

Blood pressure regulation is crucial for the maintenance of health, with hypertension being a risk factor for myocardial infarction, heart failure, stroke and renal disease. Although nitric oxide (NO) and prostacyclin trigger well-defined vasodilator pathways, when their synthesis is prevented a significant relaxation to agents such as acetylcholine remains. This remaining enigmatic relaxation, known as endothelium-derived hyperpolarizing factor (EDHF), is considered a major mechanism controlling blood pressure during health and is more prevalent in resistance than conduit vessels1,2,3,4. Hydrogen peroxide (H2O2) has been shown to be a major component of EDHF in several vascular beds in multiple species including human5,6,7,8,9,10. H2O2 also caused interprotein disulfide bond in protein kinase G (PKG) I-α, which activated the kinase independently of the NO-cyclic guanosine monophosphate (cGMP) pathway and coupled to vasodilation11. To definitively establish the importance of kinase oxidation in the EDHF mechanism and blood pressure control in vivo, we generated a ‘redox-dead’ Cys42Ser PKGI-α knock-in (KI) mouse. This single atom mutation (oxygen instead of sulfur) eliminates the cysteine-based oxidant sensor in PKG making it unable to sense or transduce oxidant signals.
We compared the dose-dependent vasorelaxation of arteries from wild-type (WT) and KI mice to H2O2. There was a significant rightward shift (insensitivity) in the dose-response curve of KI compared to WT mesenteric (Supplementary Fig. 1a, EC50 WT= 23.7±3.2 μM, EC50 KI = 79.4±13.9 μM), carotid, renal or femoral (Supplementary Fig. 1a-c) arteries. KI aortas were partially resistant to H2O2-induced relaxation, showing a 45% deficit in their maximal dilatory response which occurred at the higher doses of oxidant examined (Fig. 1b). There was no difference in the initial force of contraction generated by specific pairs of vessel types from either genotype (Table S1). The size of the vessels (Supplementary Fig. 1d) correlated with their sensitively to H2O2, with smaller size having a progressively lower EC50 for H2O2-dependent relaxation. Western immunoblotting confirmed mesenteric or aortic PKGI-α from WT mice formed a disulfide bond following H2O2 treatment whereas the KI failed to as anticipated from the mutation introduced (Fig. 1c). Pharmacological inhibition of PKG with Rp-8-Br-PET-cGMP attenuated H2O2-induced vasorelaxation of WT mesenteries (Supplementary Fig. 2a), albeit the shift in the relaxation curve by PKG inhibition was rather small. There is potential complexity in the use of Rp-8-Br-PET-cGMP as its potency or selectivity could change depending on the precise molecular state of PKG, which changes with cellular redox state or cGMP levels. However, kinase assays have confirmed that disulfide-activated PKG was inhibited by Rp-8-Br-PET-cGMP in vitro12. These potential limitations of pharmacology were addressed by comparing the responses of WT and KI mesenteries to H2O2 (Fig. 1a) where the inability to disulfide-activate PKG is clearly apparent. The deficit in H2O2-induced vasorelaxation in KI blood vessels that were also unable to form an interprotein disulfide, together with the attenuation of H2O2-induced vasorelaxation by pharmacological PKG inhibition in WT, provides robust evidence that PKGI-α oxidation is a crucial molecular event integral to vasorelaxation. H2O2-induced vasorelaxation was neither altered by the removal of endothelium (Supplementary Figs. 2b and 3b), nor by inhibition of NO synthases with NG-nitro-L-arginine methyl ester (L-NAME), or cyclooxygenase with indomethacin, or these two inhibitors in combination (Supplementary Figs. 2c-d and 3c-d).
Figure 1.

Differential response of WT and KI vessels to oxidant interventions. (a) Dose-dependent relaxation of WT or KI mesenteric vessels to H2O2. Representative traces from WT or KI mesenteric vessels following H2O2 treatment. (b) Dose-dependent relaxation of WT or KI aortas to H2O2. KI aorta had a sub-maximal (45%) relaxation to H2O2 compared to WT at the higher doses of oxidants examined, as shown in the representative trace. KI aorta relaxations are also less sensitive to H2O2 (EC50 WT= 508.4±70.9 μM, EC50 KI = 1311.3.4±703.6 μM). (c) Quantitative Western immunoblot showing that WT PKG Iα from either mesenteries or aortas forms a disulfide following H2O2 treatment, but this fails to occur in corresponding KI tissues. (d) KI mesenteric vessels showed a significantly deficient hyperpolarization in response to H2O2 compared to WTs. The H2O2-induced hyperpolarization measured in WTs was blunted individually by apamin or charybdotoxin, and was fully prevented when these potassium channel inhibitors were used in combination.
