

Abstract
Non-technical summary
A poor diet during pregnancy has been linked to long-term health outcomes for the baby, such as an increased risk of diseases of the heart and kidney. We show in an experimental model that recreates a poor diet during pregnancy, i.e. a diet low in protein with adequate energy, that kidney development in the baby is affected in such a way as to reduce the potential for new blood vessels to form. This results in a greater number of important, functional kidney cells spontaneously dying. Later in life, these effects in the kidney manifest as permanently reduced kidney function, especially if the baby subsequently becomes overweight as an adult. The research reinforces advice to pregnant mothers about the importance of eating a nutritionally balanced diet during pregnancy.
Abstract
A nutritionally poor maternal diet can reduce nephron endowment and pre-empt premature expression of markers for chronic renal disease in the offspring. A mechanistic pathway from variation in maternal diet through altered fetal renal development to compromised adult kidney structure and function with adult-onset obesity has not been described. We show that maternal protein-energy malnutrition in sheep blunts nephrogenic potential in the 0.44 gestation (65 days gestation, term ∼147 days) fetus by increasing apoptosis and decreasing angiogenesis in the nephrogenic zone, effects that were more marked in male fetuses. As adults, the low-protein-exposed sheep had reduced glomerular number and microvascular rarefaction in their kidneys compensated for, respectively, by glomerular hypertrophy and increased angiogenic support. In this study, the long-term mild anatomical deficits in the kidney would have remained asymptomatic in the lean state, but when superimposed on the broad metabolic challenge that obesity represents then microalbuminuria and blunted bilateral renal function revealed a long-term physiological compromise, that is only predicted to worsen with age. In conclusion, maternal protein-energy malnutrition specifically impacts fetal kidney vascular development and prevents full functionality of the adult kidney being achieved; these residual deficits are predicted to significantly increase the expected incidence of chronic kidney disease in prenatally undernourished individuals especially when coupled with a Western obesogenic environment.
Introduction
Chronic kidney disease (CKD) is a prevalent, age-related, non-communicable disease in Western societies (Winearls & Glassock, 2009). Obesity (Ejerblad et al. 2006; Cignarelli & Lamacchia, 2007), hypertension (Bakris & Ritz, 2009) and type 2 diabetes (Crook, 2002) compromise renal functional reserve and increase the risk of developing CKD. In 2005, 396 million people were obese (Kelly et al. 2008); in 2008, 28–35% of the US and UK population were hypertensive (Egan et al. 2010) and with each predicted to significantly increase with time then the global burden of CKD is a major public health issue. Indeed, stage 5 CKD (i.e. end-stage renal disease) is currently increasing at 5–8% per annum, globally costing $70–$75 billion US dollars per annum for dialysis (Lysaght, 2002).
Obesity, hypertension and CKD are conditions virtually absent in hunter–gatherer communities whose diet is largely grain-based, alkaline, low in sodium and thus raw, natural and unrefined (i.e. a Palaeolithic diet) (Eaton & Eaton, 2000), unlike the diet commonly consumed in Westernised societies (Cordain et al. 2005). Dietary change (from a Western toward a Palaeolithic diet), therefore, is probably the most achievable and potentially important preventative factor that could mitigate the inexorable rise in non-communicable disease (Daar et al. 2007; Narayan et al. 2010). Increased focus on diet during a woman's (or man's; Carone et al. 2010; Ng et al. 2010) reproductive years, particularly during pregnancy and lactation, has been stimulated by the Developmental Origins of Health and Disease hypothesis (Barker & Osmond, 1986; Gluckman et al. 2008) where individual variability in fetal response to maternal malnutrition (reflected as disproportionate growth (Barker et al. 2005) and/or low birth weight (Barker & Osmond, 1988)) increases risk of those individuals developing hypertension (Huang et al. 2010), coronary dysfunction (Crispi et al. 2010), type 2 diabetes (Whincup et al. 2008) and kidney disease (Woods et al. 2004; Amann et al. 2006) later in life. Increased focus on the latter is particularly pertinent as nephrogenesis is complete by term in man and non-litter-bearing mammals (Wintour & Moritz, 1997) and is therefore particularly sensitive and vulnerable to maternal malnutrition. For example, a maternal low-protein, high-glucose or high-fat diet may reduce nephron endowment (Vehaskari et al. 2001; Nehiri et al. 2008; Tran et al. 2008). Low birth weight infants have reduced nephron number (Manalich et al. 2000), increased glomerular volume (Hoy et al. 2005), increased blood pressure (Brenner et al. 1988; Mackenzie & Brenner, 1995; Keller et al. 2003) and are prone to minimal change nephrotic syndrome (Teeninga et al. 2008). In a retrospective analysis of autopsies, individuals with chronic hypertension had significantly fewer nephrons than the normotensive control group (Keller et al. 2003). Furthermore, more direct evidence has been collected from a cohort of Dutch individuals exposed to famine, as fetuses, during World War II when mean maternal energy intake reduced from 6.3 to 3.2 MJ day−1. As middle-aged adults, those individuals exposed to famine during early gestation vs. those unexposed, had evidence of programmed changes to blood pressure and an increased incidence of coronary heart disease (Roseboom et al. 1999), but also indices of early-stage renal disease such as microalbuminuria (urinary albumin:creatinine ratio, ACR ≥ 2.5, 12% vs. 7% of sampled cohort; adjusted odds ratio = 3.2, 95% confidence interval 1.4 to 7.7). These delayed developmental effects were not related to size at birth per se but rather to factors marking the quality of fetal growth or maternal macronutrient balance (Roseboom et al. 1999).
Despite small deficits (10–20%) in nephron endowment, the mammalian kidney has such functional redundancy that azotaemia only becomes evident at CKD stages 3–5, i.e. a loss of 50–70% glomeruli with glomerular filtration rate (GFR) declining to <30 ml min−1 (Reilly & Perazella, 2002). Thus, the subtle effect of maternal malnutrition on nephron endowment (the ‘first-hit’ or Brenners’ hypothesis; Brenner et al. 1988) is unlikely to be solely responsible for compromised adult renal function but coupled with the totality of potentially adverse environmental effects experienced within an individual's lifetime that may affect renal function, for example higher than average salt intake, increased dietary acid load and obesity (i.e. a ‘double-hit’), then healthspan can be significantly reduced in those prenatally compromised individuals (Griffin et al. 2008). A traditional early marker for incident CKD (i.e. before overt clinical signs present) is microalbuminuria, which can robustly predict future CKD (Fox et al. 2010). A reduced filtration barrier, allowing small amounts of albumin into the urine, is probably initially due to effacement of the podocyte foot processes, the integrity of which is supported by a number of factors, but primarily the key nexus for angiogenesis, vascular endothelial growth factor (VEGF) (Eremina et al. 2008). Obesity leads to a tissue-specific increase in VEGF, to support the concomitant local hypoxia (Wood et al. 2009) and, in obese sheep, has been shown to be positively associated with increased intra-renal endoplasmic stress, a potential early prognostic marker for CKD (Sharkey et al. 2009). The significance of developmental programming in this context is that it is pleiotrophic; that is, malnutrition during fetal development may have subtle, individual effects on nephron complement (reduced), on blood pressure (increased; Law & Shiell, 1996) and on peripheral insulin sensitivity (reduced; McMillen & Robinson, 2005) but which in an obesogenic environment may all interact in a combinatorial fashion to markedly increase any underlying susceptibility to CKD (Nehiri et al. 2008; Simonetti et al. 2008).
Epidemiological studies can highlight new and potentially important aetiologies of a multi-factorial disease like CKD, but confounding factors and their often-retrospective design can mean that the results may not be as relevant for contemporary societies, in terms of current lifestyle and diet (Huxley et al. 2002). Thus, animal models are essential to demonstrate the biological plausibility and mechanistic detail of any potential link between early life and later health. There is a considerable body of evidence in laboratory rodents and in larger animals, such as the sheep, that early maternal diet can influence renal structure of the adult offspring (McMillen & Robinson, 2005; Ojeda et al. 2008; Moritz et al. 2009; Puddu et al. 2009). Sheep are particularly relevant for such studies as the start and end periods of nephrogenesis (meso/metanephros) are virtually identical to man, ending at ∼0.90 gestation (Wintour & Moritz, 1997; Moritz & Wintour, 1999). Prenatal undernutrition of sheep increases blood pressure and decreases nephron number of the adolescent offspring (Gilbert et al. 2005), while a double-hit i.e. juvenile-onset obesity superimposed upon a background of fetal undernutrition has been shown by us to result in appreciable increases in systolic and diastolic blood pressure (∼10 mmHg) with evidence of renal dysfunction (Williams et al. 2007).
As yet no study has elucidated a pathway from maternal protein-energy malnutrition (PEM) through alterations to fetal kidney development (the first-hit) to deficits in renal structure and function in the adult offspring, especially in the context of an adult obesogenic environment (the second-hit). This is important with regard to those individuals that may have experienced a degree of PEM in utero but then later in life migrate internally to more urban, industrialised and affluent areas or for those that migrate across international borders to Westernised cultures. For example, in India, 58% of women of child-bearing age are anaemic (haemoglobin <2 SD, normal value 120 g l−1), with ∼20% children (∼110–130 million individuals) suffering PEM (WHO, 2011), whilst in the Western world, obesity has increased markedly over the last 40 years and is predicted to remain high or even increase further in the next 20 years (Wang et al. 2011). Hence, we have tested the hypothesis that restriction of maternal protein intake during early gestation negatively impacts fetal renal development resulting in a reduced nephron endowment which, later in life when coupled with an obesogenic environment, leaves the resultant adult with a structurally and functionally compromised kidney. Furthermore, we suggest that the developing renal vasculature is particularly susceptible to maternal protein-energy malnutrition, especially in males, and may provide for the first time a potential mechanistic pathway from variation in maternal diet through sex-specific fetal responses to the programming of renal disease in the resultant adult.
