

Abstract
Melanotic neuroectodermal tumor of infancy is a rare congenital neoplasm involving the head and neck region in young patients. A case of melanotic neuroectodermal tumor of infancy is presented. This tumor occurred in right maxillary alveolar ridge in a 4-month-old infant. The present case showed an increased urinary level of vanilmandelic acid, confirming that the tumor is originated from neural crest. Clinical assessment, histologic diagnosis, and laboratory findings supported the diagnosis.
Keywords: Infant, melanotic neuroectodermal tumor, neural crest cells, vanilmandelic acid
Introduction
The melanotic neuroectodermal tumor of infancy (MNTI) was first described in 1918 as a congenital melanocarcinoma. It is a rare, pigmented neoplasm of neural crest origin occurring in infants before the age of 1 year.[1] It is a rapidly growing tumor that most frequently affects the craniofacial skeleton. The most common site of occurrence is the anterior maxillary alveolar ridge (70% cases) followed by the skull, brain, and mandible. The genital organs are the most frequent extra-cranial sites.[2,3]
The tumor is usually non-ulcerated and presents as a soft tissue swelling, frequently affecting bone. Although the tumor cells produce melanin, pigmentation may not be clinically evident.[4–7] MNTI is a benign tumor, but can be locally aggressive, growing rapidly and resulting in tooth displacement as tumor cells invade bone.[8–10] With plain radiographs, MNTI appear as intrabony expansive areas of radiolucency, usually with poorly demarcated margins, probably as a result of rapid tumor growth and a tendency to be locally invasive.[12]
Its histopathological features are distinctive, with tubular or alveolar formation of large melanin-containing cells around nests of smaller neuroblastic cells possessing fibrillar cytoplasm. It usually follows a benign course but inadequate excision, occasional multicentrity, and small malignant potential result in a fairly high recurrence rate.[13] The average recurrence rate is 15–20%, but it may be as high as 50% among cases treated without wide resection.[14] High urinary excretion of vanilmandelic acid (VMA), has also been reported in cases of MNTI, indicating that the tumor is derived from neural crest cells.[15,16]
We report a case of MNTI in a 4-month-old male infant showing biphasic tumor cell population with melanin pigmentation, increased urinary excretion of VMA level, emphasizing the need for appropriate treatment.
Case Report
A 4-month-old male infant presented with a swelling arising from the upper jaw noted by his parents since 2 months, with difficulty in feeding and irritability [Figure 1]. History of pregnancy and delivery was normal and there was no history of medication during pregnancy. Growth and development of the infant were adequate for his age.
Figure 1.
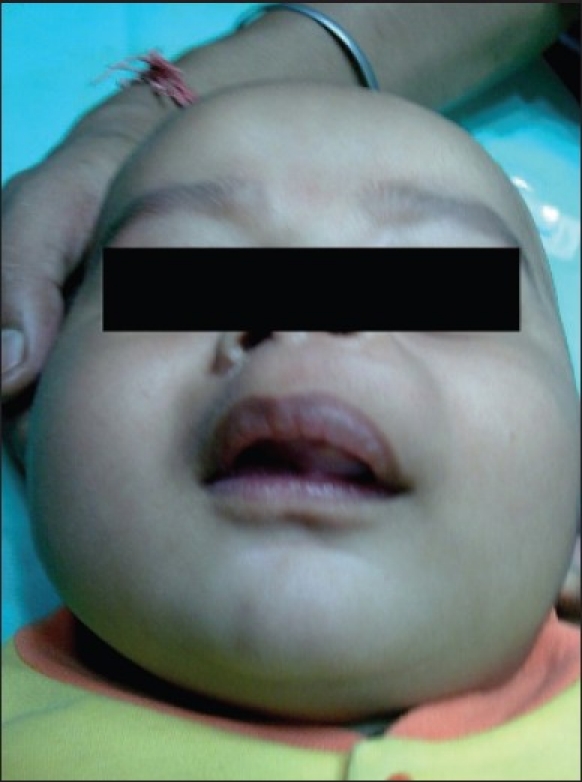
Clinical extraoral photograph of the 4-month-old infant.
