pH 2.0 buffer supplemented with 20mM EDTA for 60sec. After washing, the cells were processed for immunofluorescence, mounted and imaged. Quantification of internalized fluorescence was performed as described before19.
Alternatively, the cells were trypsinized for 5min to lift them and to remove non-internalized FAP and immediately fixed with 2% paraformaldahyde. Samples were analyzed using a FACSCanto II flow cytometer (BD San Jose, CA). All data analysis was performed using FLOWJO flow cytometry analysis software (Tree Star Inc, Ashland, OR).
In all cases, the efficiency of removal of the non-internalized probes was assessed using cells corresponding to the 0min internalization time-point subjected and not to the stripping condition. Comparison between the fluorescence intensity of treated vs. untreated cells yielded an estimation of 80–90% efficiency for the stripping procedures.
Colocalization analysis
In order to quantify the degree of colocalization, we calculated Object-Based Overlap Coefficients20 (“colocalization coefficients” or “cc”) using the ImageJ plugin JaCoP on at least 10 independent images. To quantify His6-FAP or Transferrin-AlexaFluor488 accumulation within Lamp2 structures over time, the colocalization plugin in ImageJ was used on z-projected images to detect Lamp2-overlapping FAP signal. The resulting Lamp2-colocalizing FAP values were expressed as fraction of total FAP intensity per cell.
Liposome Preparation
Liposomes were prepared by the thin film hydration method. A chloroform solution of DPPC:Chol:DSG-PEG2K-NTA:DSPE-CF™633 mixed at a 64:35:0.5:0.5 molar ratio (13 μmol total lipid) was carefully evaporated under a flow of N2 gas, producing a thin lipid film. The film was placed under a 50μm Hg vacuum for 3h to remove trace solvent impurities. This film was then hydrated in 4mL DMEM 1X media solution via 10 freeze-thaw-vortex cycles. The resulting multilamellar liposome solution was then extruded seven times at 50–55°C through three stacked polycarbonate filters of 800nm, 200nm and 50nm pore diameter using an Extruder device that was charged with 200–300psi N2 pressure.
Statistical Analysis
Normally-distributed data was represented as the mean±SD of triplicate measurements. Statistical significance of value differences was assessed by applying the t-test. An important part of the biological data analyzed within this work, however, did not follow a normal distribution (failed normality tests). Given their non-parametric nature, data distributions were shown as box-plots (constructed with Sigmaplot 11.2) depicting the median, 25–75 (bottom and top of box, respectively) and 10–90 (whiskers) percentiles. When appropriate, statistical significance was evaluated using the non-parametric Wilcoxon test. In all cases, experiments were performed at least three times with 20–50 cells analyzed per experiment and per condition unless stated otherwise.
RESULTS
Fibronectin Attachment Protein (FAP) targets fibronectin (FBN) and integrins in human fibroblasts and bladder tumor cells
Previous work has demonstrated that BCG association with bladder cells is mediated by bacterial FAP11 and that binding of radio-iodinated FAP to FBN required the RWFV sequence (residues 213–216)11, 17. In order to establish the mechanisms involved in the interaction of FAP with bladder tumor cells, we first determined the ability of recombinant purified FAP to specifically target cell-bound FBN structures by fluorescence microscopy (Fig. 1). We determined that purified FAP wt (colocalization coefficient, cc=0.83±0.09), but not FAPΔ213–216 (cc=0.33±0.14), was able to bind cell-associated FBN (Fig. 1A-B). Specifically, we found that human dermal fibroblasts, known to produce robust FBN networks, show colocalization between purified His6-FAP and both FBN and β1-Integrins (Fig. 1C). Although, less differentiated T24 bladder tumor cells showed less intricate FBN fibrilar networks, FAP was also colocalized (cc=0.65±0.13) with FBN on these cells (Fig. 1D).
Fig. 1. Purified His6-FAP Colocalizes with Fibronectin and Integrins.