Methods
Ethical approval
All procedures were performed in accordance with the UK Animals (Scientific Procedures) Act, 1986 and were approved by the relevant local ethical review committees of the James Hutton Institute and the University of Nottingham. The authors have read, and the experiments comply with, the policies and regulations of The Journal of Physiology given by Drummond (2009). The study was designed to test the primary hypothesis that a maternal low protein diet would blunt fetal renal development and therefore compromise renal structure and function in the resulting adult individual. Renal functional outcomes are subject to the most measurement error, and intra-renal transit-time represents an important variable that condenses many aspects of adult renal function. In our study we had 91% power to detect a 20% effect size for renal transit-time in the adult, with at least three male and three female animals per group (3 treatment groups), significance (α) set at 0.05 and the within- and between-animal variance of 0.005 and 0.016 min, respectively, in a split-plot design.
Animals and experimental design
Thirty-five pregnant Scottish Blackface ewes carrying singleton fetuses were randomly allocated to one of three diet groups fed either a control diet (CP; n = 13) providing adequate protein (180 g kg−1 crude protein) and energy (10.6 MJ (kg dry mass (DM))−1; protein:energy ratio 16.9 g kg−1 crude protein per MJ DM) from day 0 of gestation to term (term, ∼147 days), or a protein-restricted diet (80 g kg−1 crude protein, 9.2 MJ (kg DM)−1, protein:energy ratio 8.7 g kg−1 crude protein per MJ DM) during either early (LPE, days 0–65 gestation; n = 16) or late gestation (LPL, day 66 to term; n = 6). On an ‘as fed basis’, the diets were isocaloric with the effective level of protein restriction being 8.7 vs. 17 g crude protein MJ ME (metabolic energy). At day 65 gestation, chosen to coincide with peak nephrogenesis in the sheep (Wintour & Moritz, 1997), a proportion of CP (n = 8) and LPE (n = 10) ewes were killed and fetal kidneys snap-frozen in liquid nitrogen and stored at −80°C for further analysis. The remainder (CP, n = 6; LPE, n = 7; LPL, n = 6) carried to term and the singleton offspring were delivered naturally. After weaning (10 weeks of age) the offspring were set to pasture until 1.5 years, at which point they were transferred from The James Hutton Institute to the University of Nottingham. After a brief period of acclimatisation the sheep were exposed to an obesogenic environment, specifically designed to encourage weight gain (i.e. restricted physical activity and fed to 150% ME requirements) as previously described (Rhodes et al. 2009). After 6–7 months (i.e. 2 years of age) in vivo renography was performed on all animals as previously described (Williams et al. 2007). Thereafter all sheep were subsequently killed (electrocortical stunning with exsanguination) and kidneys either snap-frozen in liquid nitrogen and stored at –80°C (right kidney) or fixed in neutral buffered formalin (NBF) for 24 h, rinsed in 0.02 m PBS for 24 h and subsequently stored in 70% ethanol at room temperature (left kidney). Urine was sampled from the bladder at post mortem for a spot assessment of the albumin:creatinine ratio.
Renography
In brief, 100 MBq technetium−99 diethylenetri- aminepentaacetic acid (Tc99m-DTPA) was injected (i.v. 0.5 ml−1 0.9% NaCl) in restrained sheep and from 60 frames captured over a 20 min period a dynamic renogram (adjusted for movement artefact, background and bolus deconvolution) was produced for the left and right kidney. Specific measures of interest were time to peak (minutes), upslope (counts per minute) – indicative of rate of uptake of tracer into the kidney, downslope (counts per minute) – indicative of the rate of tracer clearance and transit-time (minutes) – a composite of all previous measures. In addition, at 3, 4 and 5 h after injection, 5 ml venous blood was drawn, centrifuged and 1 ml plasma measured for gamma emission from which GFR could be accurately determined (Gleadhill et al. 1999).
Renal histology and nephron number
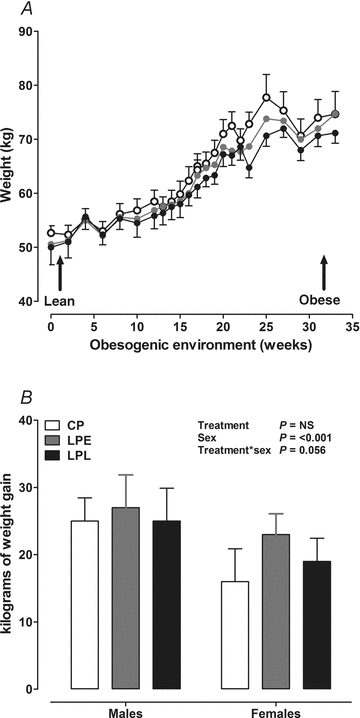
Random samples were taken from the NBF-fixed tissues, embedded in paraffin and sectioned at 5 μm (Microtome RM2255, Leica) onto polylysine slides. Slide cassettes were coded to allow for blinded analyses. Sections were stained with haematoxylin and eosin (H&E), periodic acid Schiff's reagent (PAS) or a trichrome stain as appropriate, using standard histological protocols and mounted onto coverslips with DPX mountant. Random sections from each treatment group were first examined by a consultant histopathologist to identify any obvious pathophysiological features such as monocyte infiltration, glomerular interstitial nephritus, glomerulosclerosis or basement membrane or mesangial cell thickening. Nephron number was estimated by unbiased stereology according to the random fractionator or ‘Cavalieri Principle’ technique, as previously described (Keller et al. 2003). The mean numerical density of glomeruli (Nv) was calculated as:
where 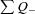 is the total number of glomeruli counted in the reference section, d is the distance between the planes and A is the area of the unbiased counting frame. The results were multiplied by the stereologically estimated cortex volume, calculated as:
is the total number of glomeruli counted in the reference section, d is the distance between the planes and A is the area of the unbiased counting frame. The results were multiplied by the stereologically estimated cortex volume, calculated as:
 |
Glomerular volume density (Vv) was calculated as:
 |
Renal immunohistochemistry
Renal sections were analysed for abundance of vascular endothelial growth factor A (VEGFA) (SC-152; Santa Cruz Biotechnology, Santa Cruz, USA) and CD34 protein (an endothelial cell marker) (AB81289; Abcam, Cambridge, UK) using an automated BOND-MAX (Leica, UK) system and primary antibodies raised against rabbit. A biotinylated ABC kit (Vector Labs, Peterborough, UK) was used to visualise VEGFA staining in fetal tissue. Appropriate negative controls were included for the primary antibody (i.e. no primary included) and for non-specific binding of the secondary antibody (i.e. using a rabbit IgG; Vector Labs, Peterborough, UK).
Detection of apoptosis
The presence and extent of apoptotic nuclei was determined using terminal deoxynucleotidyl transferase dUTP nick-end labelling and staining (TUNEL; TdT FragEL Calbiochem, Nottingham, UK) of 5 μm sections according to the manufacturer's protocol. In brief, dewaxed sections were rehydrated and samples permeabilised with 2 mg ml−1 proteinase K in 10 mm Tris pH 8 and endogenous peroxidises inactivated with 3% H2O2. Samples were equilibrated with 1× TdT equilibration buffer, labelled (TdT Labelling Reaction Mix) and incubated at 37°C for 1.5 h. The reaction was terminated, samples blocked with buffer and incubated with 1× conjugate for 30 min at room temperature. Apoptotic nuclei were detected after 10 min incubation with Diaminobenzidine buffer. After rinsing (dH2O), the samples were counterstained with methyl green, rehydrated with ethanol, cleared with xylene and mounted with DPX. Omission of primary antibody was used as a negative control.
Quantitative PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Crawley, UK) and cDNA synthesised using an Omniscript reverse transcriptase kit (Qiagen, Crawley, UK). Primers (Eurofins MWG, Ebersberg, Germany) were designed using NCBI Primer-BLAST or were as previously published (Table 1). Two micrograms of cDNA were added to each reaction, and qPCR performed using QuantiTect SYBR Green RT-PCR Kit (Qiagen, Crawley, UK) on a Roche Lightcycler 480. Melt curves were used to confirm reaction specificity, and cyclophilin was confirmed as a suitable housekeeping gene. mRNA quantities were normalised to cyclophilin using Roche Lightcycler 480 advanced relative quantification software.
Table 1.
