

Abstract
Aneuploidy, a state of having uneven numbers of chromosomes, is a form of large-effect mutation able to confer adaptive phenotypes under diverse stress conditions1,2. Here we investigate whether pleiotropic stress could in turn induce aneuploidy in budding yeast. We show that while diverse stresses can induce an increase in chromosome instability (CIN), proteotoxic stress, caused by transient Hsp90 inhibition or heat-shock, drastically elevated CIN to produce karyotypically mosaic cell population. The latter effect is linked to an evolutionarily conserved role for Hsp90 chaperon complexes in kinetochore assembly3,4. Continued growth in the presence of Hsp90 inhibitor resulted in emergence of drug-resistant colonies with chromosome XV gain. This drug-resistance phenotype is a quantitative trait involving copy number increases of at least two genes located on chromosome XV. Short-term exposure to Hsp90 stress potentiated fast adaptation to unrelated cyto-toxic compounds through different aneuploid chromosome stoichiometries. These findings demonstrate that aneuploidy is a form of stress-inducible mutation in eukaryotes, capable of fueling rapid phenotypic evolution and drug resistance, and reveal a new role for Hsp90 in regulating the emergence of adaptive traits under stress.
How cells maintain stable phenotypes and yet can adapt to diverse stress conditions through heritable change is a question with broad implications in evolution and disease progression. In prokaryotes, while the genome is propagated with high fidelity under normal conditions, extensive studies have demonstrated that different modes of genetic variation can be directly induced by stress, fueling stress adaptation 5. Recent work has revealed that one form of adaptive mutation in eukaryotic cells is the alteration of chromosome copy number, or aneuploidy 1,2,6. Aneuploid yeast has been observed in diverse laboratory1, industrial 7,8,9 and natural 8 environments. Aneuploidy leads to expression changes of many genes at levels that largely scale with gene copy number changes, bringing about dramatic phenotypic variation in a karyotype-specific manner under diverse growth conditions 6. These findings suggest that to maintain phenotypic stability, karyotype stability must be ensured, and indeed intricate mechanisms have evolved to achieve highly accurate chromosome segregation to prevent CIN during mitotic proliferation. Furthermore, as aneuploids are known to exhibit growth disadvantage compared to euploids under stress-free conditions 6,10, the pre-existing karyotype diversity in a euploid population is likely to be limited for rapid adaptation when exposed to stressful environments. This raises the question of whether the cellular mechanisms ensuring chromosome transmission fidelity may be relaxed under stress, thus allowing the emergence of karyotypic diversity to fuel rapid cellular adaptation.
To test whether stress conditions in general could increase the rate of whole chromosomal instability, we exposed haploid yeast cells to chemicals inducing various types of pleiotropic stress (Supplementary Table 1) for 12-14 hours and quantified chromosome loss rate by using the selection-neutral, chromosome fragment (CF)-based colony color assay (Fig. 1a, Supplementary Fig. 2; Supplementary Information) 11. This initial screen revealed that many stress conditions, including hydrogen peroxide (oxidative stress), cycloheximide (translational stress), tunicamycin (ER stress), etc., elevated the chromosome loss rate to a level similar to that caused by benomyl, a microtubule inhibitor (Fig. 1a). Surprisingly, radicicol, an Hsp90 inhibitor 12, was by far the most effective CIN inducer: the chromosome loss rate (7.4×10-2/cell division) was hundreds of times above the control (2×10-4/cell division), even at a radicicol concentration (10 μg/ml or 27 μM) with only minor effect on growth (Fig. 1a, Supplementary Fig. 3). Quantitative PCR (qPCR) confirmed that red colonies induced by radicicol had lost the whole CF (Supplementary Fig. 4a). Two of the 13 tested red colonies were confirmed to have also gained chromosome (Chr) X or Chr XI (Supplementary Fig. 4b, c).
Figure 1. Diverse stress conditions, especially Hsp90 inhibition, induce chromosomal instability.