On intraoral examination, a swelling measuring about 3 × 3 cm in diameter was seen in the pre-maxillary region. It was located slightly toward right side and showed extension toward hard palate. Expansion of the anterior maxillary alveolar ridge with obliteration of labial vestibule was seen [Figure 2]. On inspection, swelling was smooth and the overlying mucosa was intact and stretched. On palpation, it was bony hard, fixed, non-fluctuant, and non-tender. No lymph nodes were palpable. An intraoral periapical radiograph revealed a multilocular osteolytic lesion. Patient was planned for surgical excision under general anesthesia. Incision was made through the labial mucosa. Lesion was exposed [Figure 3]. Its enucleation was done with curettage of septa present inside the lesion with normal surrounding bone.
Figure 2.
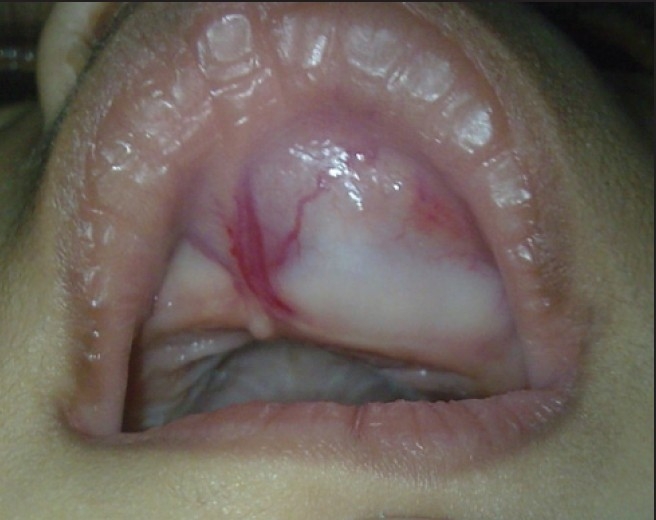
Clinical intraoral photograph showing tumor involving upper alveolus and palate.
Figure 3.
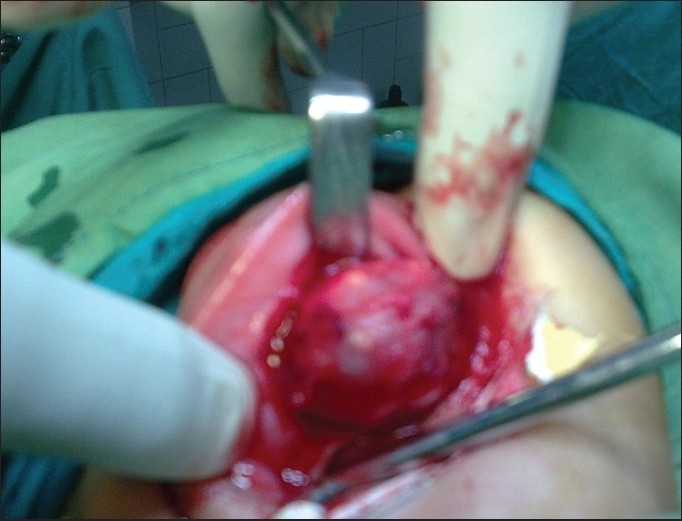
Clinical photograph of the patient at the time of surgery.
Histopathological examination of the excised tissue revealed sheets and chords of tumor cells in the background of dense fibrovascular stroma [Figure 4]. Another field showed areas of calcifications in a globular pattern with tall columnar cells showing reverse polarity, along with stellate reticulum [Figures 5 and 6].
Figure 4.
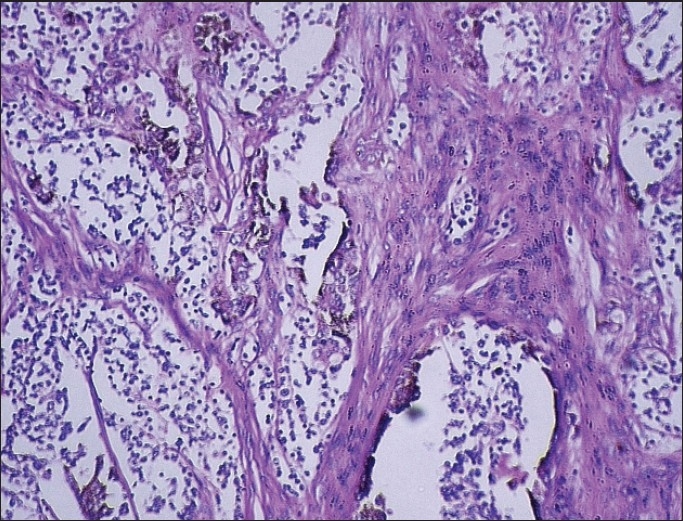
Alveolar like spaces or pseudoglandular structure with central infoldings of tumor cells in the background of fibrovascular stroma. [H & E, ×10]
Figure 5.