Primer details for quantitative PCR
| Primer | Sequence 5′–3′ | Annealing temp (°C) | |
|---|---|---|---|
| Angiopoietin 1 | F | GGGTCACACTGGGACAGCAGG | 58 |
| R | TGGGCCACAGGCATCAAACCA | ||
| ATF41 | F | AGATGACCTGGAAACCATGC | 52 |
| R | AGGGGGAAGAGGTTGAAAGA | ||
| ATF61 | F | AACCAGTCCTTGCTGTTGCT | 52 |
| R | CTTCTTCTTGCGGGACTGAC | ||
| Bax1 | F | CAGGATGCATCCACCAAGAAGC | 56 |
| R | TTGAAGTTGCCGTCGGAAAACATT | ||
| Beta actin2 | F | TGTGCGTGACATCAAGGAGAA | 55 |
| R | CGCAGTGGCCATCTCCTG | ||
| BMP 7 | F | GCCACTTCCTCACCGACGCC | 58 |
| R | CAGCTGCAGTCACGGCCTCC | ||
| CD681 | F | GTCCTGCTACCACCACCAGT | 52 |
| R | GCTGGGAACCATTACTCCAA | ||
| Cyclophilin | F | CATACAGGTCCTGGCATCTTGTC | 56 |
| R | TGCCATCCAACCACTCAGTCT | ||
| DDIT31 | F | AGGACCACCAGAGGTCACAC | 56 |
| R | TGCCACTTTCCTTTCGTTTT | ||
| bFGF | F | CTTTCCCGCCCGGCCACTTT | 58 |
| R | GTCGCTCTTCTCGCGGACCC | ||
| GAPDH2 | F | TCCGTTGTGGATCTGACCTG | 55 |
| R | TGCTTCACCACCTTCTTGATCTC | ||
| GRP781 | F | TGAAACTGTGGGAGGTGTCA | 52 |
| R | TCGAAAGTTCCCAGAAGGTG | ||
| iNOS | F | TGATGCAGAAGGCCATGTCA | 52 |
| R | TCTCCCTGTCTCTGTTGCAAAG | ||
| MCP-1 | F | CAAGACCATCCTGGGCAAA | 52 |
| R | GTCCTGGACCCATTTCAGGTT | ||
| PDGF | F | CAAGACGCGCACGGAGGTGT | 58 |
| R | GCGGTTGTTGCAGCAGCCAG | ||
| SPARC | F | CGAGGTACCCGTGGGAGCCA | 58 |
| R | CAAACTCGCCGATGGGGGCA | ||
| TGFβ1 | F | GGAGTTGTGCGGCAGTGGCT | 58 |
| R | AAGGGCCGGTTCATGCCGTG | ||
| Thrombospondin | F | TGCTGTGCGGGCGGAGAAAG | 58 |
| R | TGCCCGCCTTGCCATTGGAG | ||
| Tie-2 | F | CAGCAGACCTCGGAGGCAGGA | 58 |
| R | TGCCCTCTTCAGCTGCAGCATG | ||
| VEGFA3 | F | GGATGTCTACCAGCGCAGC | 56 |
| R | TCTGGGTACTCCTGGAAGATGTC | ||
| VEGFR13 | F | TGGATTTCAGGTGAGCTTGGA | 52 |
| R | TCACCGTGCAAGACAGCTTC | ||
| VEGFR23 | F | CTTCCAGTGGGCTGATGACC | 52 |
| R | GCAACAAACGGCTTTTCATGT |
All primers designed using NCBI Primer-BLAST; ATF, activating transcription factor; BMP 7, bone morphogenic protein 7; DDIT3, DNA damage inducible transcript 3; bFGF, basic fibroblast growth factor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; iNOS, inducible nitric oxide synthase; GRP78, 78 kDa glucose-regulated protein; MCP-1, monocyte chemotactic protein 1; PDGF, platelet-derived growth factor; SPARC, secreted protein, acidic, cysteine-rich; TGFβ-1, transforming growth factor β-1; VEGF, vascular endothelial growth factor.
Statistical analysis
All data were analysed using a General Linear Model (GLM) or general linear mixed model approach where appropriate (Genstat v13, VSNi, UK) after checking for normality (or otherwise) of the error distribution. Data with skewed errors were log transformed before analysis. Estimated marginal means are presented with SEM, SED or 95% CI as indicated, to represent the measurement error. P≤ 0.050 was accepted as indicating statistical significance. A multivariate linear discriminant analysis was used to display multiple related outcome measures (e.g. renography derived values for renal function). The linear discriminant analysis generates two linear vectors from all normalised data (i.e. z-scores for individual data) that capture <95% variation for all inputted variables and plots these as values on a 2D plot. The method is an objective means to effectively demonstrate statistically significant patterns in complex, non-independent datasets.
Results
Maternal and fetal body composition and growth
Maternal weight (64.0 ± 4.4 kg (mean ± 1 SD) and body condition score (2.6 [2.58–2.68] units, median [IQR]) were similar at conception and the increase in weight with gestation and decrease in body condition score over late gestation were also similar between groups (Fig. 1A and B). At 65 days gestation, maternal (63.8 vs. 58.1 ng ml−1; CP vs. LP) and fetal (64.7 vs. 66.1 ng ml−1; CP vs. LP) plasma cortisol were not different and after post mortem only sex of the fetus, not maternal diet, had an effect on fetal body and organ weights (male > female; Table 2). Birth weights and neonatal fractional growth rate of singleton male and female lambs were not different between diet groups, with the exception of female lambs exposed to maternal low protein during early gestation (LPE), who grew at a lower daily rate (Fig. 1C and D).
Figure 1. The effect of a maternal low protein diet on maternal and neonatal characteristics.
Data are means + SEM: for ewes; control protein (CP, n = 8), low protein early (LPE, days 0–65 gestation, n = 10) and low protein late (LPL, days 65 to term, n = 10) and for offspring; CP, 4 males and 4 females; LPE, 4 males and 6 females; LPL and 5 males and 5 females. Data were analysed by General Linear Model for the fixed effects of treatment and time (maternal data) plus gender (offspring data) with testing for suitable interactions (Genstat v13, VSNi, UK). Maternal weight and body condition were recorded at intervals before and during gestation and birth weight within 12 h of birth. BW, birth weight; dGA, days gestation. In A and B, errors bars on the two symbols indicate variation associated with time (open) and treatment (filled), respectively.
Table 2.
Effect of maternal low protein diet on maternal and fetal weights at 65 days gestation
| Sex | P value | |||||
|---|---|---|---|---|---|---|
| Treatment | Male | Female | Treatment | Sex | Treat*Sex | |
| Maternal | ||||||
| Liver (g) | CP | – | 730 ± 25 | NS | NS | NS |
| LP | – | 730 ± 35 | ||||
| Fetal | ||||||
| Body weight (g) | CP | 111 ± 4 | 89 ± 5 | NS | 0.004 | NS |
| LP | 110 ± 9 | 95 ± 3 | ||||
| Brain weight (g) | CP | 3.46 ± 0.08 | 3.32 ± 0.15 | NS | NS | NS |
| LP | 3.55 ± 0.15 | 3.52 ± 0.04 | ||||
| Liver weight (g) | CP | 8.42 ± 0.39 | 6.92 ± 0.51 | NS | 0.007 | NS |
| LP | 9.02 ± 0.37 | 7.30 ± 0.52 | ||||
| Heart weight (g) | CP | 0.97 ± 0.08 | 0.90 ± 0.09 | NS | 0.04 | NS |
| LP | 1.17 ± 0.06 | 0.90 ± 0.07 | ||||
| Lung weight (g) | CP | 5.32 ± 0.45 | 4.79 ± 0.79 | NS | NS | NS |
| LP | 5.88 ± 0.99 | 4.76 ± 0.42 | ||||
| Kidney weight (g) | CP | 1.77 ± 0.22 | 1.43 ± 0.11 | NS | 0.09 | NS |
| LP | 1.66 ± 0.16 | 1.50 ± 0.07 | ||||
Data are estimated marginal means ± SEM for ewes (CP, n = 8; LP, n = 10) fed a control protein diet (CP fetuses; male, n = 4; female, n = 4) or a low protein diet to day 65 gestation (LP fetuses; male, n = 4; female, n = 6). Data were analysed by one-way ANOVA for the fixed effects of treatment (control vs. low protein), sex (male vs. female) or their interaction using Genstat v13 (VSNi, UK). Statistical significance was accepted at P < 0.05, but P = 0.06–0.10 taken to indicate an effect falling close to the arbitrary significance boundary. NS, not significant. CP, control protein diet; LP, low protein diet.
Fetal kidney angiogenesis and apoptosis at 0.44 gestation
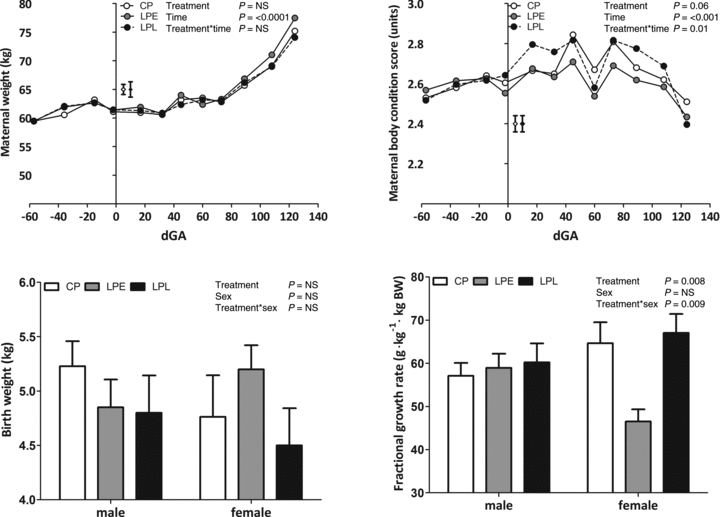
The quantity of immunopositive cells undergoing apoptosis was significantly higher (Fig. 2A and B) and positively stained for VEGFA protein significantly (P = 0.02) lower in the nephrogenic zone of the LPE fetal kidney vs. controls (Fig. 2C and D). In addition, at this time transcript expression for angiogenic markers including VEGFA, angiopoietin 1 (ANG1), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), transforming growth factor beta 1 (TGFβ1) and the Tie-2 receptor were all significantly decreased in male LPE fetuses only (P < 0.01, treatment × sex interaction for all cases; Fig. 2E–J). In addition, transcript expression of VEGFR1 showed a strong trend (P = 0.052) towards reduced expression in the LPE group in both sexes, relative to controls (17.8 ± 1.2 vs. 22.3 ± 1.36 × 10−3 units).