H2O2 also induced WT mesenteric vessel hyperpolarization, which was attenuated by either apamin or charybdotoxin alone or highly effectively blocked when these two potassium channel inhibitors were used together (Fig. 1d). There was a marked deficit in the hyperpolarization of KI mesenteries exposed to H2O2 compared to WT. The observation that apamin- or charybdotoxin-sensitive potassium channels are integral to oxidant- and EDHF-mediated vasorelaxation is in line with other studies4,13,14,15,16. Disulfide-activated PKG could potentially phosphorylate potassium channels to result in membrane hyperpolarization. The molecular basis of EDHF, not least in the context of defining the potassium channels involved, is controversial and complex, especially as responses can be species, sex or vascular bed-dependent. Potential selectivity or specificity issues associated with potassium channel inhibitors adds further intricacy. However, it is also important to consider other studies that appear inconsistent with the scheme we have presented. For example, activation of endothelial SK3 and IK1 is required for EDHF-mediated responses, as well as Ca-dependent activation of endothelial NO synthase17. However, these potassium channels are considered to be in low abundance in healthy vascular smooth muscle cells and their activation by PKG has not been described. BKCa, ATP-sensitive potassium channels and inwardly-rectifying potassium channels have also been reported to induce smooth muscle cell hyperpolarization in response to H2O2. However some of these channels or their accessory, regulatory proteins have their own redox sensitive thiols that can directly respond to H2O2, independently of kinase activity18.
To establish whether PKGI-α disulfide-activation contributes to the EDHF phenomenon we compared WT and KI vessel relaxations to acetylcholine with or without combined inhibition with L-NAME and indomethacin to remove NO and prostacyclin relaxing factors. This ‘EDHF protocol’ induced a disulfide in WT mesenteric vessels, but failed to in those from the Cys42Ser PKGI-α KI (Fig. 2a). Acetylcholine-induced vasorelaxation was markedly deficient in KI mesenteric vessels compared to WT. The acetylcholine-dependent relaxation of WT was also substantially attenuated by either L-NAME (Fig. 2b, EC50 WT= 148.9±48.9 nM, EC50 KI= 614.2±162.9 nM) or catalase (Fig. 2c) alone, or when they were combined together (Supplementary Fig. 4f). In addition, pharmacological blockade of PKG or removal of endothelium substantially attenuated acetylcholine-induced relaxation of WT mesenteries (Supplementary Fig. 4a-b). In contrast, cyclooxygenase blockade with indomethacin alone did not alter acetylcholine-induced vasorelaxation (Supplementary Fig. 4c), whereas indomethacin combined with L-NAME partial blocked relaxation (Supplementary Fig. 4d). Combining indomethacin with catalase and L-NAME resulted in full blockade of relaxation to acetylcholine (Supplementary Fig. 4e). These observations are consistent with acetylcholine triggering two primarily pathways leading to vasorelaxation in mesenteric resistance vessels; one involving synthesis of NO and another H2O2. The latter is consistent with a number of studies linking EDHF-dependent relaxation to the biosynthesis of H2O2 7,9,19, which good evidence suggests is derived from NOS-generated superoxide in resistance vessels19. Aortas of either genotype lacked an EDHF response (Fig. 2d), consistent with reports that this hyperpolarization mechanism is largely absent from conduit vessels9. In contrast an NO donor relaxed both conduit and resistance vessels from both genotypes equally (Supplementary Figs. 2a and 6a,c), as did the direct PKG activator 8-Br-cGMP (Supplementary Fig. 6b,d). Clearly in the aorta conduit vessel acetylcholine was unable to recruit the kinase oxidation pathway, evidenced by acetylcholine-induced relaxation being identical in WT and KI aorta, as well as being fully blocked by L-NAME (Fig. 2e) and insensitive to catalase (Fig. 2f). Furthermore PKGI-α in aorta did not form a disulfide with an EDHF protocol (Fig. 2d). Acetylcholine-induced relaxation of aorta was also attenuated by PKG inhibition or fully blocked by a combination of L-NAME, indomethacin and catalase, and was also endothelium-dependent (Supplementary Fig. 5a-b).