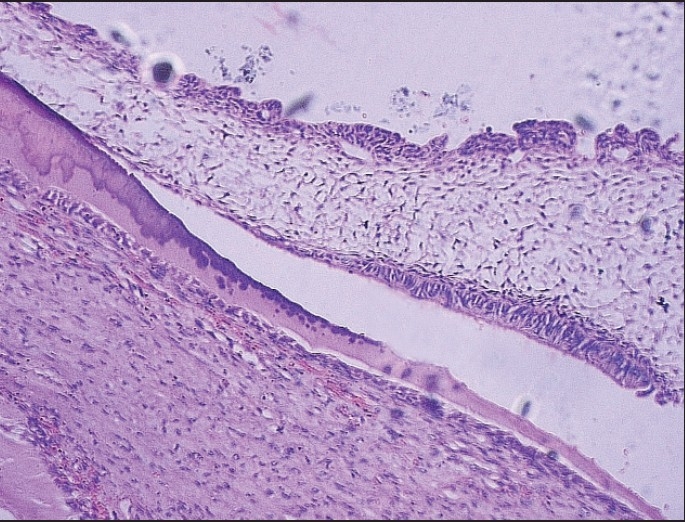
Globular pattern of calcifi cation with tall columnar cells and stellate reticulum [H and E, ×10].
Figure 6.
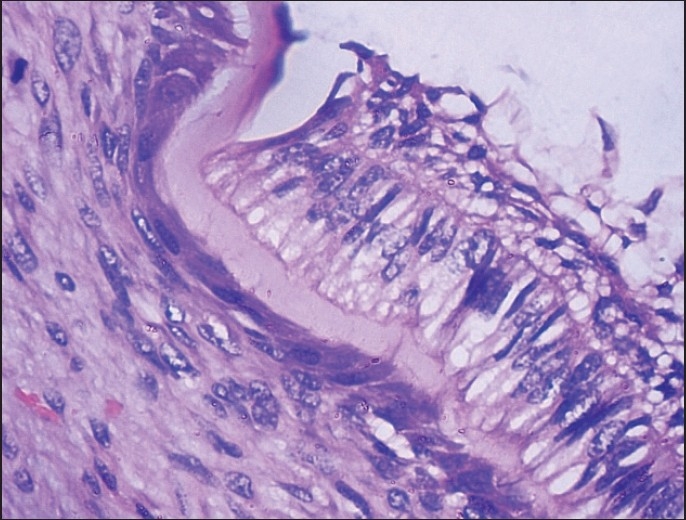
Calcifications with tall columnar cells showing reverse polarity, along with stellate reticulum [H and E, ×100].
Tumor cells were arranged in alveolus like spaces and in a pseudoglandular pattern. Individual tumor cells were recognized as two morphologically distinct types, namely large cells (densely pigmented cells, sparsely pigmented cells) and small undifferentiated cells (hyperchromatic cells) [Figure 4]. Irregular alveolar spaces are lined by larger cuboidal cells with moderate amount of cytoplasm and well-defined margins, some of which contained brownish pigment. Their nuclei were round to oval with finely dispersed chromatin and inconspicuous nucleoli. In addition, Small hyperchromatic cells with scanty cytoplasm were present within the spaces or seen as isolated nests within the fibrillar background [Figures 7 and 8]. Isolated clusters of the larger cells were also present. A characteristic biphasic pattern of cell distribution was seen. There was no evidence of atypical mitosis or necrosis in the tissue.
Figure 7.