Figure 2. The effect of a low protein maternal diet on markers of angiogenesis and apoptosis in the fetal kidney.
Data are mean ± SEM where appropriate for: A, immunohistochemical staining of TUNEL +ve cells (representative micrograph from LPE group, black arrows indicate apoptotic cells); B, quantification of apoptotic data (CP, n = 8 (4 males, 4 females); LP, n = 10 (4 males, 6 females)); C, quantification of VEGFA protein abundance (CP, n = 8 (4 males, 4 females); LP, n = 10 (4 males, 6 females)); D, immunohistochemical staining of VEGFA protein abundance (representative micrograph from nephrogenic zone of LPE group, arrows indicate +ve staining); E–J, quantification of transcript expression (CP, n = 8 (4 males, 4 females); LP, n = 10 (4 males, 6 females)). Data were analysed using General Linear Model without or with log10 transformation (% apoptotic cells) for the fixed effects of treatment, gender and their interaction (Genstat v13, VSNi, UK). Log transformed data are presented as back-transformed means. Scale bars in A and D, 100 μm.
Adult renal function
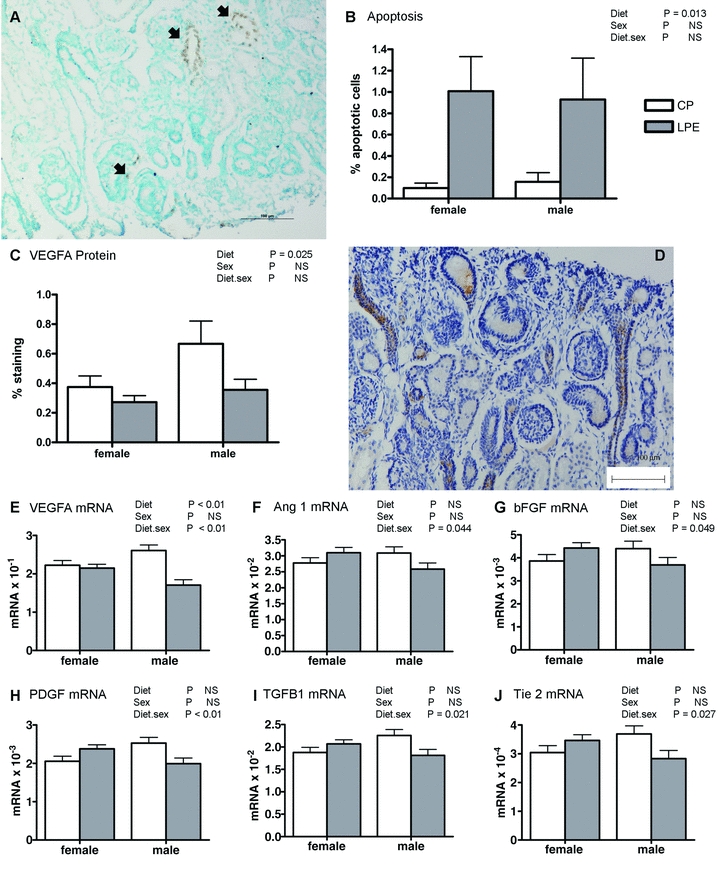
At 1.5 years of age and at a similar weight (47.1 ± 0.7 kg, all animals) the offspring were reared in an obesogenic environment to facilitate weight gain and fat deposition to a level considered obese i.e. an 8–10 unit increase in theoretical BMI (weight (kg)/height (m2)) from 22 to 32 (Fig. 3A). Offspring gained similar amounts of weight in each treatment group, but females gained less than males (Fig. 3B). Upon stabilisation of weight (weeks 25–33), all offspring underwent bilateral in vivo renography. In contrast to fetal life, we observed few treatment × sex interactions (i.e. an effect specifically in male LPE offspring) in the adult offspring, but rather revealed with obesity a number of important effects specific to both the male and female offspring exposed to a maternal low protein diet during early gestation (group LPE): relative to controls, the upslope of the renogram in LPE was unaffected (Fig. 4A) but all other indices suggested a subtle reduction in flow-based renal function; for example, the downslope (indicating clearance of tracer) tended to be decreased in LPE (Fig. 4B), the time to peak (indicating renal uptake of tracer) tended to be increased (Fig. 4C) with renal transit-time, a composite parameter of whole organ function, being significantly increased (∼20%) in LPE offspring (Fig. 4D). Measurement of glomerular filtration rate (GFR), however, indicated preservation of GFR in LPE vs. other groups in the longer term (i.e. 3–5 h, Table 3). Indeed, plasma Na+ (pooled estimate, 132 [128–143] mmol l−1; median [IQR]), plasma K+ (pooled estimate, 8.02 [7.12–8.54] mmol l−1) and plasma Cl− (pooled estimate, 108 [103–113] mmol l−1) were not different between treatment groups or sexes. However, the urinary albumin:creatinine ratio (ACR; mg μmol−1) was significantly (P = 0.03, Wilcoxons Rank sum test) increased in the LPE group, with no effect of gender (LPE, 8.88 [7.9–24.7] vs. CP, 1.73 [1.71–2.67]; LPL, 2.49 [1.63–2.98] median [IQR]), suggesting reduced glomerular barrier function. Stereological analysis of renal anatomy indicated significantly reduced (F = 3.7, P = 0.05) nephron number in the adult LPE kidney per se (LPE, 8.52 [8.04–9.28]× 106 vs. CP, 10.6 [10.5–10.9]× 106; LPL, 10.4 [8.19–11.9]× 106; Fig. 4E). Hence, using a multivariate linear discriminant analysis that predicted <99% of the variance for all renal outcome measures for all animals (e.g. upslope, time to peak, downslope, transit-time and nephron number) within just two eigenvectors (i.e. two linear orthogonals) indicates the separation of treatment groups with respect to in vivo adult renal function after exposure to a maternal low protein diet (Fig. 4F).
Figure 3. Weight gain during exposure to an obesogenic environment in adult sheep.
Data are means ± SEM for control protein (CP, n = 6), low protein early (LPE, days 0–65 gestation, n = 7) and low protein late (LPL, days 66 to term, n = 6). There were 3 males and 3 females in CP and LPL and 3 males and 4 females in LPE. A, absolute body weight gain; B, kgs of weight gain after exposure to the obesogenic diet. Data were analysed by General Linear Model for the fixed effects of treatment, gender and time with testing for suitable interactions (Genstat v13, VSNi, UK). NS, non-significant.
Figure 4. The effect of a low protein maternal diet on adult offspring renal function as assessed by renography.
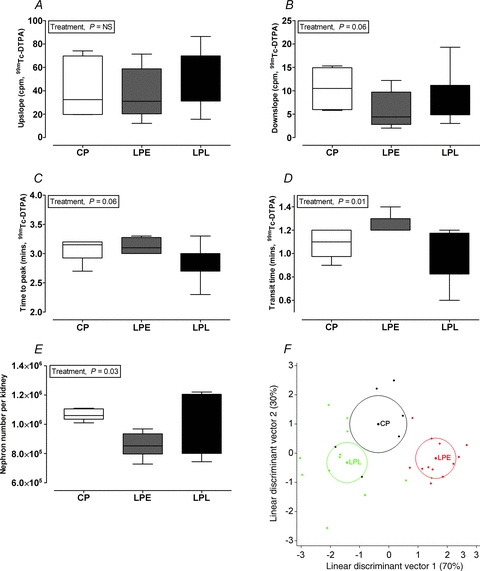
Boxplots show median (IQR with whiskers representing 5–95th centile) for control protein (CP, n = 6), low protein early (LPE, days 0–65 gestation, n = 7) and low protein late (LPL, days 66 to term, n = 6). There were 3 males and 3 females in CP and LPL and 3 males and 4 females in LPE. Data were analysed by Restricted Maximum Likelihood (REML) for the fixed effects of treatment and gender with interaction (Genstat v13, VSNi, UK). Renal function was assessed by renography using Tc99m-DTPA. The upslope (A) is defined as the positive slope of the renogram occurring before the peak and computed using the X and Y values of the renogram at 5 and 90% of the peak Y; Time to peak (B) is from bolus injection to peak counts in the right kidney, the downslope is defined as the negative slope of the renogram occurring after the peak and computed using the X and Y values of the renogram at the peak and half-peak value on the downslope of the curve (C); transit time is the mean time taken for tracer to pass through the kidney (D); nephron number was estimated by unbiased stereology (E) and F shows the mean for each treatment group (circles representing 95% confidence interval of the mean) after linear discriminant analysis of normalised (i.e. z-scores) data for upslope, time to peak, downslope, transit time, nephron number and mean glomerular volume.
Table 3.