Figure 2.

Differential response of WT and KI vessels to acetylcholine. (a) Comparison of relaxation responses of WT and KI mesenteric vessels to SpNONOate, acetylcholine or an EDHF (i.e. acetylcholine in the presence of L-NAME and indomethacin) protocol. Western immunoblot comparing EDHF-induced PKG disulfide formation in mesenteries. (b-c) Comparison of dose-response of WT and KI mesenteric vessels to acetylcholine and how this modulated by L-NAME or catalase. (d) Comparison of responses of WT and KI aortas to SpNONOate, acetylcholine or an EDHF protocol. Western immunoblot comparing EDHF-induced PKG disulfide formation in aortas. Western immunoblot comparing EDHF-induced PKG disulfide formation in aortas. (e-f) Comparison of dose-response of WT and KI aortas to acetylcholine and how this modulated by L-NAME or catalase.
Overall our observations are consistent with EDHF being a property of resistance vessels that at least in part relies on a pathway generating H2O2. This reduced ability of KI vessels to relax to an EDHF protocol is logically explained by the one atom mutation that prevents disulfide formation and associated PKG activation. In other words the Cys42Ser mutation prevents the kinase sensing H2O2 and transducing the oxidant signal into kinase activity that in WT couples to resistance vessel vasorelaxation.
To assess the importance of this disulfide oxidation pathway to blood pressure regulation in vivo we implemented telemetric monitoring. Mean, systolic and diastolic arterial pressures were significantly higher in KI mice than WT littermate controls (Fig. 3a). The blood pressure increased at night as the mice become more active during the dark cycle, but the relative hypertension in KI compared to WT mice was maintained. Both mouse genotypes increased their blood pressure in response to L-NAME, but the delta increase was greater in KI compared to WT (Fig. 3b). This is consistent with WT recruiting PKGI-α disulfide-activation during a hypertensive challenge to enable blood pressure homeostasis, but as this mechanism is not possible in KI there is a proportionately greater hypertension. Indomethacin caused only a relatively small increase in blood pressure, but despite a trend towards a greater hypertension in KI this was not significantly different than the WT response (Fig. 3c). We attribute the basal hypertension and exaggerated L-NAME pressor response of KI mice to a fundamental functional difference in the vasomotor coupling of their resistance blood vessels compared with WT. Although the responses of the mesenteries from the two genotypes to oxidants are disparate, this difference only stems from the highly conservative single atom sulfur to oxygen replacement at residue 42 of PKGI-α which removes oxidant sensing in the cell selectively from this single kinase. This has allowed us to definitively illustrate the importance of PKG-Iα Cys42 disulfide oxidation in regulating basal blood pressure during health. Potentially contrary to our observations is lower blood pressure in catalase over-expressing mice which was reversed by a catalase inhibitor20. Similarly, catalase over-expression have a reduced pressor response to vasoconstrictor agents21. However, catalase may not be anticipated to lower H2O2 concentration pertinent to vasorelaxation as this enzyme is principally located in peroxisomes. Thus spatially catalase may not compete with the highly abundant, ubiquitously-expressed peroxiredoxin proteins with a Km of □20 μM, which happens to approximate to the EC50 of WT mesenteric vessels for H2O2-dependnet relaxation. Furthermore, although catalase over-expression reduced H2O2 below WT in some tissues, it didn’t in all and the levels in resistance vessels were not studied20.
Figure 3.

KI mice are hypertense compared to WT littermates. (a) In vivo telemetric blood pressure monitoring comparing KI mice with WT littermate controls during the day and night. Data are presented as mean arterial pressure over time or as the time-averaged mean systolic and diastolic pressure in WT and KI mice. (b) Comparison of the blood pressure response to intraperitoneal injection of L-NAME in WT and KI mice. (c) Comparison of the blood pressure response to intraperitoneal injection of indomethacin in WT and KI mice.