Human dermal fibroblasts (A-C) or T24 bladder cells (D) grown on coverslips for 24h were incubated with 10μM His6-FAP, washed, fixed and immunostained with anti-His6 and either anti-FBN or anti-integrin antibodies. Arrows point to some areas of colocalization. Numbered insets refer to areas of colocalization between FAP and FBN in T24 cells (D). Scale bar: 20microns.
We next tested FAP-FBN complex formation under conditions that resemble the characteristics of the bladder lumen. Specifically, we determined that FAP interaction with FBN was resistant to acidic treatment (pH=4.8 for several hours), high dilution or the presence of urine (Supplemental Fig. 1). Furthermore, purified FAP was able to bind cell-associated FBN in the presence of a bladder instillation buffer (“Buffer 9B”) capable of controlling pH even after a 1:2 dilution with urine (see Materials and Methods and Supplemental Fig. 1B).
In addition, FAP could be observed in association with cell-bound FBN by using a Ni2+:trisNTA-AlexaFluor647 probe to bind the His6-tag present in the recombinant purified protein (Suppl. Fig. 1C).
Low FAP Internalization Rates by Bladder Tumor Cells are Enhanced by Antibody-induced Microaggregation
To assess the potential of FAP as a targeting ligand for chemotherapeutic delivery, we then studied whether purified FAP could be internalized by bladder tumor cells. Since FAP interaction with cellular FBN is resistant to low pH treatment (Supplemental Fig. 1A), standard endocytosis assays relying on acid washes to strip non-internalized ligand21 could not be used. Other approaches such as proteolytic digestion of non-internalized ligand led to severely affected morphology, adherence and viability of cells. Therefore, we relied on a different methodological approach based on the expression of a Rab5Q79L-GFP constitutively-activated mutant that is locked in its GTP-bound conformation, for the detection of endocytosed FAP. Expression of this construct does not affect the rate of ligand internalization22, but leads to the formation of an easy to visualize, GFP-positive, enlarged endosomal compartment due to enhanced early endosome fusion (Fig. 2B). Consequently, we were able to readily identify internalized FAP based on its inclusion within enlarged Rab5Q79L-GFP-decorated structures (Fig. 2). Further, this Rab5 mutant also promotes the accumulation of internalized cargo in the Rab5Q79L-containing endosomal compartment by interfering with lysosomal trafficking22. The expression of Rab5Q79L-GFP, therefore, enhances the analytical sensitivity of our assays by increasing FAP-associated fluorescence as a function of time. Unless indicated otherwise, we used this approach throughout this study.
Fig. 2. T24 Bladder Tumor Cells Internalize Antibody-crosslinked His6-FAP:trisNTA-AlexaFluor647 into Rab5Q79L-GFP Labeled Compartments.

A. Cartoon representing the antibody-induced FAP microaggregation strategy (see text for details). B. Scheme depicting the fusogenic effects of Rab5 dominant positive (Q79L) on the endosomal compartments. C. T24 cells expressing Rab5Q79L-GFP were exposed to 10μM His6-FAP for 1h at 37°C. After washing, the cells were incubated with Ni2+:trisNTA-AlexaFluor647 and either α-HA (upper row) or α-FAP (lower row) polyclonal antibodies for 45min at 10°C. After washing with cold PBS, the cells were incubated 3h at 37°C and fixed. Arrows point to some areas of colocalization. Scale bar: 20microns
In the absence of serum (to better emulate bladder lumen conditions), we found no internalization during incubation periods of several hours. Therefore, we devised an approach to trigger FAP endocytosis upon antibody-induced, microaggregation of FAP-FBN-Integrin complexes at the plasma membrane (Fig. 2A). Specifically, following FAP binding (1h at 37°C, determined to be optimal for FAP association with FBN without major incorporation into large FBN fibrilar structures, believed to be highly stable) the cells were chilled, washed and labeled with Ni2+:trisNTA-AlexaFluor647 probe. Either an anti-FAP polyclonal antibody (i.e., capable of crosslinking multiple FAP-FBN-Integrin complexes) or an irrelevant polyclonal antibody were also added to the cells and incubated for 45min at 10°C. Then, the cells were washed and incubated with serum-free DMEM at 37°C to promote clustering and internalization.
