

Abstract
Background.
Traumatic brain injury (TBI) remains a major cause of death and disability. Oxidative stress is an important element of the injury cascade following TBI. Progressive compromise of antioxidant defenses and free radical-mediated lipid peroxidation are one of the major mechanisms of secondary TBI. NR2B is a glutamate receptor and its activation is caused by TBI increasing a brain cell death, along with caspase-3 as a hall mark of apoptosis. Glutathione is a potent free radical scavenger that might prevent secondary TBI damage and inhibited apoptosis.
Materials and Methods.
In the present study, it aims to demonstrate the effect of glutathione on inhibition of brain oxidative damage in a TBI rat model.
Results.
In this study, the expressions of mRNA NR2B in placebo group and groups with glutathione administration at 0, 3, and 6 hours after TBI were 328.14, 229.90, 178.50, and 136.14, respectively (P<0.001). The highest caspase-3 expression was shown in placebo group with 66.7% showing strong positive results (>80%); as expected, glutathione administered in 0, 3, and 6 hours groups had lower strong positive results of 50%, 16.7%, and 16.7%, respectively, (P=0.025).
Conclusion.
In conclusion, this study showed that glutathione administration in a TBI rat model decreased NR2B gene- and caspase-3 protein-expression that lead to the inhibition of brain cell death. Our results suggest that glutathione, as a potent free radical scavenger, has a brain cell protective effect against oxidative damage and cell death induced by TBI in rat model.
Keywords: Caspase-3, glutathione, NR2B gene, traumatic brain injury
Introduction
Traumatic brain injury (TBI) remains a major cause of death and disability with an estimated annual occurrence of 1.4 million cases in the United States.[1] In Panti Nugroho Pakem Hospital, Yogyakarta, Indonesia, TBI cases were reported in the first quarter of year 2005, which ranked fifth of all visits to the emergency room and the second of all causes of hospitalization; of those cases, 17.8% was referred to the main-hospital with a mortality rate of 2.7%.[2] The Department of Neurosurgery at Dr. Hasan Sadikin Hospital (RSHS), Bandung, Indonesia, received 1212 cases within the 11 months of 2000 or more than 100 cases/month. The incidence was increased by 11.6% (1352) in 2006. In August 2007, 258 out of 462 accident cases were TBI (44.2%).[3]
TBI can be classified as primary, which occurs immediately following a trauma and continues to evolve during the subsequent hours and days after the initial insult in what is referred to as a secondary injury.[4] Improved outcomes might be achieved by preventing or reducing the secondary injury. Limiting a secondary injury results in lower mortality rates, improved functional outcome scores, shorter hospital stay, and decreased charges.[5,6]
Several pathogenetic mechanisms of secondary damage, including derangements in cerebral blood flow, excitotoxicity, reactive oxygen species (ROS), inflammation, and apoptosis have been described.[7] Two signaling pathways for the initiation of apoptosis are well known: One is mediated by a dead receptor, which is called an “extrinsic pathway,” and the other, the “intrinsic pathway,” is mediated by mitochondria.[8,9] In both pathways, the induction of apoptosis leads to the activation of initiator caspases: Caspase-8 for the extrinsic pathway, and caspase-9, which is activated at the apoptosome for the intrinsic pathway, which finally activates caspase's executor, caspase-3.[10]
The N-methyl-D-aspartate (NMDA) receptor is a heteromeric ligand gated ion channel that interacts with multiple proteins.[11,12] NMDA receptors are composed of association of subunits that belong to two families: A single gene product (NR1) with eight splice variants and four different NR2 subunits (NR2A, B, C, D) produced by a different gene.[13] Within the brain, the NR1, NR2A and NR2B subunits are more prominent in cortical areas and the hippocampus than in white matter and cerebellum.[14] On the other hand, the NR2B subunits are expressed at the highest levels in the hippocampus, cerebral cortex, and olfactory bulb.[15] Recently, Liu et al., reported that activation of NR2B- containing NMDA receptor results in excitotoxicity and increasing neuronal apoptosis.[16] These events are very important to promote a brain cell protection.
