

Abstract
The Human Papillomavirus (HPV) L2 capsid protein plays an essential role during the early stages of viral infection, but the molecular mechanisms underlying its mode of action remain obscure. Using a proteomic approach we have identified the adaptor protein, Sorting Nexin 17 (SNX17) as a strong interacting partner of HPV L2. This interaction occurs through a highly conserved SNX17 consensus binding motif, which is present in the majority of HPV L2 proteins analysed. Using mutants of L2 defective for SNX17 interaction, or siRNA ablation of SNX17 expression we demonstrate that the interaction between L2 and SNX17 is essential for viral infection. Furthermore, loss of the L2-SNX17 interaction results in enhanced turnover of the L2 protein and decreased stability of the viral capsids, and concomitantly there is a dramatic decrease in the efficiency with which viral genomes transit to the nucleus. Indeed, using a range of endosomal and lysosomal markers we show that capsids defective in their capacity to bind SNX17 transit much more rapidly to the lysosomal compartment. These results demonstrate that the L2-SNX17 interaction is essential for viral infection and facilitates the escape of the L2-DNA complex from the late endosomal/lysosomal compartments.
Keywords: HPV, L2, viral infection, sorting nexin, endosomes
INTRODUCTION
Human papillomaviruses (HPV) are a family of structurally similar non-enveloped double stranded DNA viruses, whose replication is strictly dependent on the terminal differentiation of the infected keratinocytes. They are the causative agents of a variety of proliferative lesions, with a subset of high-risk HPVs being capable of inducing malignant transformation and cancer. Infection with HPV-16 alone is responsible for approximately half of all the invasive cervical cancers in women worldwide (1).
HPV entry mechanisms into the host cell appear to be diverse and are still poorly understood. The viral capsid plays a key role in the infectious process and fulfils various roles that are critical for the establishment of a successful viral infection. The capsid comprises two structural proteins, the major component L1 and a more minor component, L2. The highly conserved L1 protein forms 72 pentamers, while up to 72 molecules of the minor capsid protein L2 are mostly located inside the capsid beneath the L1 pentamers (2). L1 can self-assemble into icosahedral virus-like particles that closely resemble the native structure of papillomavirus virions (3). Although L2 protein is dispensable for capsid formation, it is nonetheless critical for establishment of infection and has a number of other important non-structural functions at different stages of the viral life cycle (reviewed in 4).
There is still no consensus on the mechanisms of HPV virion entry nor the receptors involved in the process. It seems that much depends on the HPV type and the concentration at which the virions are used, as well as the particular cell type in which the assays are performed. Many HPV types appear to enter the cell via a clathrin-dependent endocytic mechanism, although different papillomavirus types seem to have evolved different entry strategies (5,6) or even take advantage of crosstalk between different routes of endocytosis (7,8). Prior to entry into basal cells, virions attach to the plasma and basement membrane via interaction of the major capsid protein, L1, with heparan sulfate moieties (9,10). This triggers conformational changes in the capsid and exposes L2, which is then N-terminally cleaved by secreted furin (11). The virions then bind to a still unknown secondary receptor for uptake (11,12). Internalisation of HPV-16 virions is thought to occur via clathrin-mediated endocytosis, with trafficking taking place through endosomal and lysosomal compartments (5,13,14). However, a recent study using non-keratinocyte cell lines demonstrated internalisation of HPV-16 through a clathrin/caveolin-independent pathway utilising tetraspanin-enriched microdomains as the entry platform (15). Regardless of the mode of entry, trafficking through the acidic compartments is thought to be necessary for HPV-16 and several other papillomavirus types, to allow effective virion uncoating and egress from the endosomes (7,13,16,17). Interestingly, a recent report showed trafficking of infectious HPV 16 virions from clathrin coated vesicles to caveolin-rich compartments, and further to the endosomal reticulum, bypassing late endosomal/lysosomal compartments (8). All these divergent reports suggest that many details remain to be clarified regarding the mode of endocytosis, intracellular trafficking and the vesicular compartments involved in uncoating.
Evidence suggests a critical role for the L2 protein in many of the above processes. L2 has been implicated in virion cell surface binding following the L2 N-terminal cleavage by furin (11) and it aids in the transport of the viral DNA to the nucleus. L2 is not essential for viral uncoating (18), but it is critical for the escape of viral DNA from the endosomal compartment, which appears to be facilitated by an L2 membrane destabilising motif (16) and γ-secretase (19). Furthermore, endosomal escape also seems to require L2 furin pre-cleavage despite this step occurring on the cell surface (20). The mechanisms by which L2 directs transport of the viral DNA to the nucleus, and by which the viral DNA enters the nucleus are also poorly understood. Interactions of the L2 protein with a microtubule motor protein dynein (21) and syntaxin 18 (22) have been reported, suggesting that syntaxin 18-positive vesicles and transport along microtubules might be involved in the trafficking of L2-DNA complexes to the nucleus. The L2 protein harbours two nuclear localisation signals, suggesting that L2 could facilitate nuclear import via nuclear pore complexes (23), however recent data indicates that nuclear envelope breakdown in early mitosis is required for efficient HPV infection (24), suggesting that the nuclear pore complex is not required for L2-DNA nuclear entry. It is generally accepted that L2 protein escorts the viral DNA to ND10 domains, where colocalisation with the major ND10 constituent, PML, has been observed (25). ND10 domains probably promote efficient early gene expression for the establishment of infection. Interestingly, L2 also plays a critical role in the late phases of the viral life cycle, aiding sequestration of the virion components during the assembly of progeny virions (26), and although virion production is not strictly L2-dependent, L2 appears to be crucial for efficient DNA encapsidation (6,27).
Given the importance of L2 in infection and the virus life cycle, surprisingly little is known of its non-structural functions or of the interactions with host cell proteins involved in these functions. We have recently shown that sumoylation regulates L2 stability and its interaction patterns, and that L2 can itself regulate the sumoylation status of the host cell (28). In this study we used a proteomic approach to search for novel interacting partners of L2. Protein mass spectroscopy revealed a strong interaction of L2 with the cellular adaptor protein sorting nexin 17 (SNX17). The sorting nexins (SNXs) are a family of proteins characterized by the presence of a phox-homology (PX) domain, which serves to localise SNXs to intracellular vesicles enriched in specific phosphatidylinositol phosphates (29). The family includes diverse cytoplasmic and membrane-associated proteins that are involved in endocytosis, endosomal trafficking and endosomal signalling (30). SNX17 has a PX domain followed by a protein 4.1, ezrin, radixin, and moesin (FERM)-like domain (31); it is localised to early endosomes and recycling tubules and is primarily involved in endosomal recycling. SNX 17 interacts with the members of low-density lipoprotein receptor (LDLR) family (32), P-selectin (33), amyloid precursor protein (34), cytosolic factors Krit1 (35) and Kif1B (36), and the scavenger receptor FEEL-1/stabilin-1 (37), modulating their internalisation or their recycling from endosomes to the cell surface or both.
In this study we show that the HPV-16 L2 protein interaction with SNX17 is crucial for HPV-16 infection. Our data suggest that SNX17 might be involved in the retention of HPV viral particles in the late endosomes or similar compartments, protecting them from lysosomal degradation, and suggests that the association between L2 and SNX17 contributes to the efficient egress of the L2-DNA complex from the late endosomes, prior to its transport to the nucleus.