Using this approach, we observed FAP uptake in an antibody-specific manner (i.e., anti-HA polyclonal antibody was unable to promote FAP uptake) (Fig. 2C). As further confirmation of this effect, an anti-His6 monoclonal antibody also induced FAP uptake when crosslinked by a polyclonal secondary antibody (Suppl. Fig. 2). As expected, the internalization of FAP as a function of antibody concentration displayed a bell-shaped relationship (Fig. 3A). Specifically, we found that increasing the antibody dose was correlated with increased amounts of crosslinked, internalized FAP up to an optimal concentration. Above this antibody dose, however, saturation of the antigen with antibody would occur, such that the increasing proportion of 1:1 antigen:antibody complexes leads to decreased crosslinking and reduced levels of FAP internalization (Fig. 3A). These studies revealed a maximal amount of internalized FAP at 1:600–1:1800 dilutions (Fig. 3A); thus, a 1:1000 antibody dose was used in all subsequent experiments.
Fig. 3. Antibody-Dose Dependence and Kinetics of Antibody-induced FAP Internalization.
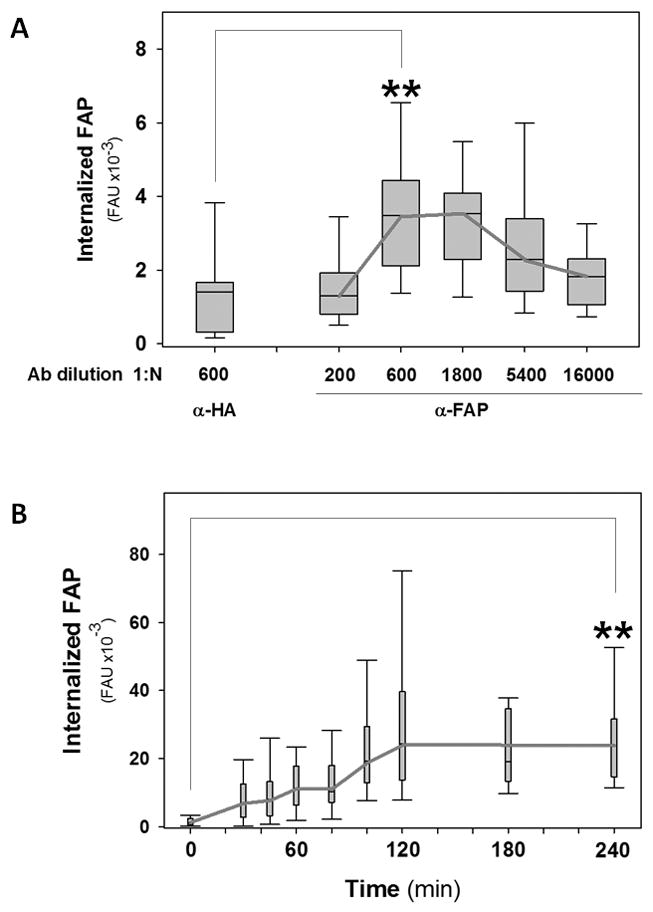
A. T24 bladder tumor cells were treated as described in Fig. 2C, but using either α-HA or α-FAP polyclonal antibodies at the indicated dilutions. FAP-associated fluorescence was quantified and results were represented as box-plots. B. Cells were treated as in Fig. 2C, but incubated for the indicated times. FAP-associated fluorescence was quantified. Statistical significance of the difference in FAP uptake between the indicated conditions was assessed using the Wilcoxon test (**: p<0.05). FAU: Fluorescence Arbitrary Units.