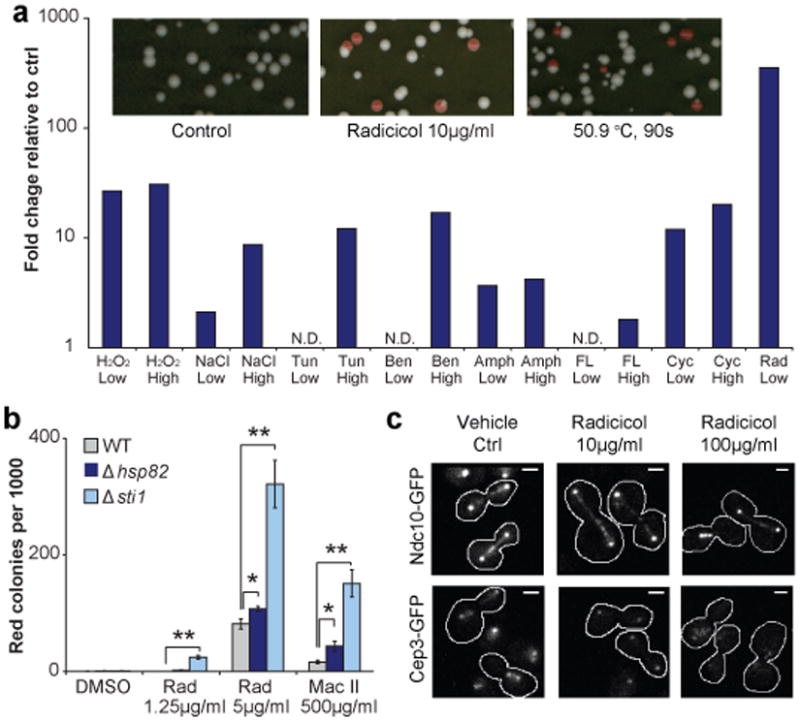
(a) Up: Colony appearance on YPD plates after cells were exposed to no stress (left), 16 hr of 10 μg/ml radicicol treatment (middle) or 90 sec heat shock at 50.9 °C. White colony color indicates retentions of CF; red indicates CF loss. Down: CF loss rates during exposure to diverse stress were inferred from red colony frequencies normalized to that of the vehicle-control population. N.D.: increase not detected over control. See Supplementary Figure 2, 3 and Supplementary Information for details. (b) Deletion of HSP82 or STI1 sensitized the CIN-inducing effect of radicicol and macbecin II. Red colony frequencies normalized to that of wild-type DMSO control were averaged among 4 replicates, shown with standard error of the mean (SEM). *, p<0.05; **, p<0.01, two-tail t-test. (c) Representative images showing kinetocore localization of Ndc10-GFP and Cep3-GFP under different conditions as indicated. Radicicol diminished Cep3-GFP localization at the kinetocore. Scale bar: 2μm. See Supplementary Fig. 5 for additional images and quantification.
A similar aneuploidy-inducing effect was also observed with macbecin II, a structurally distinct Hsp90 inhibitor (Fig.1b) 13. Deletion of one copy of Hsp90 genes, HSP82 showed enhanced CF loss compared to the wild type in the presence of radicicol or macbecin II (Fig.1b). Interestingly, deletion of STI1, the yeast homolog of mammalian Hop and a co-chaperone of Hsp90, resulted in significantly elevated CIN even at a concentration of radicicol too low to induce CIN on its own (Fig. 1b, Supplementary Fig. 5a). Heat is a common environmental stress known to tax Hsp90 function 14. Heat-shock for 90 seconds at 50.9°C induced subsequent CF loss at a rate comparable to that by pharmacological inhibition of Hsp90 (Fig. 1a). These results confirmed that Hsp90 stress is a potent inducer of aneuploidy. Hsp90 chaperon complexes are crucial facilitators of many cellular functions15. Previous biochemical studies suggested that Hsp90 is important for the activation of Ctf13 and assembly of the CBF3 inner kinetochore complex 3. Most CBF3 complex components, as well as the two co-chaperones involved in Ctf13 activation, showed haploinsufficiency toward radicicol (Supplementary Fig. 5b). Radicicol disrupted the kinetochore localization of Cep3 but had less effect on Ndc10, thus altering the stoichiometry of CBF3 complex at the kinetochore (Fig. 1c, Supplementary Fig. 5c, d, e). In addition to the CBF3 complex, Hsp90 interacts with several other pathways that could affect chromosome transmission fidelity, including the spindle assembly checkpoint 16 (see below).