RESULTS
Identification of SNX17 as a new interaction partner of HPV-16 L2 minor capsid protein
Since the non-structural roles of HPV L2 protein are poorly understood, we made an attempt to look for novel binding partners of L2 using protein mass spectroscopy. HEK293 cells were transfected with a plasmid expressing either FLAG-HA-tagged HPV-16 or HPV-11 L2 protein. Cell extracts were immunoprecipitated using anti-HA antibody-conjugated agarose beads and total immunoprecipitates were then subjected to mass spectrometric analysis. The resulting protein profiles were compared with those obtained from mock-transfected cells to exclude non-specific contaminants. As can be seen from Figure 1A, we observed a number of novel potential interacting partners of HPV-16 L2. Of these Sorting Nexin 17 (SNX17) was amongst the strongest hits, based upon the number of peptides (listed in Figure 1B) and because SNX17 is rather rarely found in proteomic analyses, according to the Global Proteome Machine (GMP) database (286 matches listed). SNX17 interacted strongly with both the HPV-16 and the HPV-11 L2 proteins, suggesting that this interaction could be a conserved feature of high-risk and low-risk HPV types. To further characterise the interaction, we performed a series of GST pulldown assays on HPV-16 L2 and SNX17 proteins expressed in HEK293 cells (Figures 1C and 1D). We confirmed that SNX17 interacts with HPV-16 L2 protein, but not with the major capsid protein, L1 (Figure 1D). Furthermore, binding between HPV-16 L2 and SNX17 was resistant to washing with 2% Triton X-100, indicating that the interaction is of high affinity (Figure 1E).
Figure 1. HPV-16 L2 protein interacts with SNX17 in vitro and in vivo.

(A) HEK293 cells were transfected with a plasmid expressing FLAG-HA-tagged HPV-16 L2. After 24 hr, extracts were immunoprecipitated using anti-HA antibody-conjugated agarose beads and the total immunoprecipitates were subjected to mass spectrometric analysis. The table shows a selection of the prominent HPV-16 L2-specific hits, together with their E-value (the base-10 log of the expectation that the assignment is stochastic), the number of unique peptide sequences (#) and total number of tandem mass spectra that can be assigned to each protein. (B) The list of unique SNX17 peptides found in the mass spectra. (C) Ectopically expressed wild type FLAG-HA-tagged HPV-16 L2 was extracted from HEK293 cells and subjected to pull-down assays with GST-SNX17 fusion protein. Bound proteins and cell extracts were analyzed by immunoblotting using anti-HA (HPV-16 L2) antibodies. (D) Endogenous SNX17 was extracted from HEK293 cells and subjected to pull-down assays with GST-HPV-16 L2 and GST-HPV-16 L1 fusion proteins. Bound proteins and cell extracts were analyzed by immunoblotting using anti-SNX17 antibodies. Arrows on the Ponceau-stained gel indicate the bands corresponding to the purified GST-fusion proteins used in the pull down-assay. (E) Endogenous SNX17 was extracted from HEK293 cells and subjected to pull-down assays with GST-HPV-16 L2. GST immunoprecipitates were then subjected to extensive washing with 0.5%, 1% or 2% Triton X-100 in PBS. Bound proteins and cell extracts were analysed by immunoblotting using an anti-SNX17 antibody. In all the GST binding experiments, non-fused GST proteins were used as a negative control. Inputs represent 10% of the extracts used for the pull-down assays. Asterisks (*) denote a non-specific band recognised by the anti-HA antibody. Numbers at the gel borders are molecular sizes in kilodaltons.
SNX17 usually associates with transmembrane cargo proteins by recognising an NPxY motif located in their cytosolic tails (33,34,37); analysis of the HPV-16 L2 protein revealed the sequence NPAY at a position 254-257. We also found that the NPAY/F motif is present in the L2 proteins from all the major HPV genera, with at least one motif present between residues 250 and 310 in all the L2 proteins analysed (Table S1), suggesting that this motif is evolutionarily highly conserved. To analyse whether the NPAY motif of the HPV-16 L2 protein is responsible for SNX17 binding, we made a single point mutation, N254A, and also changed the entire domain NPAY to AAAA. Binding of the HPV-16 L2 mutants to SNX17 was then examined by a GST pulldown (Figure 2A). As can be seen, substitution of all four residues with alanines (L2-254NPAY257AAA) almost completely abrogated binding to SNX17. However, even the single amino acid substitution of N254A markedly reduced the capacity of L2 to bind SNX17, demonstrating that the asparagine residue within the consensus recognition site is critical for this interaction, and this is in agreement with studies on other sorting nexin cargoes (33,38).
Figure 2. Mapping of the HPV-16 L2 – SNX17 interacting domains.

(A) Wild type HPV-16 L2 and two L2 mutants with mutations in the proposed SNX17 binding site (N254A and 254NPAY257AAAA) were transiently expressed in HEK293 cells. After 24 hr, proteins were extracted and subjected to a pull-down assay with GST-SNX17 fusion protein. Bound proteins and cell extracts were analyzed by immunoblotting using an anti-HA antibody. (B) Wild-type SNX17 and SNX17 truncated variants were in vitro translated and incubated with GST-HPV-16 L2 protein. Bound proteins and inputs were assessed by autoradiography and the input GST fusion proteins were visualised with Coomassie staining. In both sets of assays, non-fused GST proteins were used as a negative control and inputs corresponds to 10% of the protein that was used in the pull-down assays. Asterisks (*) denote a non-specific band recognised by the anti-HA antibody. Numbers at the gel borders are molecular sizes in kilodaltons.
To map the site of interaction on SNX17 we used truncated forms of the SNX17 protein (39), which were missing either the Phox domain (PX) or the C-terminal part of the SNX17 protein. SNX17 constructs were translated in vitro and incubated with GST-16 L2 fusion protein or with GST alone as a control. Results in Figure 2B show that in vitro-translated SNX17 interacts with GST-16 L2 and that deletion of the PX domain (construct SNX17 113-470) did not affect the interaction. This is in agreement with previous reports showing that the PX domain of SNX17 is not normally involved in the interaction with its protein partners (34,38). However, we detected no binding when the C-terminal part of SNX17 was deleted (construct SNX17 1-274), suggesting that the binding domain for HPV-16 L2 resides between residues 274-470 in the C-terminal part of SNX17.
Loss of SNX17 abolishes the infection with HPV-16 pseudovirions
Based on the function of SNX17 in endocytosis we hypothesised that the L2-SNX17 interaction might have a role in the HPV entry and trafficking processes. To investigate these aspects further, we employed HPV-16 pseudovirions (PsVs), which have been widely used for studies on HPV entry and trafficking in recent years. HPV-16 PsVs were generated, in which a luciferase reporter gene was encapsidated in HPV capsids composed of the L1 and L2 proteins. These purified PsVs were then used in infection assays, where delivery of the reporter plasmid into the nucleus was measured by the activity of the expressed firefly luciferase 48 hr post-infection. In all our infectivity assays, we routinely performed a control infection with PsVs that had been previously incubated with H16.V5 antibody, which binds to neutralising epitopes on HPV virions (40). Strong reduction of the infection with attachment-deficient PsVs showed that the luciferase activity observed in our experiments was from the luciferase construct encapsidated within PsVs and was not a result of transfection with free reporter DNA in the PsV preparations (Figure 3A).
Figure 3. Loss of SNX17 abolishes HPV-16 infection.