These results were confirmed using an independent approach. Specifically, uptake of FAP:Ni2+trisNTA-AlexaFluor647 by untransfected cells was tested using antibody-dependent microaggregation. In these experiments, cells were lifted by trypsinization (thus also eliminating non-internalized ligand) and the amount of endocytosed FAP was quantified by flow cytometry. In agreement with the results described above, this alternative approach showed FAP uptake above background levels only upon antibody-induced clustering (Suppl. Fig. 3A-B).
Next, we established the kinetics of antibody-induced FAP uptake (Fig. 3B). Our results indicate that internalized FAP started to accumulate in the Rab5Q79L-GFP positive compartment immediately following chase and reached a plateau after approximately 2h incubation (Fig. 3B). To ensure optimal FAP detection, unless indicated otherwise, we adopted a 3h-incubation scheme in our following experiments. Under these experimental conditions, T24 cells internalized 40–60% of the total cell-bound FAP.
FAP is Internalized by a Clathrin-independent, Caveolin-dependent Mechanism
We anticipated that the FAP-FBN-Integrin complex would be internalized by a mechanism similar to the one responsible for integrin uptake. However, both clathrin-dependent and –independent mechanisms have been proposed for internalization of integrin complexes23. Therefore, we first determined whether FAP uptake was dependent or independent of clathrin. Our results indicate that siRNA-mediated knock-down of clathrin did not affect FAP uptake, but it efficiently inhibited transferrin internalization (Fig. 4A). Furthermore, no colocalization was observed between cell-bound FAP and clathrin-coated areas (Fig. 4C). In contrast, (1) FAP showed strong colocalization with caveolae and (2) its uptake was inhibited by siRNA-mediated knock-down of Caveolin-1(Cav-1) (Fig. 4C). Suppl. Fig. 4 shows representative images of the knock-down cells used for the quantitations of FAP and transferrin uptake summarized in Fig. 4.
Fig. 4. FAP is Internalized by a Clathrin-independent, Caveolae-dependent Mechanism.

A. T24 bladder cells treated in the presence or absence of siRNA against either clathrin or caveolin-1 were tested for the ability to internalize FAP as in Fig. 2C. Transferrin- (“Tf”, used as a clathrin-dependent cargo control) and FAP-associated fluorescence was quantified. Results are expressed as a fraction with respect to the control (i.e., no siRNA: ”—“). Statistical significance of the difference in FAP uptake between the indicated conditions was assessed using the Wilcoxon test (**: p<0.05). B. Cells were treated with the indicated siRNAs (Cav1: Caveolin-1; Chc: Clathrin heavy chain). Samples were analyzed by Western blot with specific antibodies. Tubulin was used as a loading control. C. T24 cells were treated as in Fig. 2C, but chased after antibody binding for only 5min. Clustered FAP colocalization with clathrin and caveolae was analyzed by immunofluorescence. Numbered boxes (1–5) on the left show the location of the enlarged (4X) insets (right). Arrows point to some of the multiple areas of colocalization. Scale bar: 20microns
FAP is targeted to the lysosomal compartment
Next, we investigated the post-internalization fate of FAP. Specifically, in the absence of Rab5Q79L-GFP expression, we tested whether FAP was targeted to the lysosomal compartment following antibody-induced microaggregation. This was evaluated by assessing the extent of FAP colocalization with Lysosomal-Associated Membrane Protein 2 (Lamp2) in T24 cells pre-incubated with leupeptin to inhibit lysosomal degradation. Our results showed that FAP colocalized with Lamp2 (ccFAP=0.77±0.15, Fig. 5A-B) such that the maximal lysosomal accumulation occurred with an approximate 60min-delay relative to the internalization rate (Fig. 5C). In contrast, the transferrin-transferrin receptor complex, known to remain within non lysosomal compartments24, showed no significant colocalization with Lamp2 (ccTfr=0.13±0.08, Fig. 5A–B).
Fig. 5. FAP is Sorted to Lysosomes.