Hsp90 taxation has previous been proposed to impact evolution by releasing phenotypic variation from pre-stored genetic diversity in the population and by transposon mobilization 15,17. Does Hsp90 inhibition also promote adaptation through induction of aneuploidy? As a first test, a diploid strain was grown in the presence of high concentration of radicicol and 3 largest radicicol-resistant (Radr) colonies were selected and reconfirmed (Fig. 2a, Supplementary Figure 6 and Supplementary Information). Karyotyping revealed that all 3 Radr colonies were aneuploid with a dominant karyotype feature: all 3 Radr colonies, which adapted independently, contained one or two additional copies of Chr XV (Fig.2a). A haploid Chr XV disomy strain, generated by genetic manipulation 10, also showed strong resistance to radicicol (Fig. 2b). A previous genome-wide screen identified a set of genes exhibiting haploinsufficiency toward macbecin II, among which 2 of the top genes are located on Chr XV: STI1 and PDR5, a pleiotropic drug pump 16. We deleted a single copy of STI1 or PDR5 gene from Radr colony 3, trisomy for Chr XV. Growth measurements showed that either deletion abolished more than 50% of the growth rate gained by Chr XV trisomy over diploid in the presence of radicicol (Fig. 2c). A single copy of STI1 and/or PDR5 was then introduced into the parental diploid strain. An extra copy of each gene mildly but significantly increased radicicol resistance, but their combination drastically improved radicicol resistance (Fig. 2c, Supplementary Fig. 7). These results indicate that Chr XV gain directly confers radicicol resistance through increased copy number of STI1 and PDR5, and possibly also other genes carried on this chromosome (e.g., SGT1).
Figure 2. Aneuploidy is the predominant genetic change conferring adaptation to radicicol.

(a) Left: Replating and growth of control (ctrl) or three adapted radicicol resistant (Radr) strains on 100μg/ml radicicol plates after 3 days incubation. See the experimental scheme in Supplementary Figure 6. Right: All 3 re-confirmed Radr colonies were aneuploids with different levels of Chr XV gain. Intensity log2ratios over euploid are shown. Repetitive elements are shown as vertical lines. (b) Haploid Chr XV disomy generated by genetic manipulation shows higher growth rate than euploid in radicicol. (c) Increased gene dosages of STI1 and PDR5 encoded on Chr XV are partially required and sufficient for radicicol resistance. The maximum growth rates were averaged for 4 replicates and normalized to diploid, shown with SEM.
We next tested if the karyotype diversity produced by Hsp90 stress-induced CIN could fuel adaptation to various other stress conditions. A karyotypically mosaic yeast cell population (∼1/3 of the population were aneuploid with different karyotypes, Supplementary Fig. 8c) was generated by growing a diploid strain under moderate Hsp90 stress (20μg/ml Radicicol) for 2 days. This population was then tested for enhanced adaptability toward other stress conditions, including the presence of growth inhibiting concentrations of fluconazole, tunicamycin, or benomyl, over a control homogeneous euploid population (see experimental scheme in Supplementary Fig. 8a). The radicicol-pretreated population did not show any growth advantage over the control diploid (vehicle pre-treated) on drug-free plates (Fig. 3a, Ctrl). However, on each of the different drug-containing plates, the radicicol pre-treated populations demonstrated drastically enhanced colony viability and frequency to form large drug-resistant colonies than the vehicle-pretreated population (Fig. 3a, b, c).
Figure 3. Prior Hsp90 inhibition potentiates adaption to other stress conditions through divergent aneuploid karyotypes.