(A) HaCaT cells were transfected with siSNX17, siCTRL, different amounts of SNX17 expression plasmid or mock transfected. After 48 hr, cells were exposed to HPV-16 PsVs carrying a luciferase reporter plasmid. (B) Wild-type HaCaT and stable SNX17 knock-down HaCaT (shSNX17 HaCaT) cells were exposed to HPV-16 PsVs carrying a luciferase reporter plasmid. (C) HaCaT cells were exposed either to wild-type HPV-16 PsVs or HPV-16 PsVs with the mutated SNX17 binding site in L2 (L2 N254A mutant), both carrying a luciferase expression plasmid. In all experiments, cells were lysed 48 hr post-infection and the level of luciferase activity was evaluated in triplicate by luminometry. Obtained values were corrected for background luminescence and normalised to mock transfected HaCaT cells (A), wild type HaCaT cells (B) or HaCaT cells infected with wild type HPV-16 PsVs (C). Immunoblots show the level of SNX17 expression (AB) and input of PsVs used for the infection (C, lower panel). Alpha actinin was used as a loading control. The agarose gel in C (upper panel) shows the level of pGL3 plasmid carrying the luc+ gene extracted from PsVs used in the infectivity assay. Neutralisation assay using anti-capsid H16.V5 antibodies was performed in order to assess the level of luciferase activity due to the non-encapsidated luciferase plasmid DNA (A). Results are expressed as the means ± SD of at least three independent experiments, and the corresponding P values are: * P<0.05, **P<0.01, ***P<0.001
To understand the role of SNX17 in HPV-16 infection, we used three approaches. First, we transiently silenced SNX17 expression in immortalised human keratinocyte HaCaT cells with small interfering RNA (siRNA). Silencing of SNX17 expression resulted strikingly in up to 80% reduction in infectivity, while irrelevant control siRNA had no noticeable effect (Figure 3A, left panels). Furthermore, overexpression of SNX17 in HaCaT cells strongly enhanced the levels of infection (as determined by the levels of luciferase activity) in a dose-dependent manner (Figure 3A, right panels). To further verify a positive role for SNX17 in HPV infection, we also generated HaCaT cells that had SNX17 expression stably knocked-down using a short hairpin SNX17 RNA (shSNX17). Using these cells, we observed a greater reduction in SNX17 expression levels, and concomitantly, almost complete inhibition of HPV-16 infection (Figure 3B), with up to 95% decrease in luciferase activity in comparison with HPV-16 infection in normal HaCaT cells. To exclude the possibility that the decrease in infectivity is not due to either compromised endosomal trafficking, cell cycle progression (24) or cell viability upon SNX17 depletion, we performed a set of control experiments on the SNX17 knock-down cells. Figure S1 shows that depletion of SNX17 does not increase cytotoxicity (Figure S1A), or affect the cell cycle (Figure S1B) when compared with the control siRNA transfected cells. In addition, normal steady-state clathrin-mediated endocytosis is not adversely affected in the SNX17 depleted cells as can be seen by the levels of AlexaFluor 647-labeled transferrin internalised in shSNX17 HaCaT cells in comparison with the control cells (Figure S2).
To confirm that the effects of SNX17 loss on HPV-16 infectivity are related to its interaction with HPV-16 L2, we generated HPV-16 PsVs in which the L2 protein was mutated to the non-SNX17 binding mutant N254A. Western blot analysis of purified HPV-16 virions showed similar levels of wild type and L2-N254A HPV-16 L2, indicating that co-assembly of L1/L2 proteins into particles was not affected by the N254A substitution (Figure 3C). Likewise, extraction of DNA from the purified HPV-16 PsVs and visualisation of pGL3 vector carrying the luc+ gene by agarose gel electrophoresis confirmed that wild type and mutant PsVs contained similar amounts of encapsidated reporter luciferase plasmid (Figure 3C). We then performed an infectivity assay in HaCaT cells using the same amounts of wild type and L2 N254A PsVs. As seen in Figure 3C, the SNX17-binding deficient PsVs showed reduced infectivity in comparison with wild type PsVs, further confirming that the L2-SNX17 interaction plays an essential role in the HPV-16 infection process.
SNX17 affects the late phases of HPV-16 virion trafficking
SNX17 affects internalisation and/or intracellular trafficking of its protein binding partners (34,39,41). Based on our results showing that SNX17 is essential for efficient HPV-16 infection, we hypothesised that SNX17 would be involved in either entry and/or trafficking of HPV-16 virions. To address these questions, HPV-16 pseudovirions were labelled with amine-reactive AlexaFluor 488 dye (AF488 PsVs) and the cell uptake and intracellular trafficking were assessed by confocal microscopy. In this way, we can trace intracellular trafficking of HPV-16 capsids, although AF488 conjugation results primarily in the labeling of L1 protein due to its high copy numbers in the virion and its surface location. L2 protein is also labelled, but at a significantly lower level (42). Wild type and shSNX17 silenced HaCaT cells were exposed to wild type HPV-16 PsVs for 1 hr at 4°C to allow binding but not internalisation of PsVs. Cells were then washed and shifted to 37°C for internalisation and trafficking. At different time points, cells were fixed and immunostained either for the early endosomal marker EEA-1 (Figure 4) or the late endosomal/lysosomal marker LAMP-2 (Figure 5). In the case of EEA-1 at the 30 minute (Figure 4A) and 2.5 hr (Figure 4B) time points there is no significant difference in trafficking of HPV-16 PsVs whether SNX17 is present or not. Colocalisation of AF488-labeled PsVs with EEA-1 was 26 ± 9 % for wild type HaCaT and 30 ± 8 % for shSNX17 HaCaT (P = 0.37) at 30 min, and 23 ± 14 % for wild type HaCaT and 20 ±15 % for shSNX17 HaCaT (P = 0.78) at 2.5 hr. At both time points, HPV-16 PsVs signal partially overlapped with EEA-1 staining, although the general position of PsVs shifts from peripheral to more internal parts of the infected cells by 2.5 hr. This data suggest that SNX17 is neither involved in the internalisation nor in the early steps of HPV-16 trafficking through early endosomes. This was also confirmed by Western blot analysis of the internalised PsVs. Figure S3 shows similar levels of the internalised capsid protein L1 at 20, 40 and 90 min post-attachment in both the siSNX17-silenced and control HaCaT cells.
Figure 4. SNX17 does not affect HPV-16 trafficking to early endosomes.
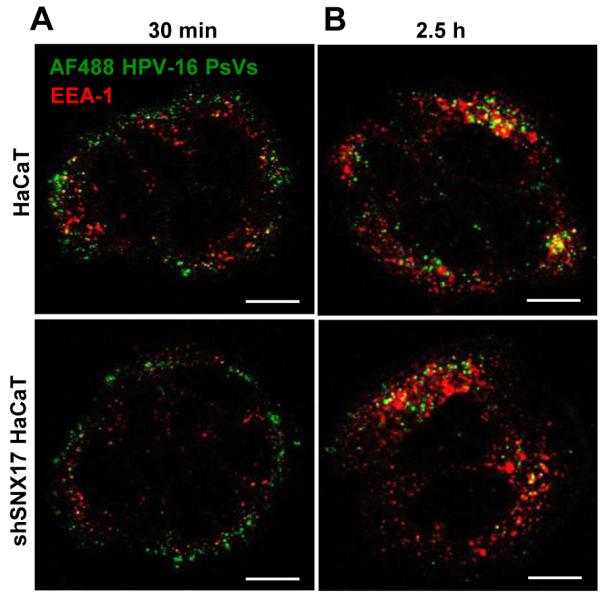
Wild-type HaCaT and stable SNX17 knock-down HaCaT (shSNX17 HaCaT) cells were exposed to AF488-labeled HPV-16 PsVs (in green) for 1 hr at 4°C. Cells were then washed and incubated at 37°C for 30 min (A) and 2.5 hr (B). After the incubation, cells were fixed and stained for endogenous EEA-1 (in red). The micrographs are representative and show the mid-cell body/nucleus focal planes. Scale bar = 10 μm.
Figure 5. SNX17 affects late phases of the HPV-16 trafficking.

Wild-type HaCaT and stable SNX17 knock-down HaCaT (shSNX17 HaCaT) cells were exposed to AF488-labeled HPV-16 PsVs (in green) for 1 hr at 4°C. Cells were then washed and incubated at 37°C for 30 min (A), 3 h (B), 8 h (C) and 16 h (D). (E) HaCaT cells were exposed to AF488-labeled HPV-16 PsVs wild type or HPV-16 L2-N254A (in green) and incubated at 37°C for 8 hr. After the incubation, cells were fixed and stained for endogenous LAMP-2 (in red). The micrographs are representative and show the mid-cell body/nucleus focal planes. Scale bar = 10 μm.