Effect of maternal low protein diet on offspring glomerular filtration rate
| Sex | P value | |||||
|---|---|---|---|---|---|---|
| Treatment | Male | Female | Treatment | Sex | Treat*Sex | |
| GFR (ml ml−1) | CP | 146 ± 16 | 147 ± 23 | NS | NS | NS |
| LPE | 114 ± 23 | 150 ± 14 | ||||
| LPL | 133 ± 23 | 139 ± 16 | ||||
| GFR (ml ml−1 (g kidney)−1) | CP | 0.97 ± 0.14 | 1.12 ± 0.21 | NS | NS | NS |
| LPE | 0.89 ± 0.21 | 1.32 ± 0.13 | ||||
| LPL | 1.02 ± 0.21 | 0.98 ± 0.14 | ||||
| GFR (ml ml−1 (kg lean mass)−1) | CP | 3.21 ± 0.37 | 3.68 ± 0.53 | NS | NS | NS |
| LPE | 3.27 ± 0.53 | 3.78 ± 0.33 | ||||
| LPL | 2.84 ± 0.53 | 3.63 ± 0.37 | ||||
Glomerular filtration rate was assessed through decay of Tc99m-DTPA in plasma samples collected after 3, 4 and 5 h post 100 MBq i.v. bolus dose. Ewes were killed as per standard operating procedures and the wet weight of organs recorded. Data are means ± SEM for offspring of ewes fed a control protein diet (CP, n = 6), or a low protein diet during early gestation (LPE, days 0–65, n = 7) or a low protein diet during late gestation (LPL, days 66 to term, n = 6). There were 3 males and 3 females in CP and LPL and 3 males and 4 females in LPE. Data were analysed by General Linear Model for the fixed effects of treatment, sex and their interaction with (Genstat v13, VSNi, UK). NS, not significant; GFR, glomerular filtration rate.
Adult renal structure
As assessed by stereology, reduced nephron number and microalbuminuria in LPE vs. controls was accompanied by increased mean glomerular volume in this group (Table 4), without effect on overall kidney mass, volume or relative volume of the renal cortex (Table 4). Histological examination of glomeruli and proximal tubules in all treatment groups indicated no obvious morphological or structural abnormality, no consistent lymphocyte infiltration or any evidence of glomerulosclerosis in LPE or LPL vs. CP (Fig. 5A–D). However, through immunohistochemistry, we found significantly decreased (P < 0.001) abundance of CD34 protein (an endothelial cell marker) in LPE vs. controls (quantification in Fig. 5H, micrograph in Fig. 5I), suggesting microvascular rarefaction in this group with increased (P = 0.02) abundance of VEGFA (quantification in Fig. 5G, micrograph in Fig. 5E). Further analysis of transcripts for angiogenic markers revealed a consistent tendency for increased expression in LPE vs. other groups (VEGFA, P = 0.07; VEGFR1, P = 0.05; VEGFR2, P = 0.09) without any consistent indication of increased intra-renal endoplasmic reticulum stress (Table 5).
Table 4.
Effect of a maternal low protein diet on offspring kidney weight and stereology
| Sex | P value | |||||
|---|---|---|---|---|---|---|
| Treatment | Male | Female | Treatment | Sex | Treat*sex | |
| Kidney weight (g (kg BW)−1) | CP | 2.63 ± 0.20 | 2.66 ± 0.28 | NS | NS | NS |
| LPE | 2.72 ± 0.29 | 2.13 ± 0.18 | ||||
| LPL | 2.44 ± 0.29 | 2.61 ± 0.20 | ||||
| Kidney volume (cm3) | CP | 79.67 ± 5.99 | 67.50 ± 7.91 | NS | NS | NS |
| LPE | 73.85 ± 7.92 | 59.20 ± 5.01 | ||||
| LPL | 73.00 ± 7.92 | 70.50 ± 5.60 | ||||
| Per cent cortex volume (%) | CP | 61.17 ± 3.37 | 67.62 ± 5.26 | NS | NS | NS |
| LPE | 70.01 ± 5.45 | 67.14 ± 3.31 | ||||
| LPL | 68.60 ± 5.34 | 67.14 ± 3.70 | ||||
| Cortex volume (cm3) | CP | 48.29 ± 4.00 | 44.82 ± 5.65 | NS | NS | NS |
| LPE | 51.55 ± 5.65 | 39.82 ± 3.57 | ||||
| LPL | 50.49 ± 5.65 | 47.32 ± 3.99 | ||||
| Total glomerular volume (cm3) | CP | 1.96 ± 0.17 | 1.79 ± 0.23 | NS | 0.011 | NS |
| LPE | 2.39 ± 0.23 | 1.70 ± 0.15 | ||||
| LPL | 2.04 ± 0.23 | 1.44 ± 0.17 | ||||
| Mean glomerular volume (mm3× 10−3) | CP | 1.81 ± 0.17 | 2.03 ± 0.24 | 0.022 | NS | NS |
| LPE | 2.61 ± 0.24 | 2.06 ± 0.15 | ||||
| LPL | 1.93 ± 0.24 | 1.47 ± 0.17 | ||||
Data are means ± SEM for offspring of ewes fed a control protein diet (CP, n = 6), or a low protein diet during early gestation (LPE, days 0–65, n = 7) or a low protein diet during late gestation (LPL, days 66 to term, n = 6). There were 3 males and 3 females in CP and LPL and 3 males and 4 females in LPE. Data were analysed by General Linear Model for the fixed effects of treatment, sex and their interaction (Genstat v13, VSNi, UK). NS, not significant.
Figure 5. The effect of a low protein maternal diet on adult offspring renal structure and vascularisation as assessed by expression and abundance of markers for renal endothelial cells.
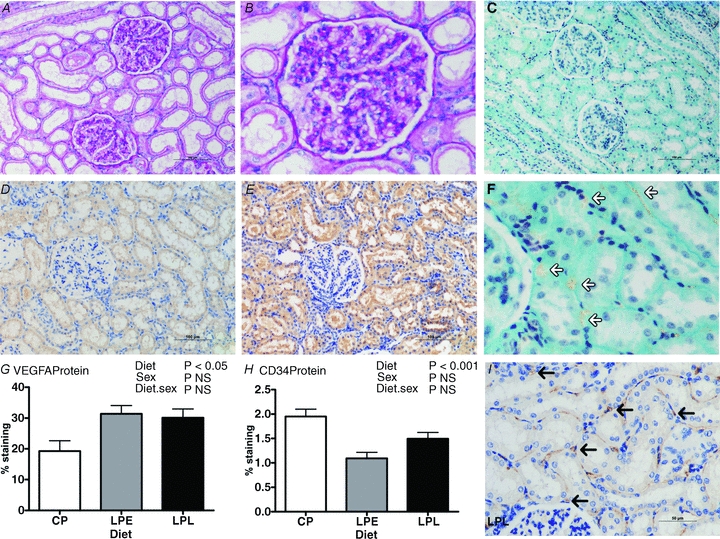
Data are means ± SEM for control protein (CP, n = 6), low protein early (LPE, days 0–65 gestation, n = 7) and low protein late (LPL, days 66 to term, n = 6). There were 3 males and 3 females in CP and LPL and 3 males and 4 females in LPE. Data were analysed by General Linear Model for the fixed effects of treatment and sex with their interaction (Genstat v13, VSNi, UK). A and B, representative histological sections from the LPE group (A, magnification ×200; B, magnification ×400) stained with periodic acid Schiffs reagent shows normal kidney structure; C and F, representative histological section (×200) from the LPE group stained with trichrome; D and E, representative histological sections with immunohistochemical staining for vascular endothelial growth factor A (VEGFA) in CP (D) vs. LPE (E), and G shows quantification of VEGFA staining (using ImagePro MC); H, quantification of CD34 stained sections (using ImagePro MC) and I, a representative section from CP, immunostained for CD34 (black arrows indicate endothelial cells). Scale bars in A, C, D and E, 100 μm; in I, 50 μm.
Table 5.
Effect of a maternal low protein diet on renal endoplasmic reticulum stress genes
| Sex | P value | |||||
|---|---|---|---|---|---|---|
| Treatment | Male | Female | Treatment | Sex | Treat*sex | |
| ATF4 | CP | 5.85 ± 0.80 | 4.95 ± 1.13 | NS | NS | NS |
| LPE | 5.00 ± 1.13 | 5.29 ± 0.72 | ||||
| LPL | 4.51 ± 1.13 | 6.33 ± 0.80 | ||||
| ATF6 | CP | 5.36 ± 0.98 | 5.86 ± 1.39 | NS | NS | NS |
| LPE | 5.90 ± 1.39 | 4.73 ± 0.88 | ||||
| LPL | 4.88 ± 1.39 | 4.83 ± 0.98 | ||||
| Bax | CP | 8.37 ± 0.98 | 9.23 ± 1.38 | NS | NS | NS |
| LPE | 8.39 ± 1.38 | 8.51 ± 0.87 | ||||
| LPL | 10.40 ± 1.38 | 8.48 ± 0.98 | ||||
| CD68 | CP | 0.09 ± 0.03 | 0.11 ± 0.04 | NS | NS | NS |
| LPE | 0.11 ± 0.04 | 0.18 ± 0.03 | ||||
| LPL | 0.22 ± 0.04 | 0.17 ± 0.03 | ||||
| DDIT3 | CP | 0.68 ± 0.16 | 0.46 ± 0.15 | 0.02 | NS | NS |
| LPE | 0.81 ± 0.26 | 1.17 ± 0.24 | ||||
| LPL | 0.62 ± 0.20 | 1.08 ± 0.25 | ||||
| HSPA5 | CP | 2.80 ± 0.47 | 2.28 ± 0.54 | NS | NS | NS |
| LPE | 2.10 ± 0.50 | 2.84 ± 0.43 | ||||
| LPL | 2.06 ± 0.49 | 2.76 ± 0.47 | ||||
| MCP-1 | CP | 1.26 ± 0.30 | 0.69 ± 0.43 | NS | NS | NS |
| LPE | 1.45 ± 0.43 | 1.40 ± 0.27 | ||||
| LPL | 1.80 ± 0.43 | 1.27 ± 0.30 | ||||
Advanced relative quantification of mRNA normalised to cyclophilin using Roche Lightcycler 480 software. Data are means ± SEM for offspring of ewes fed a control protein diet (CP, n = 6), or a low protein diet during early gestation (LPE, days 0–65, n = 7) or a low protein diet during late gestation (LPL, days 66 to term, n = 6). There were 3 males and 3 females in CP and LPL and 3 males and 4 females in LPE. Data were analysed by General Linear Model for the fixed effects of treatment, sex and their interaction (Genstat v13, VSNi, UK). NS, not significant. Gene symbols according to HUGO nomenclature (http://www.genenames.org/); ATF, activating transcription factor; DDIT3, DNA damage inducible transcript 3; HSPA5, heat shock 70 kDa Protein 5; MCP-1, monocyte chemotactic protein 1.