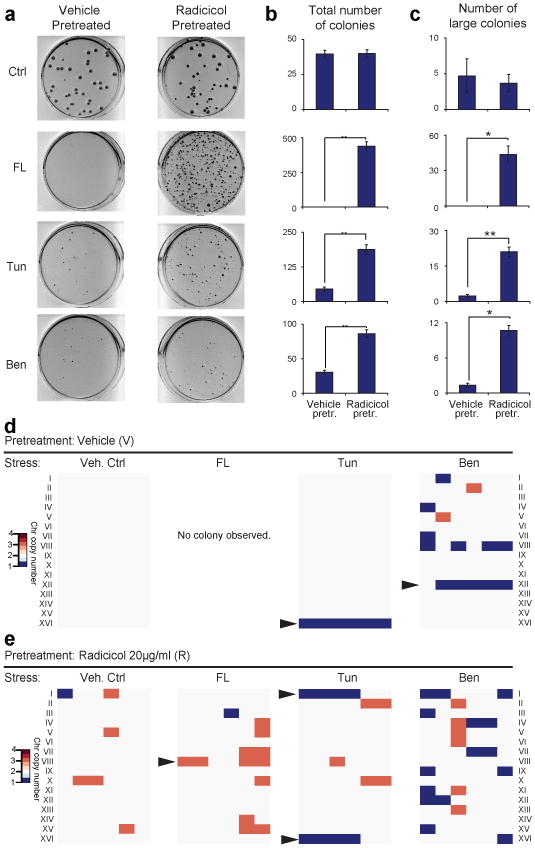
(a) Plates of vehicle-pretreated (V) group and radicicol-pretreated (R) group on different media as indicated. ∼40 cells were plated on DMSO (Ctrl); ∼40,000 cells were plated onto each drug plate. FL: 32 μg/ml fluconazole; Tun: 2.5μg/ml tunicamycin; Ben: 30μg/ml benomyl. (b) Quantification of the number of viable colonies. Shown are mean±SEM from triplicate experiments. (c) The sizes of all colonies (including both R and V groups) grown on each type of plates were measured. The distributions of top 10% largest colonies between the two groups are shown. *, p<0.05; **, p<0.01, two-tail paired t-test. (d) The karyotypes of 6 V colonies from 3 replicate experiments of each type as determined by qPCR 6. (e) The karyotypes of 6 independent R colonies from 3 replicate experiments of each type determined by qPCR. Arrowheads point to aneuploid chromosomes whose gain or loss frequency among resistant colonies was significantly higher than the starting populations (p<0.01, Mantel-Haenszel tests).
Twenty one colonies were picked from the vehicle control plates bearing the radicicol-pretreated population, and out of these 12 were aneuploid, whereas none (0/9) from the control plate bearing the vehicle-pretreated population were aneuploid (Fig.3d, e, Supplementary Fig. 8d, e). The vast majority (17/18) of the large colonies karyotyped from the drug plates bearing the radicicol-pretreated population were aneuploid (Fig. 3e). The drug-resistant colonies from the vehicle-pretreated population were also aneuploid (Fig. 3d). Importantly, the aneuploid colonies resistant to the same drug showed obvious karyotypic commonalities and tend to cluster together based on karyotype similarity (Fig.3e, Supplementary Fig. 9). For example, 4 of the 5 aneuploid colonies from fluconazole plates karyotyped gained an extra copy of Chr VIII, which carries Erg11, encoding an ergostrol biosynthetic enzyme known to confer fluconazole resistance in Candida albicans 18. Losing a copy of Chr XVI is a predominant karyotype change among the tunicamycin-resistant colonies (seen in 10/12 karyotyped, Fig. 3d). Of the 12 benomyl-resistant colonies, 10 demonstrated karyotype clustering with 6 of them losing one Chr XII, but it appears that more than one karyotypic pattern could confer benomyl resistance. This however is consistent with our previous observation of phenotypic convergence of distinct karyotypic patterns 6. All the above common karyotype features were significantly (Mantel-Haenszel tests) enriched in drug resistant colonies but not the starting radicicol-pretreated population prior to selection on drug plates (Fig. 3d, e, Supplementary Fig. 8e), suggesting an association of specific karyotypes with resistance to certain drugs.
To further assess the selective advantage of aneuploidy and karyotype dynamics under varying stress levels, two single Chr XVI monosomy colonies from a tunicamycin plate were streaked on drug-free plates. Colonies of two distinct sizes emerged, with the small ones being predominant (Fig.4a). Karyotyping showed that the small colonies represented Chr XVI monosomy, whereas the rare large colonies had gained back the missing Chr XVI and returned to diploid (Fig 4b, Supplementary Fig. 10a). Tunicamycin resistance was tightly linked to Chr XVI monosomy: all of the small colonies were tunicamycin resistant while the growth of the big colonies was abolished by tunicamycin (Fig 4c, Supplementary Fig. 10b, c, d). This result shows that an adapted aneuploid population also has the potential to return to euploid state when the stress condition is attenuated, suggesting that aneuploidy is not only a readily accessible mutation with large phenotypic impacts but is also reversible.
Figure 4. Karyotype requirement and dynamics associated with tunicamycin resistance.