The exposure of oxidative stress such as free radicals has been suggested as a major cause of neural cell death following TBI.[17,18] The brain's antioxidant mechanisms include superoxide dismutase (SOD) which converts free radicals to hydrogen peroxide (H2O2), and glutathione peroxidase which further metabolized H2O2 to H2O and O2.[19] Oxidative stress is harmful due to its high fatty acid content and proportionately large share of total body oxygen consumption.[20] SOD over expression has a protective effect against ischemic injury.[21,22] Endogenous glutathione is up regulated in the brain in response to TBI to compensate the damage.[23] We speculated that exogenous glutathione would be needed to prevent a secondary TBI damage and inhibit apoptosis.
The aim of the present study was to examine the effect of glutathione administration on the expressions of mRNA NR2B and caspase-3 as indicators of early apoptosis-sign after a brain injury on a TBI rat model. We hypothesized that glutathione administered shortly after brain injury has a protective effect against the long-term sequelae of TBI by inhibiting the expression of NR2B and caspase-3.
Materials and Methods
Animal studies
Specific pathogen-free adult male Wistar rats were obtained from Bandung Institute of Technology with standard of Animal Care Committee, Bandung, Indonesia. They were maintained on a standard laboratory feed in a 12-hour (h) light/dark cycle. This study was approved by the Animal Research Ethics Board of Universitas Padjadjaran, Bandung, Indonesia. After a one-week period of environmental adaptation, the rats (n=30) weighing~400 gm were divided into five groups: (A) Control: Without TBI and without treatment, (B) Placebo: With TBI and administration of NaCl 0.9% soon after TBI, (C) Glutathione 0 h: With TBI, administration of glutathione 10.8 mg soon after TBI, (D) Glutathione 3 h: TBI, administration of glutathione 10.8 mg 3 h after TBI, and (E) Glutathione 6 h: With TBI, glutathione 10.8 mg 6 h after TBI and euthanized 3 h later (n=6). Some parts of the brain were stored in RNA-later for subsequent RNA isolation and other parts of the brains were fixed in 10% formalin, 4 μm sections and processed for tissue staining.
Traumatic brain injury rat model
Male rats were anesthetized with pentotal intraperitoneally 40 mg/kg body weight. After a midline skin incision, a 5-7 mm craniotomy was made from 3 mm right posterior of the bregma and 2 mm right lateral to the midline. The animals were then subjected to TBI using a small iron bar, receiving a contact velocity of 571.17 Newton/mm2. The contact velocity is equal to a mild TBI.[24] After the injury, the craniotomy was closed and the rats were returned to their cages.
Drug (Glutathione) administration
A total of 10.8 mg (0.018×600 mg) of glutathione (Tationil® 600 mg/4 ml from Roche, Italy) was injected intraperitoneally. For rats weighing~400 grams, this equals to 600 mg of glutathione for human (70 kg).
RNA isolation and cDNA synthesis
Total RNA was extracted from fresh brain tissue using an RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The quantity of isolated RNA was measured using an ND-1000 Spectrophotometer (NanoDrop Tech Inc., Rockland DE). Template cDNA was synthesized from 13.5 μg of total RNA with an Omniscript Reverse Transcriptase kit (Qiagen, Hilden, Germany), Random Primer (hexadeoxyribonucleotide mixture, Takara, Shiga, Japan) and Ribonuclease Inhibitor (Porcine liver, Takara, Japan). Total RNA was reverse-transcribed with four units of Omniscript Reverse Transcriptase in a reaction volume of 20 μl (60 min at 37°C, 5 min at 93°C, and finally put on ice). The cDNA samples were stored at –30°C until analysis.