Cargo that traffics through early endosomes is typically transported to lysosomes, which has also been observed for HPV-16 virions (19,42). To investigate whether SNX17 affects this part of HPV-16 trafficking, the movement of AF488-labeled HPV-16 capsids was traced at different time points in HaCaT cells counterstained for LAMP-2. As expected, 30 minutes post-attachment no colocalisation was observed between HPV-16 PsVs and LAMP-2 (Figure 5A). At 3 hr post-attachment, we first detected the colocalisation of HPV-16 PsVs with LAMP-2, although the majority of the PsVs were still located outside the LAMP-2 staining areas (Figure 5B). At neither of these early time points did we notice any major difference between wild type and shSNX17 HaCaT cells. By eight hours post-attachment there was a degree of colocalisation between AF488-labeled PsVs and LAMP-2. However, as can be seen from Figure 5C, there are marked differences between wild type HaCaT cells and the shSNX17 cells. In the wild type cells substantial amounts of PsVs are found distributed throughout the cytoplasm outside the LAMP-2 staining areas (28 ± 10% colocalisation of AF488-labeled PsVs with LAMP-2). In contrast, in the SNX17-depleted cells the majority of the PsVs are colocalised with LAMP-2 (65 ± 9 %), showing a highly significant statistical difference of P<0,001. Only by 15 hr post-attachment are similar levels of HPV-16 PsVs co-localising with LAMP-2 in the wild type HaCaT (Figure 5D) as those seen in the SNX17 silenced cells at 8hr (59 ± 14 % in wild type HaCat vs. 73 ± 13 % in shSNX17 HaCaT at 16 hr; P = 0.2).
Monitoring of viral trafficking with LAMP-2 counterstaining was also repeated in HaCaT cells infected with the wild type and SNX17-binding deficient N254A mutant PsVs. The results of the co-staining are shown in Figure 5E. Again, as seen in the SNX17 knock-down cells, by the 8 hr time-point, the mutant PsVs show a high degree of co-localisation with LAMP-2 (63 ± 14 %). This contrasts with only a very low level of LAMP-2 co-localisation with the wild type PsVs (27 ± 11 %). These results again confirm statistically significant differences (P = 0.0041) in the trafficking of HPV-16 PsVs if they are compromised in their ability to interact with SNX17, and indicate a more rapid progress of the PsVs towards the lysosomal compartments when SNX17 interaction is not possible. Taken together, these trafficking data demonstrate that although the early steps of HPV-16 trafficking are most likely not affected by SNX17, HPV-16 virions in SNX17-silenced cells proceed to lysosomes at a considerably faster rate in comparison with wild type SNX17-expressing cells.
Interaction of HPV-16 L2 protein with SNX17 is required for transport of viral DNA to the nucleus
The trafficking experiments described above suggested that SNX17 influences the rate at which HPV-16 virions travel toward lysosomes. To investigate whether this could affect virion disassembly and the efficient egress of L2-DNA complexes from endosomal compartments, we traced the trafficking of the reporter DNA encapsidated into the HPV-16 virions. To this end, we labeled reporter plasmid DNA with a thymidine analog, 5-ethynyl-2′-deoxyuridine (EdU), during pseudovirion production. EdU-labeled wild type and SNX17-binding deficient (L2-N254A) HPV-16 PsVs were then used for the infection of HaCaT cells. Infected cells were fixed at 16 and 22 hr post-attachment and EdU was detected in an azide-alkyne reaction. Cells were then counterstained for PML, a major constituent of ND10 domains, where the HPV genomes are typically found in an established HPV infection (25). EdU staining demonstrated that EdU-labeled DNA encapsidated by wild type HPV-16 pseudovirions was detected in the nucleus after 16 hr, adjacent to PML at ND10 domains (white arrows), as well as in the perinuclear region (Figure 6A). This observation is consistent with previous reports using BrdU-labeled HPV pseudogenomes (19,20). However, when EdU-labeled reporter DNA was encapsidated in the mutant HPV-16 PsVs deficient for SNX17 binding (PsVs L2-N254A), EdU-stained DNA was undetectable in the nucleus, with no PML colocalisation detected, although we observed the normal distribution of EdU-labeled DNA in the perinuclear region. To confirm that our observation does not reflect a simple delay in the kinetics of nuclear delivery, we examined the localisation of the EdU-labeled reporter DNA at a later time-point (22 hr). Similar patterns with no major changes in the localisation of EdU-labeled DNA, as compared with 16 hr, were obtained both for wild type and L2-N254A mutant PsVs (Figure 6B). These data indicate that binding of HPV-16 L2 protein to SNX17 is crucial for the nuclear entry of HPV viral DNA, probably affecting the efficient egress of L2-viral DNA complexes from endosomal compartments.
Figure 6. SNX17 is involved in transfer of viral DNA into the nucleus.
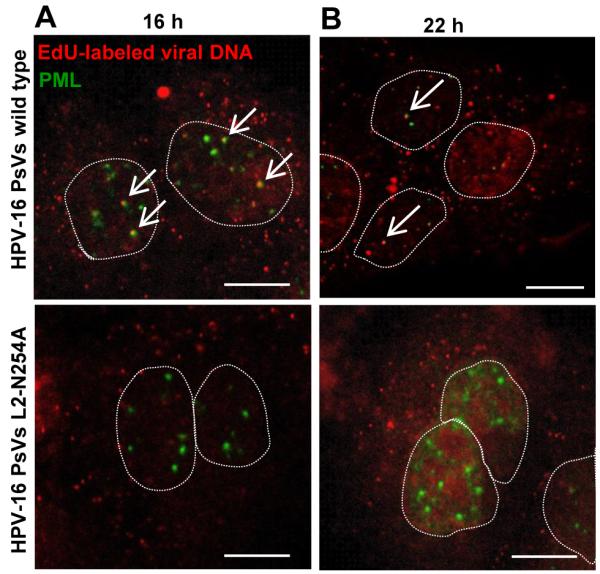
Wild type HPV-16 PsVs and HPV-16 PsVs with the mutation in the proposed SNX17 binding site (L2 N254A mutant) were generated in the presence of 5-ethynyl-2′-deoxyuridine (EdU). HaCaT cells were exposed to wild type or mutant PsVs for 1 hr at 4°C. Cells were then washed and incubated at 37°C for 16 hr (A) or 22 hr (B). After the incubation, cells were fixed and processed first for the detection of EdU-labeled DNA (in red) and then stained with anti-PML antibodies (in green). The micrographs are representative and show the mid-cell body/nucleus focal planes. Nuclei are illustrated by dotted lines. Arrows show colocalisation of EdU-labeled DNA and PML staining. Scale bar = 10 μm.
SNX17 increases the stability of the HPV-16 L2 protein
One of the common mechanisms by which SNX17 exerts its functions is to delay trafficking of its protein binding partners through the endocytotic compartments, thus reducing their lysosomal degradation (33,38,41). In the absence of SNX17, we observed a rapid progress of HPV-16 PsVs to lysosomes, as well as diminished nuclear entry of the encapsidated DNA. We hypothesised that fast lysosomal degradation of internalised PsVs could prevent HPV-16 virion disassembly and the egress of L2-viral DNA, which needs to precede lysosomal degradation (42). For this reason, we sought to determine if SNX17 affects the stability of HPV-16 PsVs. To do this, wild type and shSNX17 silenced HaCaT cells were exposed to HPV-16 PsVs for 1 h at 4°C. Cells were then washed and shifted to 37°C for internalisation and trafficking. At various times post-attachment, the cells were trypsinised and the amounts of the internalised L1 and L2 capsid proteins were assessed by Western blotting (Figure 7A). As can be seen, the internalised L1 and L2 decreased at fairly similar rates throughout the infection. In the shSNX17 HaCaT cells a similar pattern of gradual capsid protein degradation was obtained, however the degradation rate of both L1 and L2 proteins was noticeably faster, suggesting that one of the functions of the L2-SNX17 interaction is a stabilisation of the virion, prior to virion disassembly.
Figure 7. SNX17 increases stability of HPV-16 L2 protein.