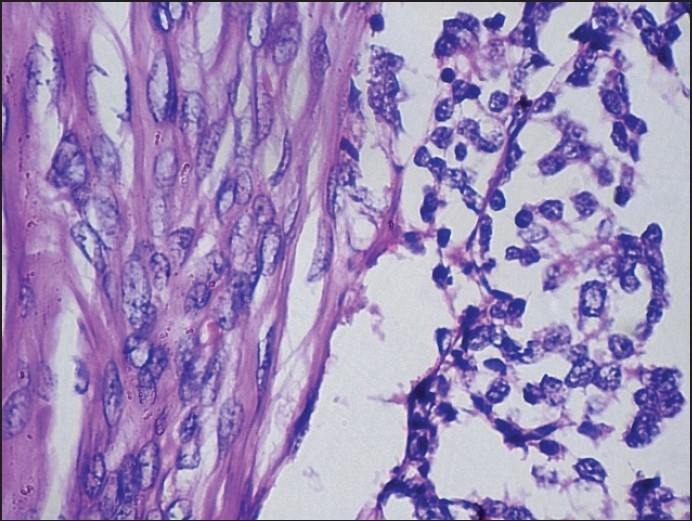
Biphasic tumor cell population, with large and small cells [H and E, ×100].
Figure 8.
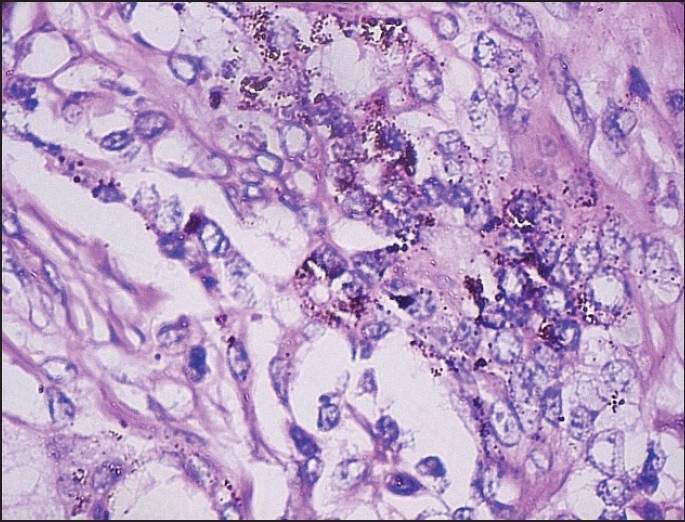
Melanin pigmented cells separated from small cells with high nuclear to cytoplasmic ratios and hyperchromatic nuclei [H and E, ×100].
Based on these findings a diagnosis of MNTI was made. For further confirmation of the diagnosis, 24 h urinary excretion of VMA was done and found to be increased, 2.9 mg/24 h, confirming the diagnosis. Post-operative course was uneventful. The 6 month follow-up of the patient showed no recurrence.
Discussion
MNTI was first described by Krompecker in 1918 as a congenital melanocarcinoma. It was known by many names as its cellular origin was not clear. These names included pigmented ameloblastoma, retinal anlage tumor, melanotic adamantinoma, retinal choristromaa, melanotic progonoma, melanotic epithelial odontoma, pigmented Teratoma, atypical melanoblastoma, pigmented epulis, and retinoblastic Teratoma.[15]
MNETI is a tumor of long debate and controversy concerning its histogenesis. As aforementioned, its histogenetic theories were various. To date, several theories have been put forward to explain its histogenesis.
Krompecher in 1918 first suggested its origin from the enclaved epithelial rest at the time of embryonic fusion of facial processes, and he considered it as a malignant tumor arising from histological findings. Other investigators proposed its histogenesis from odontogenic apparatus. Mummery and Pitts in 1926 reported a pigmented maxillary tumor of a 5-month-old female infant under the name of melanotic epithelial odontoma, an ascertaining its origin to be from odontogenic apparatus from the viewpoint that it occurs predominately in maxilla and have used the term “melanotic or pigmented ameloblastoma”, suggesting that the tumor might arise from some proliferative, and aberrant odontogenic epithelium.[17]
Bordello and Gorlin in 1966 proposed cogent objection to this theory, saying the origin of this tumor from odontogenic tissue did not explain the occurrence of the tumor in the area other than the jawbone, normal development of the teeth in the area of the tumor, and because teeth were uniformly present in that area. They also pointed out the scanty resemblance of the tumor cell to the odontogenic epithelium. They reported a pigmented maxillary tumor of 3-month-old male infant accompanied by the increased urinary secretion of 3-methoxy 4-hydromandelic acid (VMA) in the presence of tumor and VMA returned to normal after extirpation of the tumor. Since high urinary levels of VMA are common findings in the individuals with neural crest tumor such as pheochromocytoma, ganglioneuroblastoma, neuroblastoma, and retinoblastoma, they consequently concluded this tumor to be originated from the neural crest. Thereafter most reports are in agreement with Borello and Gorlin's content.[17]
Koudstaal et al, observed the similarity of the enzyme pattern of MNTI to that of certain other tumors probably derived from cells of the neural crest, concluding origin of the tumor cells from neural crest. Dooling et al, observed histologic similarity between MNTI and fetal pineal gland, suggesting MNTI might be derived from neuroectodermal tissue. Dehner et al, describe various stages of melanocytes and neuroblast like cells, suggesting neuroectodermal origin.[17] Hence, the term “melanotic neuroectodermal tumor” was coined.