Reverse transcription-polymerase chain reaction
Reverse transcription-polymerase chain reaction (RT-PCR) was performed with a one step system (Promega BioSci., San Luis, CA, USA) using NR2B primers described previously;[25] forward: GGTAGCCATGAACGAGACTG and reverse: TTCACGAAGTCCTGGTAGCC. RT-PCR mix were subjected to RT for 45 min at 45°C, followed by inactivation of RT enzyme at 94°C (2 min) and PCR (40 cycles) consisted of 94°C (30 sec), 60°C (1 min), and 68°C (2 min) with final extension at 68°C (10 min). The products were then run on a 3% agarose gel. Densitometry was performed by using Adobe Photoshop (Apple Inc., Cupertino, CA, USA) acquisition and analysis by the Quantity One (BioRad Inc., Melville, NY, USA).
Immunohistochemistry for caspase-3
Immunohistochemical staining of anti-caspase-3 antibody was performed by streptoavidin-biotin as described previously.[26] Sections of four mm thick were deparaffinized and incubated with fresh 0.3% hydrogen peroxide in methanol for 30 min at room temperature. The specimens were then incubated with anti-caspase-3 antibody (Biocare Medical, Concord, CA, USA) as the primer antibody at a 1:100 dilution. The specimens were counterstained with H and E. Negative controls were prepared by substituting normal mouse serum for each primary antibody: No detectable staining was evident. The degree of staining was scored as we previously described.[26] The sections were evaluated by two investigators without the knowledge of the pathological background.
Statistical analysis
Statistical analysis was performed using the Stat View software (ver. 5.0, SAS Institute Inc., NC, USA). The differences were considered statistically significant when P value was <0.05 (ANOVA).
Results
We examined the effects of glutathione treatment on a TBI rat model at the molecular level. Isolated RNA from the rats’ brains’ was then subjected to RT-PCR to observe NR2B gene at 341 base-pair (bp). NR2B, as shown in [Figures 1 and 2], was observed slightly in control brains (Group A, 96.25±20). In contrast, strong expressions were observed in placebo group (Group B, 328.14±24), as shown in [Figure 3], and gradually decreased in three other treated groups (Group C, 229.9±41; Group D, 178.5±41; and Group E, 136.14±63), as shown in [Figures 2 and 3]. The results showed that NR2B gene expression gradually decrease from placebo group to glutathione treated groups (P<0.001; [Figure 3]) in a time-dependent manner, it might be due to neurotoxicity-decreased (represented by decreased NR2B as glutamate receptors) effect which lead to the inhibition of brain cell death. Our results suggested that glutathione as a potent antioxidant had a brain cell protection in TBI.
Figure 1.

RT-PCR results of the NR2B gene expression. PCR products identified following RT-PCR of NR2B gene expression. The presence of 341 base pair (bp) fragment represents expression of NR2B in group A and group B (a)
Figure 2.
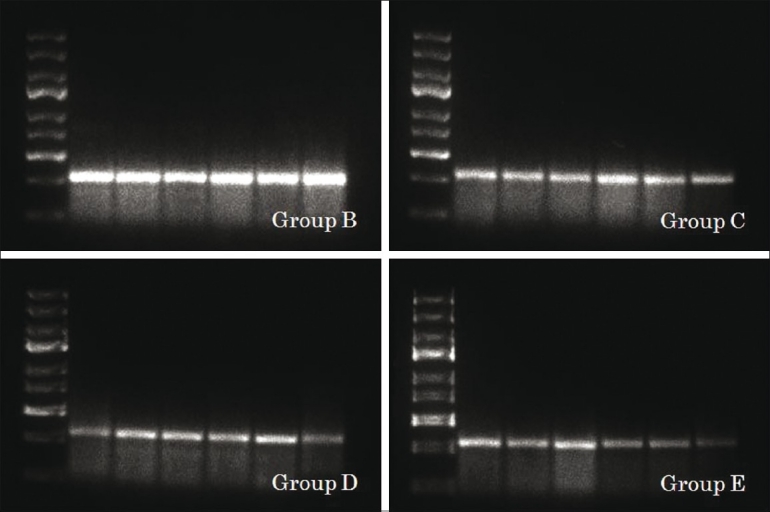
Expression of NR2B in group B, C, D and E, respectively (b)
Figure 3.