(A) Wild-type HaCaT and stable SNX17 knock-down HaCaT (shSNX17 HaCaT) cells were exposed to HPV-16 PsVs for 1 hr at 4°C. Cells were then washed and incubated at 37°C for the indicated periods of time. Cell lysates were subjected to immunoblotting using anti-L1 and anti-L2 antibodies. Zero time points represent 30% of the total sample. Alpha actinin was used as a loading control. (B) HEK293 cells were transfected with siSNX17, siCTRL or mock transfected. After 48 hr, cells were additionally transfected with HPV-16 L2 protein alone or in combination with SNX17 (+SNX17) and were grown for another 24 hr before harvesting. Cell lysates were assessed using anti-HA (HPV-16 L2) and anti-SNX17 antibodies. Beta-galactosidase was used as an internal control for monitoring the transfection efficiency. Asterisks (*) denote a non-specific band recognised by anti-HA antibody. (C) HEK293 cells were transiently transfected either with wild-type HPV-16 L2 or with HPV-16 L2 constructs carrying mutations in the proposed SNX17 binding site (L2 N254A and L2 254NPAY257AAAA). After 24 hr, cells were washed and treated with cycloheximide (CHX) for 3 hr, 6 hr, or 9 hr. Cell lysates were subjected to immunoblotting using an anti-HA antibody. The level of L2 proteins was quantitated by densitometry, corrected for differences in transfection efficiency (expression of β-galactosidase) and normalised to α-globulin as an internal loading control. Results are expressed as the means ± SD of four independent experiments. Protein half lives represent values determined from the linear regression curves.
Since L2 has other non-structural roles within the virally infected cells, we also sought to investigate whether the L2-SNX17 interaction could likewise affect L2 levels when it is not associated with viral capsids in a non-lysosomal dependent manner. To do this, HEK293 cells were transfected with HA-FLAG-tagged HPV-16 L2 either alone or in combination with an SNX17 expression plasmid or into cells that had been previously transfected with a SNX17 siRNA. Cells were extracted and the amount of L2 protein present was analysed by Western blot using anti-HA antibody. Figure 7B shows that the level of HPV-16 L2 protein in the transfected cells strongly reflects the levels of SNX17. Thus, in cells where SNX17 expression was silenced, the levels of L2 expression are also lower. Conversely, where SNX17 is overexpressed, the levels of L2 expression are also increased. To investigate how much this effect is directly related to the interaction between SNX17 and HPV-16 L2, the stability of the SNX17-binding deficient mutants of HPV-16 L2 was determined. HEK293 cells were transfected with wild type L2 and two L2 mutants (L2-254NPAY257AAA and L2-N254A respectively) and the cells were then treated with cycloheximide (CHX) to stop further protein synthesis. Cells were harvested at various times, the levels of L2 were analysed by Western blot, and the results from multiple assays were quantitated by densitometry. As can be seen in Figure 7C, the wild-type L2 had a significantly slower rate of turnover than the two SNX17-binding deficient L2 mutants. Interestingly, the N254A mutant, which retains a higher level of residual SNX17 binding capacity (Figure 2A), has a higher stability than the mutant (254NPAY257AAA) that is completely defective in its ability to bind SNX17. Taken together these results demonstrate that the interaction between L2 and SNX17 can directly contribute to the stabilisation of the L2 protein, both in the context of viral capsids and lysosomal turnover, and also in a capsid independent manner, most likely through protection of L2 from proteasome turnover.
DISCUSSION
Given the crucial role of the HPV L2 protein in virus infection, there is still comparatively little known about its non-structural functions and the identities of its host cell interacting partners. In this study, we present several lines of evidence showing that the cellular adaptor protein Sorting Nexin 17 (SNX17) is a critical component of the HPV-16 entry pathway, through its association with the L2 capsid protein. SNX17 typically associates with transmembrane cargo proteins by recognising an NPxY motif in their cytosolic tails (33,34,37). Within HPV-16 L2 the motif NPAY resides at residues 254-257, and this was indeed confirmed as being the binding site on L2 for SNX17 recognition. We also identified SNX17 as an interacting partner of HPV-11 L2 in the proteomic screen and, consistent with this, the very similar sequence NPVY resides on HPV-11 L2 at residues 250-254. Furthermore this motif appears to be highly conserved amongst different HPV types and different PV genera, although variation occurs in the presence of Y or F at the 4th position, and in the number and the exact location of the motifs in this region of L2. Taken together, this suggests that the interaction between PV L2 and SNX17 is evolutionarily conserved across a wide range of different PV types, further highlighting the fundamental importance of this interaction for the viral life cycle. Obviously further studies are required to extend these analyses to these different PV types in order to determine if binding to SNX17 is indeed a common feature of PV L2 proteins.
It remains to be determined how exactly SNX17 interacts with HPV-16 L2, which is normally buried deep inside the HPV capsid. Most of the known L2 binding domains are located in the N-terminal part of the protein, which becomes exposed after capsid conformational changes and furin cleavage. Whether this SNX17 binding region on L2 is exposed at these times is currently not known, however, the C-terminal region of L2 has been found to be involved in late endosomal membrane integration and destabilisation (16). This suggests that the interaction of L2 with SNX17 most likely occurs at times in infection when L2 is more readily accessible, either prior to viral encapsidation or following partial viral uncoating. Likewise, a recently described transmembrane domain towards the N-terminus of L2 (Bronnimann et al., manuscript in preparation) may provide a mechanism by which L2 could span the endosomal membrane to interact with cytosolic trafficking factors like SNX17. Again future studies will aim to address these issues.
Previous reports have shown that the C-terminus of SNX17 contains a canonical F3 module of the FERM-like domain, which has structural similarity to a phosphotyrosine-binding (PTB) domain (31). PTB domains recognise a variety of peptide signals including NPxY (43) and we mapped the location of the L2 interacting domain on SNX17 to this C-terminal region spanning residues 274-470, where the F3 module is located, indicating that the PTB domain of SNX17 is most likely involved in HPV-16 L2 binding. So far no tyrosine phosphorylation of L2 protein has been reported, and consistent with this, it has also been shown that SNX17 binds preferentially to non-phosphorylated ligands (43).
To investigate the requirement for SNX17 during viral infection we used HPV-16 L2 PsVs harbouring a luciferase reporter construct. We observed that both transient and stable silencing of SNX17 in HaCaT cells dramatically reduced viral infectivity, whilst ectopically expressed SNX17 enhanced viral infectivity in a dose-dependent manner. To exclude the possibility that SNX17 depletion simply impedes normal endosomal trafficking, we performed infection assays with PsVs harbouring a mutant L2 defective for SNX17 binding. Again, a great decrease in infectivity was obtained. Taken together, these results demonstrate that the L2-SNX17 interaction is essential for HPV-16 infection.
SNX17 affects recycling of its binding partners either by increasing internalisation or by decreasing lysosomal degradation (33,37,38,41). We found no differences in the rate with which HPV-16 virions associated with EEA-1-positive early endosomal compartments, regardless of the capacity to associate with SNX17, suggesting that SNX17 does not play a role in viral entry or localisation to early endosomes. However, in the absence of an ability to bind SNX17, HPV-16 PsVs showed a dramatically increased rate of progression towards the LAMP-2 positive lysosomal compartments. Delayed delivery to lysosomes and decreased degradation has already been reported for the SNX17 binding partner P-selectin (41). Therefore it seems that SNX17 mediates the retention of HPV virions in late endosomes, preventing their rapid lysosomal degradation and thereby allowing L2-DNA complexes to egress from the endosomes. Indeed, by following the fate of the internalised PsVs capsid components we observed increased turnover of both the L1 and L2 proteins in SNX17-silenced cells (Figure 7C). This suggests that the L2-SNX17 interaction serves to stabilise the partially disassembled capsids, allowing time for L2-DNA egress. Indeed, previous studies have shown that failure of L2-DNA complexes to exit endo/lysosomal compartments results in lysosomal degradation and loss of HPV infectivity (19,20,42). Consistent with this, our experiments using EdU-labeled DNA showed that PsV DNA does not reach PML bodies when they are compromised for their ability to bind SNX17.