Although the hypertensive phenotype of the KI mouse is explained by an intrinsic deficit in their resistance blood vessels ability to sense, transduce and relax to H2O2, we also assessed several other components that could potentially contribute to the hypertension in the KI. Using echocardiography we found that the KI mice have a slightly depressed cardiac output, despite heart weights being the same (Fig. 4a-b). If the cardiac output of the KI was elevated to match the WT then the observed hypertension would likely be markedly greater. Thus, the reduced cardiac output may be an adaptive mechanism in the KI to limit hypertension. The blood vessels from WT and KI mice were no different in terms of hypertrophy or fibrosis (Fig. 4c-d), and neither was kidney function between genotypes in terms of plasma renin (Fig. 4e) or inulin clearance (Fig. 4f). We assessed possible central effects that may result from the Cys42Ser mutation by indexing heart rate variability and environmental stress responses as a non-invasive measure of autonomic function. There were no genotype differences in these metrics (Supplementary Fig. 7a, b), meaning the hypertension in KI is not associated with a centrally-mediated decrease in parasympathetic activity.
Figure 4.
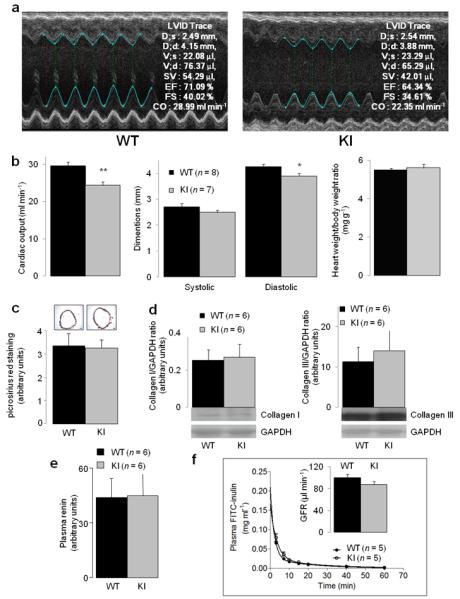
Comparison of cardiac output, vascular fibrosis and renal function in WT and KI mice. (a) Representative M-mode echocardiography recordings from WT and KI mice. (b) Comparison of indices of cardiac function in WT and KI as determined by echocardiography. (c-d) Comparison of blood vessel fibrosis, indexed by measuring picrosirius red staining of aorta (scale bar = 100 μm) or collagen content of mesenteries measured by Western immunoblotting. Renin activity was compared in the plasma of WT and KI mice. (f) Plasma inulin clearance was compared in WT and KI mice.
We conclude PKGI-α disulfide formation is a significant mechanism contributing to blood pressure homeostasis and a major component of the EDHF phenomenon. Acetylcholine, in addition to an NO-dependent pathway, also recruits an H2O2-dependant mechanism which leads to oxidation and activation by PKG, which phosphorylates potassium channels to cause vessel hyperpolarization and relaxation. This regulatory mechanism operates basally to control blood pressure in healthy animals. Elimination of this mechanism by the genetic removal of the thiol oxidant sensor in PKG results in hypertension. These observations are contrary to the historical perspective that oxidants are principally harmful. Instead this evidence supports an evolving paradigm shift in which oxidants are important regulators of homeostasis during health22.
Supplementary Material
Acknowledgements
We would like to acknowledge support from the Medical Research Council, the British Heart Foundation, the Leducq Foundation and the Department of Health via the NIHR cBRC award to Guy’s & St Thomas’ NHS Foundation Trust.
Appendix
Online Methods
Animals
Mice were maintained as stated in ‘Principles of Laboratory Animal Care’ as published by the National Institutes of Health (NIH Publication no.85-23).
Generation of Cys42Ser PKGIα knock-in mouse
Mice constitutively expressing PKGIα Cys42Ser were generated for us on a pure C57BL/6 background by TaconicArtemis. A targeting vector was constructed, which involved PCR amplification of the murine Prkg1, introducing the Cys42Ser mutation into exon 1a (which is specific for the alpha isoform) by site directed mutagenesis and inserting an FRT-flanked neomycin selection marker (to allow for selection of transfected embryonic stem (ES) cells) close to the mutation to favour homologous recombination. Then screening by southern blot was carried out to identify if homologous recombination had occurred followed by validation of the positive clones. ES cell transfection was then carried out followed by chimera generation. The chimeras were directly bred with an Flp deletor for the in vivo deletion of the selection marker. As the ES cells always go germline, chimeras can be directly bred to the deletor in order to obtain germline transmission and selection marker deletion at the same time.