Discussion
In this study we have traced a pathway from mild, maternal protein-energy malnutrition (PEM) acting during early gestation to blunt the microvascular development of the fetal kidney in a sex-specific manner to a structurally- and functionally-compromised adult kidney with evidence of microvascular rarefaction in the face of an obesogenic environment. The potential implications of the work are relevant for those individuals born to mothers with varying degrees of PEM that may retain residual, asymptomatic effects of the maternal nutritional imbalance within their kidneys that manifest later in life as an earlier than expected onset of CKD, especially when coupled with the ever-increasing likelihood of having to metabolically compensate to an obesogenic adult environment.
In this study of PEM in sheep, despite a 50% decrease in the availability of protein (8.7 vs. 16.9 g kg−1 MJ) fed to pregnant ewes we observed few effects of the diet on maternal weight or her body condition score (a subjective marker of overall fat mass) or on fetal body or organ weights at 0.44 gestation or at term (e.g. Fig. 1, Table 1), suggesting the overall dietary regimen to be modest. This contrasts with studies in laboratory rodents in which a similar degree of protein restriction often results in disproportionate fetal growth (Langley-Evans et al. 1996), low birth weight (Zeman, 1967; Levy & Jackson, 1993; Desai et al. 1996) and a reported 20–50% deficit in nephron endowment in the permanent kidney (Vehaskari et al. 2001; McMullen et al. 2004; Hoppe et al. 2006). In this study, we observe only a small, ∼15% reduction in nephron number, but a large (∼45%) compensatory increase in mean glomerular volume in LPE. The likely explanation is first due to allometry; larger animals like sheep and man have greater metabolic reserve capacity (and lower surface area:volume ratio) compared to smaller mammals and are thus more able to metabolically adjust to nutritional deficit. Second, being monotocous, the anabolic demands required to support the uteroplacental unit in sheep are much lower relative to the polytocous rodent (McCance & Widdowson, 1986). Hence, nutritional protocols in laboratory rodents probably present a more severe phenotype, due to the relatively greater impact of malnutrition. Nevertheless, in this study, mild maternal protein:energy malnutrition translated to the fetal environment with significant impact upon an organ undergoing hyperplastic growth at that time e.g. the fetal kidney. This is emphasised by the observation that, for the LPL group, which experienced a longer period of maternal protein restriction, spanning almost the entire period of hypertrophic growth of the fetus (weights at 65 days gestation are only ∼3–4% weight at term), there was no consistent renal phenotype. To our knowledge, this study is the first to demonstrate a renal phenotype in large animals after changes to the macronutrient composition of the maternal diet and suggests that the protein:energy ratio of the maternal diet (i.e. diet quality) is more important in terms of developmental programming than a balanced reduction in nutrient intake per se (i.e. diet quantity) (Brennan et al. 2005). This clearly has relevance when considered in context to the marked changes in nutritional quality that have occurred over the last 50 years, in the UK (Prynne et al. 1999) but also elsewhere.
The small reduction in the full complement of nephrons in LPE illustrates an important consequence of maternal protein-energy deprivation but the reported absolute numbers are also of academic value and interest and deserve comment. Epidemiological studies have illustrated how an optimal fetal environment is a prerequisite for adequate kidney development and for good blood pressure control later in life (Manalich et al. 2000; Hughson et al. 2003; Keller et al. 2003). Our stereologically derived estimate of nephron number in control sheep (∼1,064,611) was determined according to the methodology of Keller (Keller et al. 2003), but contrasts markedly with other sterologically derived estimates for sheep (e.g. from 289,000 to 420,000; Bains et al. 1996; Gray et al. 2008). Nephron number may subtly vary within a species (Zimanyi et al. 2009; McNamara et al. 2010), but this is usually due to size and lean mass, as the number of glomeruli are highly correlated with both birth weight (up to 3 kg, 250,000 glomeruli (kg BW)−1; r = 0.870, P < 0.0001) (Manalich et al. 2000) and weight-adjusted metabolic mass between species (Kunkel, 1930). Using log10 data for published values of nephron number from a mouse (20 g) to an elephant (3700 kg) generates an equation of y = 0.613x + 3.116, therefore accurately predicting an adult sheep (50–70 kg) to have between 994,050 and 1,221,839 nephrons per kidney. This estimate encompasses the reported values in this manuscript, but not others (Bains et al. 1996; Gray et al. 2008) and emphasises, perhaps, that prenatal effects on nephron number are best considered relative to appropriate controls rather than to species-specific values.
In rodents, Welham et al. (2002) were the first to suggest a potential mechanistic pathway to explain loss of nephrons. They just with maternal protein restriction – increased loss (apoptotic) of mesenchymal (either nephron progenitor or interstitial supportive) cells, but the determining factor remained elusive. Here, we replicate these findings for the first time in a large animal model and demonstrate a potential mechanism: fetal hyperplastic kidneys exposed to maternal low protein had increased apoptosis and decreased angiogenesis in the nephrogenic zone. While previous work has shown how one or other of these pathways may be influenced by maternal nutrition (Welham et al. 2002; Cox et al. 2006) we show a coherent effect on both. Taken together, the data suggest that maternal protein-energy malnutrition specifically limits fetal intrarenal vascularity, resulting in blunted nephrogenesis and restricted nephron endowment of the permanent kidney, effects that are asymptomatic until superimposed on an adult obesogenic environment when renal dysfunction is revealed.
The extent of the renal dysfunction in LPE (microalbuminuria and blunted intra-renal transit of radionuclide) is subtle, but considering the age of the animals (1.5–2 years of age of an expected ∼10–12 year lifespan) is significant. Microalbuminuria is a good early predictor of later renal disease (Painter et al. 2005) but without aged animals, one can only make the assumption that the programmed phenotype would deteriorate with age. From epidemiological studies, those human individuals exposed to famine during early (but not late) gestation were at higher risk of developing obesity (Ravelli et al. 1976) with concomitant microalbuminuria in middle age (Painter et al. 2005). In our study, microalbuminuria in LPE, marking reduced renal barrier function, is probably due to single-nephron hyperfiltration as this occurs with obesity per se, as shown by us in a previous study (Williams et al. 2007) but also may be exacerbated in this study by (1) reduced nephron number, (2) microvascular rarefaction (marked by reduced intra-renal endothelial CD34+ abundance) and (3) increased intra-renal pressure (indicated by reduced Tc-99m DTPA time to peak and intra-renal transit-time). With no clear difference in gross renal morphology, inflammatory infiltration or indices of intra-renal stress then the functional deficits in LPE (both males and females) suggests minimal change disease, perhaps due to podocyte effacement (Haraldsson et al. 2008; Veron et al. 2010). However, lack of appropriately fixed tissue for transmission electron microscopy precludes this possibility being explored. Nevertheless, the molecular and immunohistochemical data (increased VEGFA) suggest greater support for a compromised renal barrier – VEGFA is highly expressed in podocytes, plays an important role in establishing and maintaining the glomerular barrier (Eremina et al. 2008) and when overexpressed, is associated with glomerular disease in mice (Veron et al. 2010).
Finally, in fetal life, many aspects of the adverse renal phenotype described were sex-specifically altered, that is, the effect was greater in males vs. females (witness the reduction in angiogenic factors in the male LPE fetal kidney as an example). A sex effect in the experimental endpoints of studies examining relationships between maternal diet and offspring phenotype (males adversely affected more often than females) is not uncommon (Grigore et al. 2008). These effects are unlikely due to differences in the plasma concentration of sex hormones (very low in the fetus, male adult offspring were castrates) but could be related to growth since males (fetuses and offspring) grow at a faster rate than females (reflected at day 65 gestation here, and in fractional growth rate postnatally). Faster growing animals may be more susceptible to deficits and/or changes in the pattern of substrate supply (in particular of amino acids). Alternatively, the effect may be a product of nutritionally induced sex-specific epigenetic programming – a phenomenon recently described in bovine blastocysts (Bermejo-Alvarez et al. 2010) but also in adult sheep (Sinclair et al. 2007). However, in this study at 2 years of age, the sex-specific programming of the kidney was less evident. Our study was powered to reveal sex-specific differences in renal function (e.g. transit time) as our primary outcome. Thus, we are confident that if any clear sex-specific differences in renal function (and molecular end-points) were to exist we would have been able to reveal them with our study design. However, for alternative outcomes in which a treatment × sex interaction appears likely (e.g. for mean glomerular volume the effect size was +27%, male LPE vs. controls) but for which the measurement error increases, then we acknowledge a potential Type II error (in this example we have 85% power to detect a 27% difference at the treatment level, but only 56% at the treatment × sex level). Nevertheless, an adult renal phenotype has been described that indicates a specific effect of maternal low protein acting during early development of the fetal kidney. We have described a potential mechanism, at this time, that is mediated through the key nexus for control of angiogenesis, VEGF, and that the blunting of renal development appears more marked in male fetuses. Further, mechanistic studies aimed at determining the nature of the relationship between maternal protein-energy malnutrition, the fetal nutritional environment and renal growth (microvascular, nephrogenesis) in male and female fetuses is warranted.