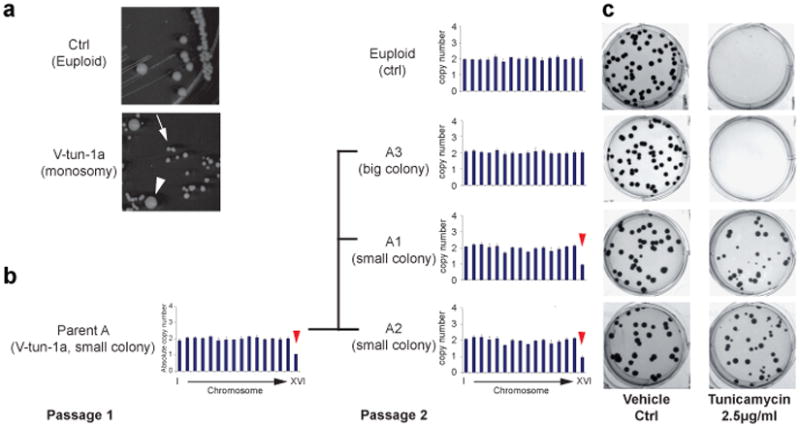
(a, b) Chr XVI monosomy (small colonies (arrow)) is unstable and produces large euploid progenies (arrowhead). Shown are a representative image of the colonies (observed after 3 day growth on YPD) (a) and karyotypes of the Parent A and the progeny colonies (A1-3) determined by qPCR (b). (c) Chr XVI monosomy progenies (A1 and A2) but not euploid progeny (A3) displayed tunicamycin resistance. Note that the size difference between small and large colonies on control plates was no longer apparent after 7-day growth.
Taken together, the above results demonstrated that stress-induced chromosome instability, leading to aneuploidy, is a mechanism of stress-induced mutagenesis in eukaryotes with high adaptive value to diverse perturbations (Supplementary Figure 1). By far Hsp90 inhibition is the most potent inducer of aneuploidy among the stress conditions tested. This may be due to a broad but critical involvement of Hsp90 in pathways governing chromosome transmission fidelity and cell division 16. For example, the mitotic checkpoint gene MAD2 is a genetic interaction hub sensitive to Hsp90 perturbation 16. MAD2 deletion was also sufficient to lead to rapid emergence of fluconazole-resistant colonies bearing an extra copy of Chr VIII (Supplementary Figure 11). As Mad2 requires the CBF3 complex for its activity at the kinetochore 19, the exceptionally high-level CIN induced by Hsp90 inhibitors may be explained by a combined effect of interference with both kinetochore assembly and the checkpoint monitoring spindle defects. It is presently unknown whether the other stress conditions induce CIN through similar or different cellular targets.
The Hsp90 chaperon complex specializes in modulating the stability and function of many important regulatory and structural proteins 16. As a result, Hsp90 acts as a capacitor facilitating evolutionary adaptation by unleashing the effects of pre-existing mutations when Hsp90 activity is taxed under mild stress 14,15. Strong Hsp90 inhibition also induces phenotypic variation through transposon activation in Drosophila 17. The results presented in this work reveal a new role for Hsp90 in adaptive evolution - as the guardian of chromosomal stability, the inhibition of which could trigger de novo karyotypic diversity leading to rapid adaptation through aneuploidy. We note that our observed induction of aneuploidy required more potent Hsp90 inhibition than that required to reveal phenotypic effects of pre-existing mutations 14. As the function of Hsp90 chaperon complex in kinetochore assembly is conserved in mammalian species 4,20, the Hsp90 stress-induced aneuploidy may be a mechanism of cellular adaptation affecting a wide range of organisms.
Methods Summary
Yeast strains are listed in Supplementary Table 2. Standard genetic techniques were used for yeast strain construction. All deletions were verified by genomic PCR, and all aneuploid transformants were re-karyotyped by qPCR, and those retaining the original karyotype were used for experiments. Yeast qPCR karyotyping was performed as previously described 6. Briefly, the chromosome copy number was inferred from qPCR with sets of primers located on peri-centrimeric regions. aCGH was performed on either a home-made spot array.
A detailed description of all methods is provided in Supplementary Information.