Myography
Vascular rings were isolated from the thoracic aorta, carotid, renal (second order), femoral or mesenteric (second order) arteries. They were mounted in a tension myograph (Danish Myo Technology), stretched to the optimal pre-tension conditions (using DMT Normalization Module), bathed in Krebs solution maintained at 37°C and gassed with 95% CO2:5% O2. During the vessel ‘wake up’ phase, if vessels failed to generate >1mN of force they were rejected and the myograph channel was switched off. No vessels were rejected during subsequent data analysis of responses. Vasotone measurements of aortic rings were made essentially as before1, determining the responses of phenylephrine- (1 μM) or U46619-contracted (0.1 μM) vessels to a number of agents. Vasotone measurements of mesenteric vessels were made by determining the responses of U46619-contracted (0.1 μM) vessels to various vasoactive agents, which included hydrogen peroxide (0-1000 μM), spermine-NONOate (0-10 μM), 8-Br-cGMP (0-30 μM) or acetylcholine (0-10 μM). Inhibitors were pre-incubated with vessels for 30min before dose-response curves were constructed. Rp-8-Br-PET-cGMP inhibitor was used at 100 μM and peg-catalase at 1000units/ml. If endothelium was removed from vessels this was achieved by passing a human hair twice through the lumen. The EDHF protocol involved pre-incubating vessels with NO synthetase and cyclooxygenase inhibitors NG-nitro-L-arginine methyl ester (L-NAME, 300 μM) and indomethacin (10 μM) for 30 minutes respectively, then treating with acetylcholine (0.1 μM).Tension experiments were carried out using one or two vessels per treatment intervention derived from at least 5 different WT of KI animals. The initial force of contraction generated by vessels under different conditions used in these studies are shown in Table S1.
Membrane hyperpolarization
Membrane hyperpolarization was measured using established methods2,3. Bis-(1,3- dibutylbarbituric acid)trimethine oxonol (DiBAC4) was loaded (30min, 2μM) into mesenteric vessels and fluorescence (excitation 495 nm, emission 520 nm) output monitored by microscopy. Hyperpolarization following hydrogen peroxide treatment (10min, 30μM) resulted in loss of DiBAC4 fluorescence and was compared in wild-type or knock-in vessels. In some studies the potassium channel inhibitors apamin (0.1 μM) or charybdotoxin (0.1 μM) were utilised. Quantitative analysis of images was carried out using ImageJ software (NIH).
Telemetric blood pressure monitoring in vivo
Blood pressure was assessed by remote radio-telemetry in conscious freely moving mice as described 4. Briefly, mice were anesthetised with 2% isoflurane in 1l of oxygen per minute with pre and post-operative analgesia (buprenorphine, 0.1mg/kg). Radio-telemetry probe catheter (TA11PA-C10, O.D 0.4 mm, Data Science International Inc.) was implanted into the aortic arch via the left carotid artery. Following one week recovery, mice housed in individual cages were placed above the telemetric receivers with output to a computer. Blood pressure was recorded by scheduled sampling for 10 seconds every 5min (Dataquest LabPRO Acquisition System version 3.01, Data Sciences International, St Pauls, MN). 8-10 animals were studied per group to produce average blood pressure data.
Heart rate variability and environmental stress test
The heart rate variability (HRV) parameters were determined as a measure of autonomic function5,6. Briefly, HRV were analysed from a 300sec continuous telemetric blood pressure record made between 0900 and 1000hrs in undisturbed telemetered animals in a quiet room. Data sets recorded in a sinus rhythm were used. Time and frequency domain of HRV analysis were performed using HRV module of Chart 5.0 analysing software (ADInstruments, Colorado Springs, CO). Ectopics and visible short artefacts were manually excluded or replaced by intervals linearly interpolated from the nearest normal interval in order to avoid discontinuity in the record. Integrated boundaries for spectral bands were set at 0.4-1.5Hz for low frequency (LF) and 1.5-4Hz for high-frequency (HF) component.