A. T24 cells were treated with the lysosomal inhibitor leupeptin and allowed to internalize Transferrin (Tfn)-AlexaFluor488 or FAP clustered with anti-FAP antibodies for 3h. Colocalization between Tfn-AlexaFluor488 or FAP and the lysosomal marker Lamp2 was revealed by immunofluorescence. Scale bar: 20microns. B. FAP accumulation within Lamp2 structures was quantified as a function of incubation time. Arrows point to some areas of colocalization.
Since integrin complexes are known to recycle back to the plasma membrane23, we also evaluated the contribution of different recycling pathways to the intracellular trafficking of FAP using an unbiased recycling quantitative assay25. Briefly, we allowed T24 cells (in the presence of lysosomal inhibitors) to undergo microaggregation-induced internalization of His6-FAP: Ni2+trisNTA-AlexaFluor647. Then, non-internalized Ni2+trisNTA-AlexaFluor647 probe was stripped and the cells were incubated at 37°C for different periods of time (chase). The time-course scheme was designed to encompass standard (i.e., 30min26) as well as longer times (up to 90min) to also cover the possibility of extremely slow recycling kinetics.
At each chase time-point, cells were trypsinized to produce a cell suspension and to eliminate signal from putative membrane recycled FAP; following immediate fixation the amount of cell-associated fluorescence was determined by FACS analysis. Recycling of FAP is expected to produce a decrease in the amount of cell-associated fluorescence relative to chase time=025. Our data showed that following internalization, the amount of intracellular FAP in these lysosome-inhibited cells remained approximately constant for at least 1.5h (Suppl. Fig. 3). This result indicated that FAP recycling is negligible. Similar results were obtained by monitoring internalized FAP with an anti-FAP antibody instead of the Ni2+trisNTA-AlexaFluor647 probe.
In addition, we quantified the intracellular accumulation of FAP upon overexpression of the dominant negative mutants of the GTPases that control different recycling pathways. Dominant negative versions of Rab4 or Rab11 did not exert any discernible effect on internalized FAP accumulation (data not shown). Expression of an ARF6 dominant negative mutant only produced very subtle effects on FAP trafficking (data not shown).
Taken together, our data indicate that internalized FAP was mainly targeted to the lysosome, with a negligible extent of recycling.
Multivalent FAP-ligated Liposomes Undergo FAP-mediated Internalization in T24 Bladder Tumor Cells
Since our results indicate that antibody-induced microaggregation substantially accelerated the uptake of FAP under bladder-like, serum-free conditions, we hypothesized that other FAP-FBN-integrin crosslinking strategies would also enhance FAP internalization. To test this concept, we incubated T24 cells with 50nm-liposome suspension bearing multiple Ni2+NTA His6-binding sites per nanoparticle as FAP-FBN-integrin crosslinker instead of antibody. We found that multivalent Ni2+NTA-liposomes, were internalized in the presence, but not in the absence of recombinant purified FAP (Fig. 6A-B). Importantly, FAP-Liposome uptake was impaired in Cav-1 knock-down cells (Fig. 6C). These results strongly support the concept of FAP as a targeting agent for nano-carrier based therapeutic approaches for bladder cancer treatment.
Fig. 6. FAP-mediated Internalization of Liposomes.

T24 cells (with or without Cav-1 siRNA treatment) expressing Rab5Q79L-GFP were incubated in the presence (+) or absence (−) of purified FAP for 1h at 37°C. Then, the coverslips were rinsed and labeled with Ni2+:trisNTA-CF633-liposomes at 10°C and shifted to 37°C for 3h. Internalized CF633-liposomes present in enlarged endosomes were observed (A: arrows) and quantified, with results represented as box-plots (B and C). Statistical significance of the difference in uptake between the indicated conditions was assessed using the Wilcoxon test (**: p<0.05).
DISCUSSION
This study establishes for the first time the endocytosis mechanism and intracellular trafficking route of purified FAP (fibronectin attachment protein from Bacillus Calmette-Guerin) in bladder tumor cells. Importantly, we developed a clustering-based strategy to accelerate the uptake of FAP in serum-free environments (e.g., the lumen of the bladder) and demonstrated its suitability to induce liposome internalization. Overall, our data indicate that FAP is a suitable agent for targeting bladder cancer cells.