In summary, we have shown that the L2-SNX17 interaction plays a critical role in at least one step of the viral infection entry process: the maintenance of viral capsids in the endosomal compartment and their protection from premature lysosomal degradation. However it is also tempting to speculate that the L2-SNX17 interaction plays a role in aiding the L2-DNA complex to egress from the endosomal compartment, since we found no evidence of viral DNA reaching nuclear ND10s when the L2-SNX17 interaction was compromised. Clearly further studies are required to fully dissect the overall role that SNX17 plays in HPV infectious entry.
Materials and methods
Plasmids
FLAG-HA-tagged HPV-11 and HPV-16 plasmids were described previously (28). The SNX17 expression plasmid SNX17-GFP (44) was a kind gift from Daniel F. Cutler (University College London, UK). SNX17-FLAG expression plasmids were described previously (39) and were kindly provided by Ann K. Soutar (Imperial College of Medicine, London, UK). pXULL, coexpressing both codon-optimized HPV16 L1 and L2, was generously provided by John Schiller (National Institutes of Health, Bethesda, US). pGL3 construct encoding luc+ gene was purchased from Promega. For expression as glutathione S-transferase (GST) fusion proteins, HPV-16 L2, HPV-16 L1 and SNX17 were cloned into pGEX2T vector. The HA-FLAG-tagged HPV-16 L2s with the mutations in the SNX17 binding site (HPV-16 L2-N254A and 254NPAY255AAAA) were generated using the Gene Tailor Mutagenesis Kit (Invitrogen), while N254A substitution in the context of pXULL plasmid (PsVs L2-N254A) was done using QuikChange XL site-directed mutagenesis kit (Stratagene). The mutants were confirmed by DNA sequencing.
Antibodies
Mouse antibodies against HPV-16 L2 (16.D4 64-81) were generated and generously provided by Martin Müller (German Cancer Research Center, Heidelberg, Germany). The following antibodies were also used: anti-SNX17 (H-10, Santa Cruz), anti-EEA-1 (N-19, Santa Cruz), anti-PML (H-238, Santa Cruz), anti-LAMP-2 (H4B4, Abcam), anti-L1 (CamVir 1, Abcam), anti-HA (12CA5, Roche), anti-β-galactosidase (Z3781, Promega), anti-α-actinin (H-300, Santa Cruz), and appropriate secondary antibodies conjugated to horseradish peroxidase (HRP; Dako), fluorescein, or rhodamine (Molecular Probes). We gratefully thank Neil D. Christensen (Pennsylvania State University College of Medicine, USA) for providing H16.V5 antibody used for PsVs neutralisation assays (40).
Cells and transfections
HEK293 (human embryonic kidney cells), HaCaT (spontaneously immortalised human keratinocyte cells) and 293TT cells (45) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin–streptomycin (100U/ml) and glutamine (300μg/ml). For stable knockdown, HaCaT cells were transfected with a pool of short hairpin (sh) RNA against SNX17 (HuSh 29mer shRNA constructs against SNX17, Locus ID = 9784, OriGene) using Lipofectamine 2000 (Invitrogen). Cells were maintained in medium containing 500 μg/ml of puromycin. For transient small interfering RNA (siRNA) experiments, cells were transfected using Lipofectamine 2000 (Invitrogen) with siRNA against S N X 1 7 (ON-TARGETplus SMARTpool, 5′CAUGCAAGCUGUUCGGCAA, 5′AUGCGAUGCUGGCGGGUCA, 5′CUUUAUGCUCAGACGGUAU, 5′CCAGUGAUGUCCACGGCAA, Dharmacon) and siRNA against HPV-18 E6/E7 (5′CAUUUACCAGCCCGACGAG, Dharmacon) as a control. When applicable, cells were transfected with DNA constructs using calcium phosphate (CaPO4) precipitation ((46); HEK293 and 293TT) or Lipofectamine 2000 (HaCaT), according to the manufacturer’s protocol.
Protein mass spectrometry
HEK293 cells were transfected with FLAG-HA-tagged HPV-11 and HPV-16 L2 expression plasmids. After 24 hr cells were extracted in mass spectrometry lysis buffer (50 mM HEPES, pH 7.4 [at 4°C], 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 0.25% NP-40), and extracts were incubated with EZview Red Anti-HA Affinity Gel beads (Sigma) for 2 to 3 hr on a rotating wheel at 4°C. The beads were then extensively washed and subjected to proteomic analysis as already described (47). Briefly, proteins were eluted directly from the affinity beads using 50 ng of sequencing grade trypsin (Promega) in 20mM diammonium phosphate pH 8.0, for 6 h at 37 1C. The supernatant was removed from the beads and the cysteines were reduced and alkylated by boiling for 2 min in the presence of 10mM Tris(2-carboxyethyl)phosphine (Pierce) followed by incubating with 20mM acetaminophen (Sigma) for 1 h at 37 1C. The reactions were stopped by the addition of acetic acid (Sigma) to 0.1%. The resulting mixture was desalted using C18 Ziptips (Millipore) and lyophylized to dryness.
Nanobore columns were constructed using Picofrit columns (NewObjective) packed with 15 cm of 1.8mm Zorbax XDB C18 particles using a homemade highpressure column loader. The desalted samples were injected onto the nanobore column in buffer A (10% methanol/0.1% formic acid) and the column was developed with a discontinuous gradient and sprayed directly into the orifice of an LTQ ion trap mass spectrometer (Thermo Electron). A cycle of one full scan (400–1700 m/z) followed by eight data-dependent MS/MS scans at 25% normalized collision energy was performed throughout the LC separation.
RAW files from the LTQ were converted to mzXML files by READW (version 1.6) and searched against the Ensembl human protein database and the NCBInr Viral database using the Global Proteome Machine interfaced to the XTandem algorithm (version 2006.06.01.2).
Fusion protein purification and binding assays
GST-tagged fusion proteins were expressed and purified as described previously (48). SNX17-FLAG proteins were translated in vitro using a Promega TNT kit and radiolabeled with [35S]methionine (Perkin Elmer). Equal amounts of in vitro translated proteins were added to GST fusion proteins bound to glutathione agarose (Sigma) and incubated for 1 hr at 4°C. After extensive washing with PBS containing 1% Triton-X100, the bound proteins were analyzed by SDS-PAGE and autoradiography.
GST pulldowns using cellular extracts were performed by incubating GST fusion proteins immobilized on glutathione agarose with cells extracted in E1A buffer (25 mM HEPES, pH 7.0, 0.1% NP-40, 150 mM NaCl, plus protease inhibitor cocktail set I (Calbiochem) for 1 hr at 4°C on a rotating wheel. After extensive washing in binding buffer, or with increasing concentrations of Triton X-100 as indicated in the text, the bound proteins were detected using SDS-PAGE and Western blotting
Pseudovirion production and labeling
Luciferase reporter transducing HPV-16 pseudovirions (PsVs) were generated in 293TT cells as previously described (14,45). Purity and L1 protein content were determined by SDS-PAGE and Coomassie Brilliant Blue staining against bovine serum albumin (BSA) standards. To assess the amount of encapsidated DNA, purified PsVs preparations were incubated with DNA extraction buffer (20 mM Tris-pH8, 20 mM DTT, 20 mM EDTA, 0.2% SDS and 0.2% Proteinase K) for 15 min at 50°C and the DNA then analysed by agarose gel electrophoresis.
For PsVs fluorophore labeling, freeze/thawed lysates were cleared by centrifugation and protein concentration was determined using Bio Rad Protein assay (Bio Rad). Lysates were diluted with water to 4-6 mg/ml and 1M sodium bicarbonate (pH 8.4) was added at 10%. 500-1000 μl of lysate was labeled with 300 μg of AlexaFluor 488 (AF488) carboxylic acid succinimidyl ester (Molecular Probes) in the dark for 60 min at room temperature with constant stirring. After the conjugation reaction, pH neutralisation was obtained by adding 1M NaPO4 (pH 6.5). Further purification and concentration of AF488-labeled PsVs was performed as for non-labeled PsVs. Pseudovirions that contained packaged 5-ethynyl-2′-deoxyuridine (EdU)-labeled plasmid were prepared by addition of 25 μM EdU to the 293TT cells at 12 hr post-transfection. Harvesting and purification of EdU-labeled PsVs were performed using the procedure described above. AF488-labeled PsVs and EdU-labeled PsVs were stored at −80°C in HSB.