Mice were exposed to the environmental stress as described7. Briefly, mice were removed from their home cages and placed into a new cage without bedding for 2 hours. MAP and HR were recorded continuously while the mice were exposed to the new environment. Blood pressure and heart rate changes were expressed as response in % of the average baseline recorded for 1.5 hours before conducting the stress-test.
Plasma renin
This was measured using a SensoLyte 520 mouse renin assay kit (Anaspec), according to the manufacturer’s instructions.
Inulin clearance
This was carried out by measuring plasma clearance of FITC-inulin (Sigma) essentially as described,8 using anaesthetised mice to avoid animal stress. Plasma FITC-inulin levels were serially monitored using a Spectramax Gemini XS microplate fluorometer (Molecular Devices).
Fibrosis
Picrosirius red was used to monitor fibrosis in aorta using a commercial kit (Polysciences Inc.), according to the manufacturer’s instructions. Western immunoblotting (details below) was used to compare Type I and III collagen content in WT and KI mesenteric vessels.
Western immunoblotting
Immunoblotting was carried out as before 9, utilizing maleimide (100mM) in preparation buffers to alkylate thiols and prevent thiol disulfide exchange. cGKIα (E-17) primary antibody (Santa Cruz), Type I collagen (Abcam), Type II collagen (Abcam), HRP-linked secondary antibody (Dako) and ECL reagent (GE Healthcare) were utilized. Digitized immunoblots were quantitatively analyzed using Gel-Pro Analyzer 3.1.
Statistics
Results are presented as mean ± SEM. Differences between groups were assessed using a one or two way ANOVA where appropriate followed by a t test. Differences were considered significant at the 95% confidence level.
References for methods
- 1.Burgoyne JR, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 2.Epps DE, Wolfe ML, Groppi V. Characterization of the Steady-State and Dynamic Fluorescence Properties of the Potential-Sensitive Dye Bis-(1,3-Dibutylbarbituric Acid)Trimethine Oxonol (Dibac(4)(3)) in Model Systems and Cells. Chemistry And Physics Of Lipids. 1994;69:137–150. doi: 10.1016/0009-3084(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka M, et al. Cytochrome P-450 metabolites but not NO, PGI(2), and H2O2 contribute to ACh-induced hyperpolarization of pressurized canine coronary microvessels. American Journal Of Physiology-Heart And Circulatory Physiology. 2003;285:H1939–H1948. doi: 10.1152/ajpheart.00190.2003. doi:10.1152/ajpheart.00190.2003. [DOI] [PubMed] [Google Scholar]
- 4.Huetteman DA, Bogie H. Direct blood pressure monitoring in laboratory rodents via implantable radio telemetry. Methods Mol Biol. 2009;573:57–73. doi: 10.1007/978-1-60761-247-6_4. [DOI] [PubMed] [Google Scholar]
- 5.Elghozi JL, Julien C. Sympathetic control of short-term heart rate variability and its pharmacological modulation. Fundamental & Clinical Pharmacology. 2007;21:337–347. doi: 10.1111/j.1472-8206.2007.00502.x. doi:10.1111/j.1472-8206.2007.00502.x. [DOI] [PubMed] [Google Scholar]
- 6.Thayer JF, Yamamoto SS, Brosschot JF. The relationship of autonomic imbalance, heart rate variability and cardiovascular disease risk factors. International Journal of Cardiology. 2010;141:122–131. doi: 10.1016/j.ijcard.2009.09.543. doi:10.1016/j.ijcard.2009.09.543. [DOI] [PubMed] [Google Scholar]
- 7.Gross V, et al. Autonomic nervous system and blood pressure regulation in RGS2-deficient mice. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2005;288:R1134–R1142. doi: 10.1152/ajpregu.00246.2004. doi:10.1152/ajpregu.00246.2004. [DOI] [PubMed] [Google Scholar]
- 8.Qi ZH, et al. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. American Journal of Physiology-Renal Physiology. 2004;286:F590–F596. doi: 10.1152/ajprenal.00324.2003. doi:DOI 10.1152/ajprenal.00324.2003. [DOI] [PubMed] [Google Scholar]
- 9.Brennan JP, et al. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. doi: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
Footnotes
Author Contributions
O.P. and conducted myography and protein analysis experiments and wrote the manuscript; O.R. undertook in vivo blood pressure and inulin clearance studies and wrote the manuscript; P.E. supervised the project and wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests
Methods
Methods are available online.