This study shows that FAP can be used to successfully address two important challenges for bladder cancer therapeutics:
The presence of an unfavorable extracellular environment: low pH (<6)27 and constant dilution of the lumen content.
The relative stasis of bladder tumor cells toward internalizing lumen content.
FAP as a targeting agent for tumor bladder cells
Our results demonstrate that purified FAP can surmount the first of these challenges. Specifically, purified His6-FAP recognized FBN fibrils and, as expected, was found to colocalize with integrins as revealed by immunofluorescence with an anti-activated integrin antibody (Fig. 1). FAP association with cells was resistant to the low pH characteristic of patients’ urine27, to the ionic strength conditions imposed by the intravesical instillation buffer and the presence of human urine (Suppl. Fig. 1). Our experiments also showed that FAP binding was resistant to extreme dilutions. Indeed, Suppl. Fig. 1A suggests no decrease in the amount of cell-bound FAP following complete media replacement and incubation with 10ml of FAP-free pH 4.8 buffer for several hours. Although we have not explored the effect of mechanical stress on FAP binding (as during bladder’s filling/voiding cycles), it should be noted that in vivo T cell-dependent antitumor response to intravesical bladder instillation of purified FAP indicated that FAP was bound and internalized despite the adverse conditions of the bladder18.
We speculate that FAP targeting of bladder tumor cells is superior to other targeting ligands because it binds to FBN-integrin complexes rather than attempting to compete off integrin ligands that are already engaged in FBN complexes. Importantly, in addition to its targeting activity, FAP is uniquely advantageous because it is capable of eliciting the immune responses that are thought to play an important role in BCG’s therapeutic mechanism18.
FAP internalization
Since our results demonstrate that purified FAP was internalized by bladder tumor cells (Fig. 2C), we envision that it may be a suitable reagent for the development of intracellular delivery strategies for cytotoxic cargo. In view of this, a clustering-based approach was developed to overcome the slow endocytic kinetics of T24 bladder tumor cells (challenge #2, see above). The findings of this study establish the importance of multivalency as a clear determinant for controlling FAP endocytosis. In fact, either polyclonal anti-FAP antibodies or multivalent Ni2+NTA-liposomes were capable of undergo endocytosis upon FAP binding (Fig. 2–6).
Since the integrin-bound FBN fibrilar structures are massive (they can be a few microns long Fig. 1), we believe is highly unlikely that they can mediate FAP internalization via 60nm-Cav1 vesicles. Therefore, we speculate that the best candidates to mediate FAP uptake are: 1) FBN-Integrin complexes not yet incorporated into fibrils and 2) small-enough FBN fibril fragments (resulting from fibril turnover). Interestingly, the caveolae-dependent mechanism for FAP internalization reported in this study is similar to that previously described for FBN turnover in fibroblasts28. Our results showing routing of the FAP-FBN-integrin complexes to degradative compartments (Figs. 4–5) are also in agreement with a recent report describing the interaction of FBN-integrin complexes with the machinery responsible for protein sorting into late endosomal/lysosomal compartments29.
In summary, our data show that FAP follows the normal FBN-integrin complex trafficking route. The fact that the FAP-FBN-integrin complexes are resistant to pH lower than those typically found in endosomes (Supplemental Fig. 1A) is also consistent with this hypothesis.
Interestingly, our results also indicate that in contrast to the RhoA-dependent compensatory endocytosis mechanism described in umbrella cells30, T24 bladder tumor cells internalize FAP via a different clathrin-independent pathway. We hypothesize that tumor cells may regain common endocytic mechanisms absent or downregulated in highly-differentiated umbrella cells. An alternative possibility is that the FAP microaggregation strategy used here promotes the activation of an endocytic pathway that is not normally used in bladder cells. Further experiments will be needed to evaluate these possible explanations.