To the best of our knowledge, 254 cases have been reported in the English literature.[14,18,19] Characteristically, MNTI arises from maxilla, more so from the intraoral side. Bone destruction and displacement of teeth often occur because of the intraosseous location of the maxilla. Other sites are skull, mandible, and brain. The lesion is usually solitary and the mucosa over the lesion is usually intact. It is typically bluish in color due to the presence of melanin; it is usually benign but is locally aggressive. A majority of MNTI patients present are in the first year of life. The children present with the swelling in the oral cavity, which often hinders feeding. In a survey of 179 cases, done by Joseph et al, a majority of lesions arise in the maxilla (68.8%), followed by the skull (10.8%), mandible (5.8%), and brain (4.3%). Although the majority of tumors are benign, a local recurrence rate of 10% to 15% and a malignancy rate of 3.2% is reported.[20] In the present case location of MNTI is in the anterior maxillary alveolar ridge. It was bony solitary, hard bony hard, fixed and non-fluctuant. The mucosa over the tumor was intact.
Clinical differential diagnosis includes ameloblastoma, odontoma, odontogenic myxoma, fibroma, rhabdomyo-sarcoma, Ewing's sarcoma, Langerhans cell histiocytosis, and non-Hodgkin's lymphoma. The plain radiograph of MNTI shows a well-circumscribed radiolucent lesion, and advancing tumors shows excessive bone destruction, suggesting a malignant neoplasm.[1] The present case showed a multilocular osteolytic lesion, pretending its aggressive behavior.
Histopathologically, the biphasic neoplastic population and polyphenotypic immunohistochemical expression are quite distinctive and unique from most of the other pediatric “small blue round cell” neoplasms.”[12,20] Histopathological differential diagnosis of MNTI is quite broad, but must be separated from other pediatric “small round cell” neoplasms such as neuroblastoma, Ewing's sarcoma, peripheral neuroepithelioma, rhabdomyosarcoma, peripheral primitive neuroectodermal tumor, desmoplastic small round cell tumor, malignant melanoma, and lymphoma.[12]
In the present case, histopathological findings demonstrate tall columnar cell showing reverse polarity, stellate reticulum with induction of hard tissue formation in a globular pattern. Borello and Gorlin have postulated that maxillary and mandibular varieties of MNTI may arise from neural crest elements which develop in association with odontoblasts that eventually form dentin of primary anterior teeth. MNTI tumor cell, lying in close association with normal odontoblasts, may induce to activity by the normal functioning of their neighboring odontoblasts.[15]
MNTI may share a common histologic and immunophenotypic expression with cellular blue nevus, melanoma, neuroblastoma, and rhabdomyosarcoma, but MNTI does not express diffuse reactivity with S-100 protein, lacks other markers of neuroendocrine differentiation, and lacks myo-D1, myoglobin, myogenin, and muscle specific actin reactivity. Melanoma is distinctly rare in pediatric patients and even more so if the “mucosal” sites of development of MNTI are considered. Cellular blue nevus characteristically has a spindle cell population, the cytoplasm of which is filled with pigment, while MNTI does not contain spindle cells. Teratoma, especially immature types, may show isolated foci of MNTI, but these foci are of no prognostic significance. Curiously, even though there is a histologic similarity with neuroblastoma and other pediatric neoplasms, there is no genetic basis at present to link them.[21,22]
An increased urinary VMA levels (2.9 mg/24 h) was present in this case, which was in accordance with the findings of Borello and Gorlin[15] and Anil et al,,[16] (2.8 mg/24 h). Although high levels of VMA are common findings in the individuals with neural cell tumors such as pheochromocytoma, ganglioneuroblastoma, neuroblastoma, and retinoblastoma, indicating tumor originating from neural crest.[17]
There are many interesting aspects of the lesion which requires further consideration. These are its clinical behavior pattern, its tissue of origin, and clinical management of the patient from the infancy through adulthood. Clinical behavior of MNTI has two features which makes it serious and difficult to manage. First, though classified as a benign lesion, it is often clinically aggressive because of its rapid onset and alarming local growth rate and secondly, an increased propensity for recurrence.[14]
The significant feature of MNTI is that recurrence has been observed in maxilla and mandible. No recurrence has been reported from a tumor of extraoral origin. The recurrence of MNTI may occur either as a development from the cells of the original tumor that were missing at the initial surgery or that were seeded into intra-trabecular space during excision of the original tumor. An average recurrence rate of 15–20% was observed, but it may be as high as 50% among cases treated without wide resection.[14] The present case was followed up for a period of 6 with no recurrence.