References
- 1.Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann Med. 2007;39:495–516. doi: 10.1080/07853890701491000. [DOI] [PubMed] [Google Scholar]
- 3.Weston AH, Edwards G, Feletou M. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Archiv-European Journal Of Physiology. 2010;459:863–879. doi: 10.1007/s00424-010-0817-1. doi:10.1007/s00424-010-0817-1. [DOI] [PubMed] [Google Scholar]
- 4.Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Britsh Journal of Pharmacology. 2010;164:839–52. doi: 10.1111/j.1476-5381.2010.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimokawa H, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. Journal of Clinical Investigation. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimokawa H, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochemical and Biophysical Research Communications. 2002;290:909–913. doi: 10.1006/bbrc.2001.6278. doi:10.1006/bbrc.2001.6278. [DOI] [PubMed] [Google Scholar]
- 7.Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol. 2005;39:725–732. doi: 10.1016/j.yjmcc.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Yada T, Shimokawa H, Hiramatsu O, Goto M, Kajiya F. Protective role of hydrogen peroxide, an endogenous EDHF, in ischemia-reperfusion injury in canine coronary microcirculation in vivo. Circulation. 2003;108:226–226. [Google Scholar]
- 9.Shimokawa H. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pflugers Archiv-European Journal Of Physiology. 2010;459:915–922. doi: 10.1007/s00424-010-0790-8. doi:10.1007/s00424-010-0790-8. [DOI] [PubMed] [Google Scholar]
- 10.Miura H, et al. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 11.Burgoyne JR, et al. Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 12.Burgoyne JR. Thiol-dependent redox regulation in the cardiovascular system: An investigation of protein S-nitrosylation and a novel oxidant-induced activation of protein kinase G. King’s College; London: 2008. PhD Thesis. [Google Scholar]
- 13.Huang Y, et al. Endothelial mediators of the acetylcholine-induced relaxation of the rat femoral artery. Vascular Pharmacology. 2006;44:299–308. doi: 10.1016/j.vph.2006.01.010. doi:10.1016/j.vph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Takaki A, et al. Genetic disruption of all nitric oxide synthase isoforms abolishes EDHF-mediated responses in mice. Journal of Hypertension. 2006;24:48–48. [Google Scholar]
- 15.Busse R, et al. EDHF: bringing the concepts together. Trends In Pharmacological Sciences. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 16.Dora KA, Garland CJ. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. American Journal Of Physiology-Heart And Circulatory Physiology. 2001;280:H2424–H2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- 17.Dalsgaard T, Kroigaard C, Simonsen U. Calcium-activated potassium channels - a therapeutic target for modulating nitric oxide in cardiovascular disease? Expert Opinion on Therapeutic Targets. 2010;14:825–837. doi: 10.1517/14728222.2010.500616. doi:Doi 10.1517/14728222.2010.500616. [DOI] [PubMed] [Google Scholar]
- 18.Yi L, Morgan JT, Ragsdale SW. Identification of a Thiol/Disulfide Redox Switch in the Human BK Channel That Controls Its Affinity for Heme and CO. Journal Of Biological Chemistry. 2010;285:20117–20127. doi: 10.1074/jbc.M110.116483. doi:DOI 10.1074/jbc.M110.116483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimokawa H, et al. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. Journal of Experimental Medicine. 2008;205:2053–2063. doi: 10.1084/jem.20080106. doi:10.1084/jem.20080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suvorava T, et al. Endogenous vascular hydrogen peroxide regulates arteriolar tension in vivo. Circulation. 2005;112:2487–2495. doi: 10.1161/CIRCULATIONAHA.105.543157. doi:Doi 10.1161/Circulationaha.105.543157. [DOI] [PubMed] [Google Scholar]
- 21.Yang H, et al. Reduction of pressor response to vasoconstrictor agents by overexpression of catalase in mice. American Journal of Hypertension. 2003;16:1–5. doi: 10.1016/s0895-7061(02)03086-8. [DOI] [PubMed] [Google Scholar]
- 22.Eaton P. Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic Biol Med. 2006;40:1889–1899. doi: 10.1016/j.freeradbiomed.2005.12.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.