Supplementary Material
Acknowledgments
Financial Support was provided by the Purdue University Center for Cancer Research.
We thank to the members of the Aguilar, Ratliff and Thompson labs for useful discussions and critical reading of the manuscript and to Claudia B. Hanna for excellent technical assistance. This work was supported by the Purdue University Center for Cancer Research through funds provided by Indiana Elks charities.
Abbreviations
- BCG
Bacillus Calmette-Guerin
- FAP
fibronectin attachment protein
- PBS
Phosphate Buffer Saline
- FBN
fibronectin
Footnotes
Conflicts of Interest: None
References
- 1.Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 2.Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R. The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics. 2003;21:1315–30. doi: 10.1007/BF03262330. [DOI] [PubMed] [Google Scholar]
- 3.Romih R, Korosec P, de Mello W, Jr, Jezernik K. Differentiation of epithelial cells in the urinary tract. Cell Tissue Res. 2005;320:259–68. doi: 10.1007/s00441-004-1005-4. [DOI] [PubMed] [Google Scholar]
- 4.Truschel ST, Wang E, Ruiz WG, Leung SM, Rojas R, Lavelle J, Zeidel M, Stoffer D, Apodaca G. Stretch-regulated exocytosis/endocytosis in bladder umbrella cells. Mol Biol Cell. 2002;13:830–46. doi: 10.1091/mbc.01-09-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lilly JD, Parsons CL. Bladder surface glycosaminoglycans is a human epithelial permeability barrier. Surg Gynecol Obstet. 1990;171:493–6. [PubMed] [Google Scholar]
- 6.Liang FX, Riedel I, Deng FM, Zhou G, Xu C, Wu XR, Kong XP, Moll R, Sun TT. Organization of uroplakin subunits: transmembrane topology, pair formation and plaque composition. Biochem J. 2001;355:13–8. doi: 10.1042/0264-6021:3550013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ratliff TL. Role of the immune response in BCG for bladder cancer. Eur Urol. 1992;21 (Suppl 2):17–21. doi: 10.1159/000474916. [DOI] [PubMed] [Google Scholar]
- 8.Becich MJ, Carroll S, Ratliff TL. Internalization of bacille Calmette-Guerin by bladder tumor cells. J Urol. 1991;145:1316–24. doi: 10.1016/s0022-5347(17)38622-6. [DOI] [PubMed] [Google Scholar]
- 9.Brandau S, Suttmann H. Thirty years of BCG immunotherapy for non-muscle invasive bladder cancer: a success story with room for improvement. Biomed Pharmacother. 2007;61:299–305. doi: 10.1016/j.biopha.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Lamm DL, McGee WR, Hale K. Bladder cancer: current optimal intravesical treatment. Urol Nurs. 2005;25:323–6. 31–2. [PubMed] [Google Scholar]
- 11.Ratliff TL, McCarthy R, Telle WB, Brown EJ. Purification of a mycobacterial adhesin for fibronectin. Infect Immun. 1993;61:1889–94. doi: 10.1128/iai.61.5.1889-1894.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schorey JS, Holsti MA, Ratliff TL, Allen PM, Brown EJ. Characterization of the fibronectin-attachment protein of Mycobacterium avium reveals a fibronectin-binding motif conserved among mycobacteria. Mol Microbiol. 1996;21:321–9. doi: 10.1046/j.1365-2958.1996.6381353.x. [DOI] [PubMed] [Google Scholar]
- 13.Schorey JS, Li Q, McCourt DW, Bong-Mastek M, Clark-Curtiss JE, Ratliff TL, Brown EJ. A Mycobacterium leprae gene encoding a fibronectin binding protein is used for efficient invasion of epithelial cells and Schwann cells. Infect Immun. 1995;63:2652–7. doi: 10.1128/iai.63.7.2652-2657.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hudson MA, Brown EJ, Ritchey JK, Ratliff TL. Modulation of fibronectin-mediated Bacillus Calmette-Guerin attachment to murine bladder mucosa by drugs influencing the coagulation pathways. Cancer Res. 1991;51:3726–32. [PubMed] [Google Scholar]