Infectivity assays
HaCaT cells were seeded in a 12-well plate at a density 0.5 × 105/well. After adherence, HPV-16 pseudovirions were added at a concentration 12 ng/ml. Infection was monitored at 48 hr by luminometric analysis of firefly luciferase activity using Luciferase Assay System kit (Promega). When HaCaT cells were transfected with siRNA or plasmid DNA prior to infection, HaCaT cells were seeded at a density 0.3 × 105/well and infected 48 hr post-transfection. Neutralisation assays were performed by incubation of PsVs with neutralising antibody H16.V5 at the final concentration 1:4000 for 1 hr at 4°C. Neutralised PsVs were then added to HaCaT cells and infectivity was determined 48 hr later as described above.
Pseudovirion trafficking assay
HaCaT cells were seeded on sterile glass coverslips at a density of 0.75 × 105/well and grown overnight. Cells were prechilled to 4°C and 200–300 ng of AF488-labeled PsVs were bound to cells for 1 hr at 4°C with agitation to allow viral attachment. Cells were then washed and transferred to 37°C. At different time points, cells were washed with PBS and fixed in PBS + 3.7 % paraformaldehyde for 15 min at room temperature. After washing in PBS, the cells were permeabilized in PBS/0.1 % Triton for 5 min, washed extensively with PBS and incubated in anti-EEA-1 (Santa Cruz, 1:100) or anti-LAMP-2 (Calbiochem, 1:100) for 1 hr at 37°C. Cells were washed and incubated in AF548-labeled secondary antibodies (Molecular Probes, 1:700) diluted in PBS for 1 hr at 37°C.
For detection of EdU-labeled reporter DNA, cells were exposed to EdU-labeled PsVs as described above. At various times post-attachment, cells were washed with PBS and fixed in PBS + 3.7 % paraformaldehyde. After washing in PBS/3 % BSA, the cells were permeabilized in PBS/0.5 % Triton for 20 min. The cells were then washed in PBS/3 % BSA and incubated with freshly prepared Click-iT reaction cocktail (Clik-iT EdU Imaging Kit, Molecular Probes) for 30 min at room temperature to detect EdU-labeled DNA. Cells were again washed in PBS/3 % BSA and subjected to immunofluoresce using anti-PML primary antibodies (Santa Cruz, 1:100) and AF488-conjugated secondary antibodies (Molecular Probes, 1:700). Cells were then counterstained with DAPI, washed in water and mounted on glass slides. Slides were visualized using a Zeiss Axiovert 100 M microscope (Zeiss) attached to a LSM 510 confocal unit.
Colocalisation analysis was performed using ImageJ software (49) with JACoP plug-in (50). Percentages of PsVs colocalising with either EEA-1 or LAMP-2 were determined with Mander’s Colocalization coefficients for two channels. Statistical analysis was performed using GraphPad Prism 5 software. Results are presented as means ± SD of at least 30 cells per condition.
In order to quantify the uptake of PsVs, HaCaT cells were transfected with siSNX17 or control siRNA for 72 hr. Attachment at 4°C and internalisation at 37°C were performed as described above. To assess internalised capsid protein levels, cells were trypsinised, extensively washed and then lysed in SDS buffer. Cell lysates were analysed by Western blotting using anti-L1 (Abcam, 1:5000) and anti-SNX17 (Santa Cruz) antibodies. Alpha-actinin was used as an internal loading control.
Protein stability assays
HEK293 cells were transfected with wild type HPV-16 L2 and two SNX17-deficient L2 mutants and cotransfected with β-galactosidase-expresion plasmid. 24 hr later, cells were incubated for different times with cycloheximide (50 μg/ml in dimethyl sulfoxide [DMSO]) to block protein synthesis. DMSO-treated cells were used as a control. Total cellular extracts were analyzed by Western blotting, and the band intensities were analysed using ImageJ software. Statistical analysis of four independent experiments was performed using GraphPad Prism 5 software.
For the measurements of capsid protein levels during infection, HaCaT cells were seeded in a 12-well plate at a density 0.75 × 105/well and grown overnight. Cells were prechilled to 4°C and 200–300 ng of PsVs were bound to cells for 1 hr at 4°C with agitation. Cells were then washed to remove unbound PsVs and either lysed in SDS buffer (0 time point) or replaced with fresh medium and incubated at 37°C for the indicated periods of time. To assess internalised capsid protein levels, cells were first trypsinised, washed and then lysed in SDS buffer. Cell lysates were analysed by Western blotting using anti-L1 (Abcam) and anti-L2 antibodies (16.D4 64-81). α-actinin was used as an internal loading control.
Cytotoxicity and cell cycle assays
HaCaT cells were transfected with siSNX17, control siRNA or left untreated. After 96 hours, cells were trypsinised and separately processed for cytotoxicity and cell cycle analyses. Citotoxicity was evaluated by FACS analysis of apoptosis using Annexin-V-FLUOS (Roche Diagnostics) and propidium iodide staining, following the manufacturer’s instructions. Cell cycle analysis was done by measuring DNA content by propidium iodide staining and FACS analysis as described previously (51).
Transferrin uptake assay
HaCaT cells were seeded on sterile glass coverslips at a density 0.75 × 105/well and grown overnight. Cells were then incubated with 10 μg/ml AlexaFluor 647 transferrin conjugate (Invitrogen) in serum-free DMEM for 30 minutes at 37°C. After extensive washing, cells were fixed and counterstained with anti-EEA-1 (Santa Cruz, 1:100). Samples were visualized using a Zeiss Axiovert 200 M microscope (Zeiss) attached to a LSM 510 META confocal unit.
Supplementary Material
Acknowledgements
We are most grateful to Martin Müller who provided the anti-L2 antibody and L1/L2 expression constructs, Neil Christensen for the anti-L1 H16.V5 antibody, Daniel F. Cutler for GFP-tagged SNX17 and Anne K. Soutar for FLAG-tagged SNX17 truncated constructs. We are also very grateful to Miranda Thomas and David Pim for comments on the manuscript. This work was supported in parts by research grants from the NIH to MAO: NIH PHS RO1CA132136 and from the American Cancer Society to SKC: RSG 117469.