Supplementary Material
Acknowledgments
We thank N. Pavelka, B. Rubinstein and H. Li for help with data analysis, J. Zhu, B. Fleharty and J. Haug for experimental assistance, S. Lindquist for helpful discussion, and T. potapova and B. Slaughter for comments on the manuscript. CF strains and Chr XV disomy are kind gifts from F. Spencer and A. Amon, respectively. This work was supported by NIH grant RO1GM059964 to R.L.
Footnotes
Author Contributions G.C. and R.L. designed and G.C. performed the experiments. W.D.B performed qPCR karyotyping. C.W.S. performed aCGH data analysis. G.C. and R.L. prepared the manscrript. R.L. conceived and supervised the project. All authors read and agreed with the paper content.
References
- 1.Pavelka N, Rancati G, Li R. Dr Jekyll and Mr Hyde: role of aneuploidy in cellular adaptation and cancer. Current Opinion in Cell Biology. 2010;22:809–815. doi: 10.1016/j.ceb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selmecki A, Forche A, Berman J. Genomic Plasticity of the Human Fungal Pathogen Candida albicans. Eukaryotic Cell. 2010;9:991–1008. doi: 10.1128/EC.00060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stemmann O, Neidig A, KÃcher T, Wilm M, Lechner J. Hsp90 enables Ctf13p/Skp1p to nucleate the budding yeast kinetochore. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8585–8590. doi: 10.1073/pnas.082223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies AE, Kaplan KB. Hsp90-Sgt1 and Skp1 target human Mis12 complexes to ensure efficient formation of kinetochore-microtubule binding sites. The Journal of Cell Biology. 2010;189:261–274. doi: 10.1083/jcb.200910036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SM. Evolving responsively: adaptive mutation. Nat Rev Genet. 2001;2:504–515. doi: 10.1038/35080556. [DOI] [PubMed] [Google Scholar]
- 6.Pavelka N, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Infante JJ, Dombek KM, Rebordinos L, Cantoral JM, Young ET. Genome-Wide Amplifications Caused by Chromosomal Rearrangements Play a Major Role in the Adaptive Evolution of Natural Yeast. Genetics. 2003;165:1745–1759. doi: 10.1093/genetics/165.4.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kvitek DJ, Will JL, Gasch AP. Variations in Stress Sensitivity and Genomic Expression in Diverse S. cerevisiae Isolates. PLoS Genet. 2008;4:e1000223. doi: 10.1371/journal.pgen.1000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borneman AR, et al. Whole-Genome Comparison Reveals Novel Genetic Elements That Characterize the Genome of Industrial Strains of Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1001287. doi: 10.1371/journal.pgen.1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torres EM, et al. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 11.Spencer F, Gerring SL, Connelly C, Hieter P. Mitotic Chromosome Transmission Fidelity Mutants in Saccharomyces cerevisiae. Genetics. 1990;124:237–249. doi: 10.1093/genetics/124.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene. 1998;16:2639–2645. doi: 10.1038/sj.onc.1201790. [DOI] [PubMed] [Google Scholar]
- 13.Bohen SP. Genetic and Biochemical Analysis of p23 and Ansamycin Antibiotics in the Function of Hsp90-Dependent Signaling Proteins. Mol Cell Biol. 1998;18:3330–3339. doi: 10.1128/mcb.18.6.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarosz DF, Lindquist S. Hsp90 and Environmental Stress Transform the Adaptive Value of Natural Genetic Variation. Science. 2010;330:1820–1824. doi: 10.1126/science.1195487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 16.McClellan AJ, et al. Diverse Cellular Functions of the Hsp90 Molecular Chaperone Uncovered Using Systems Approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 17.Specchia V, et al. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature. 2010;463:662–665. doi: 10.1038/nature08739. [DOI] [PubMed] [Google Scholar]
- 18.Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Molecular Microbiology. 2008;68:624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 19.Gardner RD, et al. The Spindle Checkpoint of the Yeast Saccharomyces cerevisiae Requires Kinetochore Function and Maps to the CBF3 Domain. Genetics. 2001;157:1493–1502. doi: 10.1093/genetics/157.4.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niikura Y, et al. 17-AAG, an Hsp90 inhibitor, causes kinetochore defects: a novel mechanism by which 17-AAG inhibits cell proliferation. Oncogene. 2006;25:4133–4146. doi: 10.1038/sj.onc.1209461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.