- 15.Kavoussi LR, Brown EJ, Ritchey JK, Ratliff TL. Fibronectin-mediated Calmette-Guerin bacillus attachment to murine bladder mucosa. Requirement for the expression of an antitumor response. J Clin Invest. 1990;85:62–7. doi: 10.1172/JCI114434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuroda K, Brown EJ, Telle WB, Russell DG, Ratliff TL. Characterization of the internalization of bacillus Calmette-Guerin by human bladder tumor cells. J Clin Invest. 1993;91:69–76. doi: 10.1172/JCI116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W, Schorey JS, Groger R, Allen PM, Brown EJ, Ratliff TL. Characterization of the fibronectin binding motif for a unique mycobacterial fibronectin attachment protein, FAP. J Biol Chem. 1999;274:4521–6. doi: 10.1074/jbc.274.8.4521. [DOI] [PubMed] [Google Scholar]
- 18.Sinn HW, Elzey BD, Jensen RJ, Zhao X, Zhao W, Ratliff TL. The fibronectin attachment protein of bacillus Calmette-Guerin (BCG) mediates antitumor activity. Cancer Immunol Immunother. 2008;57:573–9. doi: 10.1007/s00262-007-0397-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coon BG, Mukherjee D, Hanna CB, Riese DJ, Lowe M, Aguilar RC. Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5b phosphatase. Human Molecular Genetics. 2009;18:4478–91. doi: 10.1093/hmg/ddp407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 21.McGraw TE, Subtil A. Endocytosis: biochemical analyses. Curr Protoc Cell Biol. 2001;Chapter 15(Unit 15):3. doi: 10.1002/0471143030.cb1503s03. [DOI] [PubMed] [Google Scholar]
- 22.Dinneen JL, Ceresa BP. Expression of dominant negative rab5 in HeLa cells regulates endocytic trafficking distal from the plasma membrane. Exp Cell Res. 2004;294:509–22. doi: 10.1016/j.yexcr.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 23.Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10:843–53. doi: 10.1038/nrm2799. [DOI] [PubMed] [Google Scholar]
- 24.Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiological Reviews. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 25.Naslavsky N, Rahajeng J, Sharma M, Jovic M, Caplan S. Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol Biol Cell. 2006;17:163–77. doi: 10.1091/mbc.E05-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts M, Barry S, Woods A, van der Sluijs P, Norman J. PDGF-regulated rab4-dependent recycling of alpha v beta 3 integrin from early endosomes is necessary for cell adhesion and spreading. Current Biology. 2001;11:1392–402. doi: 10.1016/s0960-9822(01)00442-0. [DOI] [PubMed] [Google Scholar]
- 27.Alguacil J, Pfeiffer RM, Moore LE, Del Fresno MR, Medina-Lopez R, Kogevinas M, Vermeulen R, Dosemeci M, Silverman DT, Rothman N, Garcia-Closas M. Measurement of urine pH for epidemiological studies on bladder cancer. Eur J Epidemiol. 2007;22:91–8. doi: 10.1007/s10654-006-9101-2. [DOI] [PubMed] [Google Scholar]
- 28.Shi F, Sottile J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J Cell Sci. 2008;121:2360–71. doi: 10.1242/jcs.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lobert VH, Brech A, Pedersen NM, Wesche J, Oppelt A, Malerod L, Stenmark H. Ubiquitination of alpha 5 beta 1 integrin controls fibroblast migration through lysosomal degradation of fibronectin-integrin complexes. Dev Cell. 19:148–59. doi: 10.1016/j.devcel.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 30.Khandelwal P, Ruiz WG, Apodaca G. Compensatory endocytosis in bladder umbrella cells occurs through an integrin-regulated and RhoA- and dynamin-dependent pathway. Embo J. 29:1961–75. doi: 10.1038/emboj.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.