REFERENCES
- 1.Smith JS, Lindsay L, Hoots B, Keys J, Franceschi S, Winer R, Clifford GM. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer. 2007;121:621–632. doi: 10.1002/ijc.22527. [DOI] [PubMed] [Google Scholar]
- 2.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. Arrangement of L2 within the papillomavirus capsid. J Virol. 2008;82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira R, Hitzeroth II, Rybicki EP. Insights into the role and function of L2, the minor capsid protein of papillomaviruses. Arch Virol. 2009;154:187–197. doi: 10.1007/s00705-009-0310-3. [DOI] [PubMed] [Google Scholar]
- 5.Bousarghin L, Touze A, Sizaret PY, Coursaget P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J Virol. 2003;77:3846–3850. doi: 10.1128/JVI.77.6.3846-3850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmgren SC, Patterson NA, Ozbun MA, Lambert PF. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J Virol. 2005;79:3938–3948. doi: 10.1128/JVI.79.7.3938-3948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J Virol. 2008;82:9505–9512. doi: 10.1128/JVI.01014-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laniosz V, Dabydeen SA, Havens MA, Meneses PI. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. J Virol. 2009;83:8221–8232. doi: 10.1128/JVI.00576-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem. 1999;274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 10.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. Human papillomavirus infection requires cell surface heparan sulfate. J Virol. 2001;75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day PM, Lowy DR, Schiller JT. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol. 2008;82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang R, Day PM, Yutzy WHt, Lin KY, Hung CF, Roden RB. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J Virol. 2003;77:3531–3541. doi: 10.1128/JVI.77.6.3531-3541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Day PM, Lowy DR, Schiller JT. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology. 2003;307:1–11. doi: 10.1016/s0042-6822(02)00143-5. [DOI] [PubMed] [Google Scholar]
- 14.Smith JL, Campos SK, Ozbun MA. Human papillomavirus type 31 uses a caveolin 1- and dynamin 2-mediated entry pathway for infection of human keratinocytes. J Virol. 2007;81:9922–9931. doi: 10.1128/JVI.00988-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. Clathrin- and caveolin-independent entry of human papillomavirus type 16--involvement of tetraspanin-enriched microdomains (TEMs) PLoS One. 2008;3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol. 2006;80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dabydeen SA, Meneses PI. The role of NH4Cl and cysteine proteases in Human Papillomavirus type 16 infection. Virol J. 2009;6:109. doi: 10.1186/1743-422X-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laniosz V, Nguyen KC, Meneses PI. Bovine papillomavirus type 1 infection is mediated by SNARE syntaxin 18. J Virol. 2007;81:7435–7448. doi: 10.1128/JVI.00571-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karanam B, Peng S, Li T, Buck C, Day PM, Roden RB. Papillomavirus infection requires gamma secretase. J Virol. 2010;84:10661–10670. doi: 10.1128/JVI.01081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards RM, Lowy DR, Schiller JT, Day PM. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A. 2006;103:1522–1527. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Florin L, Becker KA, Lambert C, Nowak T, Sapp C, Strand D, Streeck RE, Sapp M. Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J Virol. 2006;80:6691–6696. doi: 10.1128/JVI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bossis I, Roden RB, Gambhira R, Yang R, Tagaya M, Howley PM, Meneses PI. Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J Virol. 2005;79:6723–6731. doi: 10.1128/JVI.79.11.6723-6731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darshan MS, Lucchi J, Harding E, Moroianu J. The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol. 2004;78:12179–12188. doi: 10.1128/JVI.78.22.12179-12188.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009;5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A. 2004;101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Day PM, Roden RB, Lowy DR, Schiller JT. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol. 1998;72:142–150. doi: 10.1128/jvi.72.1.142-150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okun MM, Day PM, Greenstone HL, Booy FP, Lowy DR, Schiller JT, Roden RB. L1 interaction domains of papillomavirus l2 necessary for viral genome encapsidation. J Virol. 2001;75:4332–4342. doi: 10.1128/JVI.75.9.4332-4342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marusic MB, Mencin N, Licen M, Banks L, Grm HS. Modification of human papillomavirus minor capsid protein L2 by sumoylation. J Virol. 2010;84:11585–11589. doi: 10.1128/JVI.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seet LF, Hong W. The Phox (PX) domain proteins and membrane traffic. Biochim Biophys Acta. 2006;1761:878–896. doi: 10.1016/j.bbalip.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Cullen PJ. Endosomal sorting and signalling: an emerging role for sorting nexins. Nat Rev Mol Cell Biol. 2008;9:574–582. doi: 10.1038/nrm2427. [DOI] [PubMed] [Google Scholar]
- 31.Ghai R, Mobli M, Norwood SJ, Bugarcic A, Teasdale RD, King GF, Collins BM. Phox homology band 4.1/ezrin/radixin/moesin-like proteins function as molecular scaffolds that interact with cargo receptors and Ras GTPases. Proc Natl Acad Sci U S A. 2011;108:7763–7768. doi: 10.1073/pnas.1017110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stockinger W, Sailler B, Strasser V, Recheis B, Fasching D, Kahr L, Schneider WJ, Nimpf J. The PX-domain protein SNX17 interacts with members of the LDL receptor family and modulates endocytosis of the LDL receptor. Embo J. 2002;21:4259–4267. doi: 10.1093/emboj/cdf435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knauth P, Schluter T, Czubayko M, Kirsch C, Florian V, Schreckenberger S, Hahn H, Bohnensack R. Functions of sorting nexin 17 domains and recognition motif for P-selectin trafficking. J Mol Biol. 2005;347:813–825. doi: 10.1016/j.jmb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Lee J, Retamal C, Cuitino L, Caruano-Yzermans A, Shin JE, van Kerkhof P, Marzolo MP, Bu G. Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J Biol Chem. 2008;283:11501–11508. doi: 10.1074/jbc.M800642200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czubayko M, Knauth P, Schluter T, Florian V, Bohnensack R. Sorting nexin 17, a non-self-assembling and a PtdIns(3)P high class affinity protein, interacts with the cerebral cavernous malformation related protein KRIT1. Biochem Biophys Res Commun. 2006;345:1264–1272. doi: 10.1016/j.bbrc.2006.04.129. [DOI] [PubMed] [Google Scholar]
- 36.Seog DH, Han J. Sorting Nexin 17 Interacts Directly with Kinesin Superfamily KIF1Bbeta Protein. Korean J Physiol Pharmacol. 2008;12:199–204. doi: 10.4196/kjpp.2008.12.4.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adachi H, Tsujimoto M. Adaptor protein sorting nexin 17 interacts with the scavenger receptor FEEL-1/stabilin-1 and modulates its expression on the cell surface. Biochim Biophys Acta. 2010;1803:553–563. doi: 10.1016/j.bbamcr.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 38.van Kerkhof P, Lee J, McCormick L, Tetrault E, Lu W, Schoenfish M, Oorschot V, Strous GJ, Klumperman J, Bu G. Sorting nexin 17 facilitates LRP recycling in the early endosome. Embo J. 2005;24:2851–2861. doi: 10.1038/sj.emboj.7600756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burden JJ, Sun XM, Garcia AB, Soutar AK. Sorting motifs in the intracellular domain of the low density lipoprotein receptor interact with a novel domain of sorting nexin 17. J Biol Chem. 2004;279:16237–16245. doi: 10.1074/jbc.M313689200. [DOI] [PubMed] [Google Scholar]
- 40.Christensen ND, Dillner J, Eklund C, Carter JJ, Wipf GC, Reed CA, Cladel NM, Galloway DA. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology. 1996;223:174–184. doi: 10.1006/viro.1996.0466. [DOI] [PubMed] [Google Scholar]
- 41.Williams R, Schluter T, Roberts MS, Knauth P, Bohnensack R, Cutler DF. Sorting nexin 17 accelerates internalization yet retards degradation of P-selectin. Mol Biol Cell. 2004;15:3095–3105. doi: 10.1091/mbc.E04-02-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campos SK, Ozbun MA. Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes. PLoS One. 2009;4:e4463. doi: 10.1371/journal.pone.0004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith MJ, Hardy WR, Murphy JM, Jones N, Pawson T. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol Cell Biol. 2006;26:8461–8474. doi: 10.1128/MCB.01491-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Florian V, Schluter T, Bohnensack R. A new member of the sorting nexin family interacts with the C-terminus of P-selectin. Biochem Biophys Res Commun. 2001;281:1045–1050. doi: 10.1006/bbrc.2001.4467. [DOI] [PubMed] [Google Scholar]
- 45.Buck CB, Pastrana DV, Lowy DR, Schiller JT. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med. 2005;119:445–462. doi: 10.1385/1-59259-982-6:445. [DOI] [PubMed] [Google Scholar]
- 46.Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 47.Tomaic V, Gardiol D, Massimi P, Ozbun M, Myers M, Banks L. Human and primate tumour viruses use PDZ binding as an evolutionarily conserved mechanism of targeting cell polarity regulators. Oncogene. 2009;28:1–8. doi: 10.1038/onc.2008.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas M, Massimi P, Banks L. HPV-18 E6 inhibits p53 DNA binding activity regardless of the oligomeric state of p53 or the exact p53 recognition sequence. Oncogene. 1996;13:471–480. [PubMed] [Google Scholar]
- 49.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 50.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. Journal of Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 51.Banks L, Barnett SC, Crook T. HPV-16 E7 functions at the G1 to S phase transition in the cell cycle. Oncogene. 1990;5:833–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.