An aggressive surgical approach consisting of complete surgical excision is advocated. Carnevale et al, suggested the use of an operating microscope for the surgical excision of MNTI to remove pigmented remnants of the lesion.[19] In continuity, reconstruction is recommended in the newborns and infants. Adjuvant radiation and/or chemotherapy are occasionally required. The usefulness of DNA ploidy by flow cytometry in predicting tumor behavior is controversial although aneuploidy is associated with its recurrence.[24]
Conclusion
MNTI is a rare benign neoplasm that occurs in infancy before the age of 1 year. Its distinguishing features which allows its separation from other pediatric neoplasms is its biphasic tumor cell population with melanin pigmentation. An aggressive surgical approach consisting of complete surgical excision is required with a close clinical follow-up suggested for the first few years after presentation to identify its recurrence or the rare development of metastatic disease.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Sailukar M, Bhagwat R, Seth T. Melanotic neuroectodermal tumor of infancy. J Indian Assoc Pediatr Surg. 2007;12:136–7. [Google Scholar]
- 2.Enzinger FM, Weiss SW. Soft Tissue Tumors. St. Louis: Mosby-Year Book Inc; 1995. Benign tumors of peripheral nerves; pp. 879–81. [Google Scholar]
- 3.Agarawal P, Agarawal V, Raina V K. Melanotic neuroectodermal tumor of infancy: Case report of an unusual tumor. Indian J Plastic Surg. 2003;36:123–5. [Google Scholar]
- 4.Kapadia SB, Frisman DM, Hitchcock CL, Ellis GL, Popek EJ. Melanotic neuroectodermal tumor of infancy.Clinicopathological, immunohistochemical, and flow cytometric study. Am J Surg Pathol. 1993;17:566–73. doi: 10.1097/00000478-199306000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Regezzi JA, Scuibba JJ. Philadelphia, PA: Saunders; 1989. Oral Pathology.Clinical-Pathologic Correlations; 163 pp. [Google Scholar]
- 6.Bouckaert MM, Raubenheimer EJ. Gigantiform melanotic neuroectodermal tumor of infancy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:569–72. doi: 10.1016/s1079-2104(98)90347-x. [DOI] [PubMed] [Google Scholar]
- 7.Johnson RE, Scheithauer BW, Dahlin DC. Melanotic neuroectodermal tumor of infancy.A review of seven cases. Cancer. 1983;52:661–6. doi: 10.1002/1097-0142(19830815)52:4<661::aid-cncr2820520416>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 8.Howell RE, Cohen MM., Jr Pathological case of the month.Melanotic neuroectodermal tumor of infancy. Arch Pediatr Adolesc Med. 1996;150:1103–4. doi: 10.1001/archpedi.1996.02170350105021. [DOI] [PubMed] [Google Scholar]
- 9.Hoshina Y, Hamamoto Y, Suzuki I, Nakajima T, Ida-Yonemochi H, Saku T. Melanotic neuroectodermal tumor of infancy in the mandible: Report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:594–9. doi: 10.1067/moe.2000.105519. [DOI] [PubMed] [Google Scholar]
- 10.Eckardt A, Swennen G, Teltzrow T. Melanotic neuroectodermal tumor of infancy involving the mandible: 7-Year follow-up after hemimandibulectomy and costochondral graft reconstruction. J Craniofac Surg. 2001;12:349–54. doi: 10.1097/00001665-200107000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Sefa Kaya a, Ömer Faruk Ünal. Melanotic neuroectodermal tumor of infancy: Report of two cases and review of literature. Int J Pediatr Otorhinolaryngol. 2000;52:169–72. doi: 10.1016/s0165-5876(99)00302-x. [DOI] [PubMed] [Google Scholar]