To conclude, protein-energy malnutrition (PEM) is highly prevalent in developing countries with up to 20%, 8% and 0.8% children in India, China and the USA, respectively, being affected (equating to ∼130, 18 and 1.1 million individuals; WHO, 2011). Whilst it is less prevalent in developed, Westernised societies the incidence of pregnancy-induced nausea (75–90% women) and, in extremis, hyperemesis gravidarum (1% women) during early pregnancy means that many women (and their fetuses) may experience some degree of macro/micronutrient deficiency at this time (Fejzo et al. 2009). Here we show that PEM at this time may specifically impact the developing, hyperplastic fetal kidney to limit renal vascularisation and nephrogenesis with functional consequences later in life that are predicted to exacerbate the age-related decline in renal function. With the prevalence of renal morbidity increasing by ∼5% per annum largely through the increased prevalence of obesity and T2D then a clinical focus on a good, balanced and high-quality maternal diet as a potentially modifiable risk factor may help to mitigate a proportion of the expected cases of CKD in the future.
Acknowledgments
The authors would like to acknowledge the contributions of: Dr Ali Mobasheri (University of Nottingham) and Dr Cindy Wong (Abcam) for the gift of the CD34 antibody, Dr Tom McCulloch (Consultant Pathologist, Nottingham University Hospitals Trust) for advice concerning renal histopathology and Dr Jim Craigon for statistical advice on the manuscript. Mrs. Louise Lloyd was supported by a British Heart Foundation PhD studentship (FS/09/011/26562).
Glossary
Abbreviations
- ACR
albumin:creatinine ratio
- CKD
chronic kidney disease
- PEM
protein-energy malnutrition
- Tc99m-DTPA
technetium−99 diethylenetriaminepentaacetic acid
- VEGF
vascular endothelial growth factor
Author contributions
The experiments were conducted at the School of Veterinary Medicine and Science, Sutton Bonington Campus, University of Nottingham. D.S.G., P.R. and S.M.R. conceived and designed the experiments; L.J.L., D.S.G., P.R., T.F. and S.M.R. conducted research; L.J.L. and D.S.G. analysed and interpreted research and L.J.L. and D.S.G wrote the paper. D.S.G. has primary responsibility for its final content and all authors have approved the final version of the manuscript.
References
- Amann K, Wanner C, Ritz E. Cross-talk between the kidney and the cardiovascular system. J Am Soc Nephrol. 2006;17:2112–2119. doi: 10.1681/ASN.2006030204. [DOI] [PubMed] [Google Scholar]
- Bains RK, Sibbons PD, Murray RD, Howard CV, Van Velzen D. Stereological estimation of the absolute number of glomeruli in the kidneys of lambs. Res Vet Sci. 1996;60:122–125. doi: 10.1016/s0034-5288(96)90005-3. [DOI] [PubMed] [Google Scholar]
- Bakris GL, Ritz E. The message for World Kidney Day 2009: hypertension and kidney disease: a marriage that should be prevented. Kidney Int. 2009;75:449–452. doi: 10.1038/ki.2008.694. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C. Low birth weight and hypertension. BMJ. 1988;297:134–135. doi: 10.1136/bmj.297.6641.134-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Forsen TJ, Kajantie E, Eriksson JG. Trajectories of growth among children who have coronary events as adults. N Engl J Med. 2005;353:1802–1809. doi: 10.1056/NEJMoa044160. [DOI] [PubMed] [Google Scholar]
- Bermejo-Alvarez P, Rizos D, Rath D, Lonergan P, Gutierrez-Adan A. Sex determines the expression level of one third of the actively expressed genes in bovine blastocysts. Proc Natl Acad Sci U S A. 2010;107:3394–3399. doi: 10.1073/pnas.0913843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan KA, Gopalakrishnan GS, Kurlak L, Rhind SM, Kyle CE, Brooks AN, et al. Impact of maternal undernutrition and fetal number on glucocorticoid, growth hormone and insulin-like growth factor receptor mRNA abundance in the ovine fetal kidney. Reproduction. 2005;129:151–159. doi: 10.1530/rep.1.00229. [DOI] [PubMed] [Google Scholar]
- Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens. 1988;1:335–347. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cignarelli M, Lamacchia O. Obesity and kidney disease. Nutr Metab Cardiovasc Dis. 2007;17:757–762. doi: 10.1016/j.numecd.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- Cox LA, Nijland MJ, Gilbert JS, Schlabritz-Loutsevitch NE, Hubbard GB, McDonald TJ, et al. Effect of 30 per cent maternal nutrient restriction from 0.16 to 0.5 gestation on fetal baboon kidney gene expression. J Physiol. 2006;572:67–85. doi: 10.1113/jphysiol.2006.106872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispi F, Bijnens B, Figueras F, Bartrons J, Eixarch E, Le Noble F, et al. Fetal growth restriction results in remodelled and less efficient hearts in children. Circulation. 2010;121:2427–2436. doi: 10.1161/CIRCULATIONAHA.110.937995. [DOI] [PubMed] [Google Scholar]
- Crook ED. Diabetic renal disease in African Americans. Am J Med Sci. 2002;323:78–84. doi: 10.1097/00000441-200202000-00004. [DOI] [PubMed] [Google Scholar]
- Daar AS, Singer PA, Persad DL, Pramming SK, Matthews DR, Beaglehole R, et al. Grand challenges in chronic non-communicable diseases. Nature. 2007;450:494–496. doi: 10.1038/450494a. [DOI] [PubMed] [Google Scholar]
- Desai M, Crowther NJ, Lucas A, Hales CN. Organ-selective growth in the offspring of protein-restricted mothers. Br J Nutr. 1996;76:591–603. doi: 10.1079/bjn19960065. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton SB, Eaton SB., 3rd Paleolithic vs modern diets – selected pathophysiological implications. Eur J Nutr. 2000;39:67–70. doi: 10.1007/s003940070032. [DOI] [PubMed] [Google Scholar]
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA. 2010;303:2043–2050. doi: 10.1001/jama.2010.650. [DOI] [PubMed] [Google Scholar]
- Ejerblad E, Fored CM, Lindblad P, Fryzek J, McLaughlin JK, Nyren O. Obesity and risk for chronic renal failure. J Am Soc Nephrol. 2006;17:1695–1702. doi: 10.1681/ASN.2005060638. [DOI] [PubMed] [Google Scholar]
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo MS, Poursharif B, Korst LM, Munch S, MacGibbon KW, Romero R, Goodwin TM. Symptoms and pregnancy outcomes associated with extreme weight loss among women with hyperemesis gravidarum. J Womens Health (Larchmt) 2009;18:1981–1987. doi: 10.1089/jwh.2009.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CS, Gona P, Larson MG, Selhub J, Tofler G, Hwang S-J, et al. A multi-marker approach to predict incident CKD and microalbuminuria. J Am Soc Nephrol. 2010;21:2143–2149. doi: 10.1681/ASN.2010010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol. 2005;565:137–147. doi: 10.1113/jphysiol.2005.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleadhill A, Marlin D, Harris PA, Michell AR. Use of a three-blood-sample plasma clearance technique to measure GFR in horses. Vet J. 1999;158:204–209. doi: 10.1053/tvjl.1999.0385. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SP, Kenna K, Bertram JF, Hoy WE, Yan EB, Bocking AD, et al. Repeated ethanol exposure during late gestation decreases nephron endowment in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2008;295:R568–R574. doi: 10.1152/ajpregu.90316.2008. [DOI] [PubMed] [Google Scholar]
- Griffin KA, Kramer H, Bidani AK. Adverse renal consequences of obesity. Am J Physiol Renal Physiol. 2008;294:F685–F696. doi: 10.1152/ajprenal.00324.2007. [DOI] [PubMed] [Google Scholar]
- Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gend Med. 2008;5:S121–S132. doi: 10.1016/j.genm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- Hoppe CC, Evans RG, Moritz KM, Cullen-McEwen LA, Fitzgerald SM, Dowling J, Bertram JF. Combined prenatal and postnatal protein restriction influences adult kidney structure, function, and arterial pressure. Am J Physiol Regul Integr Comp Physiol. 2006;292:R462–R469. doi: 10.1152/ajpregu.00079.2006. [DOI] [PubMed] [Google Scholar]
- Hoy WE, Hughson MD, Bertram JF, Douglas-Denton R, Amann K. Nephron number, hypertension, renal disease, and renal failure. J Am Soc Nephrol. 2005;16:2557–2564. doi: 10.1681/ASN.2005020172. [DOI] [PubMed] [Google Scholar]
- Huang C, Li Z, Wang M, Martorell R. Early life exposure to the 1959–1961 Chinese famine has long-term health consequences. J Nutr. 2010;140:1874–1878. doi: 10.3945/jn.110.121293. [DOI] [PubMed] [Google Scholar]
- Hughson M, Farris AB, III, Douglas-Denton R, Hoy WE, Bertram JF. Glomerular number and size in autopsy kidneys: The relationship to birth weight. Kidney Int. 2003;63:2113–2122. doi: 10.1046/j.1523-1755.2003.00018.x. [DOI] [PubMed] [Google Scholar]
- Huxley R, Neil A, Collins R. Unravelling the fetal origins hypothesis: is there really an inverse association between birthweight and subsequent blood pressure? Lancet. 2002;360:659–665. doi: 10.1016/S0140-6736(02)09834-3. [DOI] [PubMed] [Google Scholar]
- Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes. 2008;32:1431–1437. doi: 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- Kunkel PA. The number and size of the glomeruli in the kidney of several mammals. Bulletin of the Johns Hopkins Hospital. 1930;47:285–291. [Google Scholar]
- Langley-Evans SC, Gardner DS, Jackson AA. Association of disproportionate growth of fetal rats in late gestation with raised systolic blood pressure in later life. J Reprod Fertil. 1996;106:307–312. doi: 10.1530/jrf.0.1060307. [DOI] [PubMed] [Google Scholar]
- Law CM, Shiell AW. Is blood pressure inversely related to birth weight? The strength of evidence from a systematic review of the literature. J Hypertens. 1996;14:935–941. [PubMed] [Google Scholar]
- Levy L, Jackson AA. Modest restriction of dietary protein during pregnancy in the rat: fetal and placental growth. J Dev Physiol. 1993;19:113–118. [PubMed] [Google Scholar]
- Lysaght MJ. Maintenance dialysis population dynamics: Current trends and long-term implications. J Am Soc Nephrol. 2002;13:S37–S40. [PubMed] [Google Scholar]
- McCance RA, Widdowson EM. Glimpses of comparative growth and development. In: Falkner F, Tanner JM, editors. Developmental Biology and Prenatal Growth. London: Plenum Press; 1986. pp. 133–151. [Google Scholar]
- Mackenzie HS, Brenner BM. Fewer nephrons at birth: a missing link in the etiology of essential hypertension? AmJ Kidney Dis. 1995;26:91–98. doi: 10.1016/0272-6386(95)90161-2. [DOI] [PubMed] [Google Scholar]
- McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- McMullen S, Gardner DS, Langley-Evans SC. Prenatal programming of angiotensin II type 2 receptor expression in the rat. Br J Nutr. 2004;91:133–140. doi: 10.1079/bjn20031029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara BJ, Diouf B, Douglas-Denton RN, Hughson MD, Hoy WE, Bertram JF. A comparison of nephron number, glomerular volume and kidney weight in Senegalese Africans and African Americans. Nephrol Dial Transplant. 2010;25:1514–1520. doi: 10.1093/ndt/gfq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manalich R, Reyes L, Herrera M, Melendi C, Fundora I. Relationship between weight at birth and the number and size of renal glomeruli in humans: a histomorphometric study. Kidney Int. 2000;58:770–773. doi: 10.1046/j.1523-1755.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Singh RR, Probyn ME, Denton KM. Developmental programming of a reduced nephron endowment: more than just a baby's birth weight. Am J Physiol Renal Physiol. 2009;296:F1–F9. doi: 10.1152/ajprenal.00049.2008. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Wintour EM. Functional development of the meso- and metanephros. Pediatr Nephrol. 1999;13:171–178. doi: 10.1007/s004670050587. [DOI] [PubMed] [Google Scholar]
- Narayan KMV, Ali MK, Koplan JP. Global noncommunicable diseases – where worlds meet. N Engl J Med. 2010;363:1196–1198. doi: 10.1056/NEJMp1002024. [DOI] [PubMed] [Google Scholar]
- Nehiri T, Duong Van Huyen J-P, Viltard M, Fassot C, Heudes D, Freund N, et al. Exposure to maternal diabetes induces salt-sensitive hypertension and impairs renal function in adult rat offspring. Diabetes. 2008;57:2167–2175. doi: 10.2337/db07-0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S-F, Lin RCY, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature. 2010;467:963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- Ojeda NB, Grigore D, Alexander BT. Intrauterine growth restriction: fetal programming of hypertension and kidney disease. Adv Chronic Kidney Dis. 2008;15:101–106. doi: 10.1053/j.ackd.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RC, Roseboom TJ, van Montfrans GA, Bossuyt PM, Krediet RT, Osmond C, et al. Microalbuminuria in adults after prenatal exposure to the Dutch famine. J Am Soc Nephrol. 2005;16:189–194. doi: 10.1681/ASN.2004060474. [DOI] [PubMed] [Google Scholar]
- Prynne CJ, Paul AA, Price GM, Day KC, Hilder WS, Wadsworth ME. Food and nutrient intake of a national sample of 4-year-old children in 1950: comparison with the 1990s. Public Health Nutr. 1999;2:537–547. doi: 10.1017/s1368980099000725. [DOI] [PubMed] [Google Scholar]
- Puddu M, Fanos V, Podda F, Zaffanello M. The kidney from prenatal to adult life: perinatal programming and reduction of number of nephrons during development. Am J Nephrol. 2009;30:162–170. doi: 10.1159/000211324. [DOI] [PubMed] [Google Scholar]
- Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- Reilly RF, Perazella MA. Chronic kidney disease: a new approach to an old problem. Conn Med. 2002;66:579–583. [PubMed] [Google Scholar]
- Rhodes P, Craigon J, Gray C, Rhind SM, Loughna PT, Gardner DS. Adult-onset obesity reveals prenatal programming of glucose-insulin sensitivity in male sheep nutrient restricted during late gestation. PLoS One. 2009;4:e7393. doi: 10.1371/journal.pone.0007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, van der Meulen JH, Ravelli AC, van Montfrans GA, Osmond C, Barker DJ, Bleker OP. Blood pressure in adults after prenatal exposure to famine. J Hypertens. 1999;17:325–330. doi: 10.1097/00004872-199917030-00004. [DOI] [PubMed] [Google Scholar]
- Sharkey D, Fainberg HP, Wilson V, Harvey E, Gardner DS, Symonds ME, Budge H. Impact of early onset obesity and hypertension on the unfolded protein response in renal tissues of juvenile sheep. Hypertension. 2009;53:925–931. doi: 10.1161/HYPERTENSIONAHA.108.122812. [DOI] [PubMed] [Google Scholar]
- Simonetti GD, Raio L, Surbek D, Nelle M, Frey FJ, Mohaupt MG. Salt sensitivity of children with low birth weight. Hypertension. 2008;52:625–630. doi: 10.1161/HYPERTENSIONAHA.108.114983. [DOI] [PubMed] [Google Scholar]
- Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci U S A. 2007;104:19351–19356. doi: 10.1073/pnas.0707258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeninga N, Schreuder MF, Bökenkamp A, Delemarre-van de Waal HA, van Wijk JA. Influence of low birth weight on minimal change nephrotic syndrome in children, including a meta-analysis. Nephrol Dial Transplant. 2008;23:1615–1620. doi: 10.1093/ndt/gfm829. [DOI] [PubMed] [Google Scholar]
- Tran S, Chen YW, Chenier I, Chan JSD, Quaggin S, Hebert MJ, et al. Maternal diabetes modulates renal morphogenesis in offspring. J Am Soc Nephrol. 2008;19:943–952. doi: 10.1681/ASN.2007080864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehaskari VM, Aviles DH, Manning J. Prenatal programming of adult hypertension in the rat. Kidney Int. 2001;59:238–245. doi: 10.1046/j.1523-1755.2001.00484.x. [DOI] [PubMed] [Google Scholar]
- Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010;77:989–999. doi: 10.1038/ki.2010.64. [DOI] [PubMed] [Google Scholar]
- Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378:815–825. doi: 10.1016/S0140-6736(11)60814-3. [DOI] [PubMed] [Google Scholar]
- Welham SJ, Wade A, Woolf AS. Protein restriction in pregnancy is associated with increased apoptosis of mesenchymal cells at the start of rat metanephrogenesis. Kidney Int. 2002;61:1231–1242. doi: 10.1046/j.1523-1755.2002.00264.x. [DOI] [PubMed] [Google Scholar]
- Whincup PH, Kaye SJ, Owen CG, Huxley R, Cook DG, Anazawa S, et al. Birth weight and risk of type 2 diabetes: a systematic review. JAMA. 2008;300:2886–2897. doi: 10.1001/jama.2008.886. [DOI] [PubMed] [Google Scholar]
- WHO. Nutrition Landscape Information System. World Health Organisation; 2011. [Google Scholar]
- Williams PJ, Kurlak LO, Perkins AC, Budge H, Stephenson T, Keisler D, et al. Hypertension and impaired renal function accompany juvenile obesity: the effect of prenatal diet. Kidney Int. 2007;73:279–289. doi: 10.1038/sj.ki.5002276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winearls CG, Glassock RJ. Dissecting and refining the staging of chronic kidney disease. Kidney Int. 2009;75:1009–1014. doi: 10.1038/ki.2009.49. [DOI] [PubMed] [Google Scholar]
- Wintour EM, Moritz KM. Comparative aspects of fetal renal development. Equine Vet J Suppl. 1997;24:51–58. doi: 10.1111/j.2042-3306.1997.tb05078.x. [DOI] [PubMed] [Google Scholar]
- Wood IS, de Heredia FP, Wang B, Trayhurn P. Cellular hypoxia and adipose tissue dysfunction in obesity. Proc Nutr Soc. 2009;68:370–377. doi: 10.1017/S0029665109990206. [DOI] [PubMed] [Google Scholar]
- Woods LL, Weeks DA, Rasch R. Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int. 2004;65:1339–1348. doi: 10.1111/j.1523-1755.2004.00511.x. [DOI] [PubMed] [Google Scholar]
- Zeman FJ. Effect of the young rat of maternal protein restriction. J Nutr. 1967;93:167–173. doi: 10.1093/jn/93.2.167. [DOI] [PubMed] [Google Scholar]
- Zimanyi MA, Hoy WE, Douglas-Denton RN, Hughson MD, Holden LM, Bertram JF. Nephron number and individual glomerular volumes in male Caucasian and African American subjects. Nephrol Dial Transplant. 2009;24:2428–2433. doi: 10.1093/ndt/gfp116. [DOI] [PMC free article] [PubMed] [Google Scholar]