- 12.Gaiger de Oliveira M, Thompson LD, Chaves AC, Rados PV, Lauxin IS, Filho MS. Management of Melanotic Neuroectodermal of infancy. Ann Diagn Pathol. 2004;8:207–12. doi: 10.1053/j.anndiagpath.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Mast BA, Kapadia SB, Yunis E, Bentz M. Subtotal maxillectomy for melanotic neuroectodermal tumor of infancy. Plast Reconstr Surg. 1999;103:1961–3. doi: 10.1097/00006534-199906000-00022. [DOI] [PubMed] [Google Scholar]
- 14.Kumari NR, Sreedharan S, Balachandran D. Melanotic neuroectodermal tumor of infancy: A case report. J Indian Soc Pedod Prevent Dent. 2007;25:148–51. doi: 10.4103/0970-4388.36568. [DOI] [PubMed] [Google Scholar]
- 15.Broello ED, Gorlin RJ. Melanotic neuroectodermal tumor of infancy- A neoplasm of neural crest origin.Report of a case associated with high urinary excretion of Vanilmandelic acid. Cancer. 1966;19:196–206. doi: 10.1002/1097-0142(196602)19:2<196::aid-cncr2820190210>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Anil S, Raji MS, Beeana VT. Melanotic neuroectodermal tumor of infancy.A case report. Indian J Dent Res. 1993;4:65–7. [PubMed] [Google Scholar]
- 17.Lee CH, Hong SP, Lim CY. Melanotic neuroectodermal tumor of infancy. J Korean med sci. 1986;1:63–7. doi: 10.3346/jkms.1986.1.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selim H, Shaheen S, Barakat K, Selim A. Melanotic neuroectodermal tumor of infancy: Review of literature and case report. J Pediatr Surg. 2008;43:e25–9. doi: 10.1016/j.jpedsurg.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 19.Butt FMA, Guthua SW, Chindia ML, Rana F, Osundwa TM. Early outcome of three cases of melanotic neuroectodermal tumour of infancy. J Craniomaxillofac Surg. 2009;37:434–7. doi: 10.1016/j.jcms.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 20.Cutler LS, Chaudhry AP, Topazian R. Melanotic neuroectodermal tumor of infancy: An ultrastructural study, literature review, and reevaluation. Cancer. 1981;48:257–70. doi: 10.1002/1097-0142(19810715)48:2<257::aid-cncr2820480209>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Sharma MC, Mahapatra AK, Sudha K, Gailwad S. Melanotic neuroectodermal tumour of infancy: Immunohistochemical and histogenetic consideration. J Assoc Physicians India. 1996;44:278–80. [PubMed] [Google Scholar]
- 22.Khoddami M, Squire J, Zielenska M, Thorner P. Melanotic neuroectodermal tumor of infancy: A molecular genetic study. Pediatr Dev Pathol. 1998;1:295–9. doi: 10.1007/s100249900042. [DOI] [PubMed] [Google Scholar]
- 23.Carnevale GG, Mortelliti AJ. The operating microscope in the management of Melanotic neuroectodermal tumor of infancy. Am J Otolaryngol. 2001;22:76–9. doi: 10.1053/ajot.2001.20696. [DOI] [PubMed] [Google Scholar]
- 24.Pettinato G, Manivel JC, d’Amore ES, Jaszcz W, Gorlin RJ. Melanotic neuroectodermal tumor of infancy.A reexamination of a histogenetic problem based on immunohistochemical, flow cytometric, and ultrastructural study of 10 cases. Am J Surg Pathol. 1991;15:233–45. [PubMed] [Google Scholar]