Densitometric estimation of NR2B in each group (c). For group-explanation see Materials and Methods. Line 1: DNA ladder marker 100 bp. Lanes 2-6: Six rats brain tissue in each group
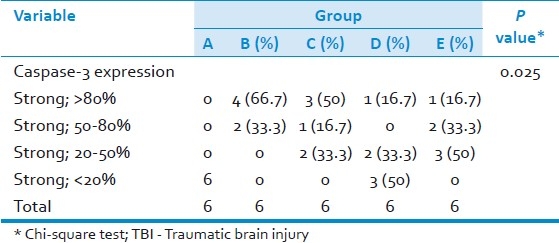
Furthermore, we observed caspase-3 expression in the cytoplasm of brain cells [Figures 4 and 5]. Expression of caspase-3 was not observed in control brains (Group A). By comparison, strong expression of caspase-3 was observed in placebo group (Group B, 66.7% was strong positive >80%), as shown in [Figure 4] and gradually decreased in three other treated groups (Group C, 50% was strong positive >80%; Group D, 16.7%; and Group E, 16.7%) as shown in [Figure 5]. The results showed that caspase-3 protein expressions gradually decrease from placebo group to glutathione treated groups (P=0.025; Table 1). The expressions of caspase-3 on a TBI rat model treated with glutathione were significantly lower than the expressions observed among those in the placebo group; the presence of this antigen was found to be strongly associated with glutathione treatment (P=0.025; Table 1). Glutathione administered shortly after brain injury as a single dose of 10.8 mg prevented the increase in cell death (represented by decreased caspase-3 as hall mark of apoptosis) in traumatized brain hemispheres.
Figure 4.
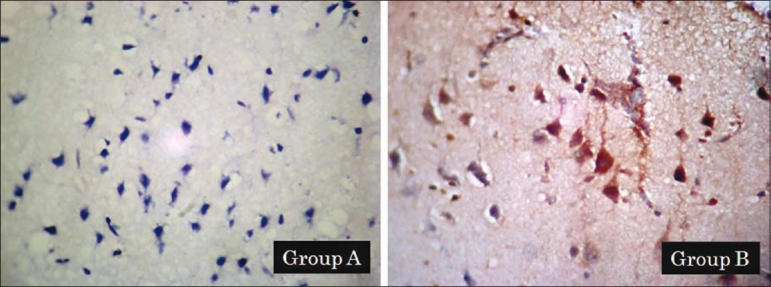
Immunohistochemistry results of the caspase-3 protein expression. The brown color represents a positive staining of caspase-3. Expression of caspase-3 in group A and group B (a)
Figure 5.

Expression of caspase-3 in each group B, C, D and E, respectively (b). For group-explanation see Materials and Methods
Table 1.
Distribution of caspase-3 protein expression in TBI rat model treated with Glutathione
Discussion
Our study demonstrated a temporal sequence of event following TBI, which included a mark increase of NR2B and caspase-3 expression levels in placebo group of TBI rat model, indicating that peroxidation of lipid membrane as well as oxidative stress produced by head trauma induced damage of Deoxyribonucleic acid (DNA) and proteins.[27] The oxidative stress played a key role in mediating secondary brain injury induced cell death by TBI.[24,28] When the tissues are exposed to oxidative stress, they increase the activity and expression of antioxidant enzymes as a compensatory mechanism against free radical-mediated damage. Nevertheless, the increased activity of the antioxidant enzymes may be inadequate to counteract the potential damage in many conditions of oxidative stress.[29] Moreover, antioxidant enzyme activities have been found to be diminished under highly elevated oxidative stress conditions as a result of molecular damage. On the other hand, over expression of the glutathione enzyme as one of the antioxidant and anti peroxidase which strongly decrease the cell death from brain injury.[30]
The brain is particularly vulnerable to oxidative injury because of its high rate of oxygen consumption and intense production of reactive radicals. NMDA receptor (NMDAR), a glutamate receptor, mediates neural plasticity and synaptic transmission in the mammalian central nerves system and also neuronal cell death.[31,32] Recently, NR2B receptor was shown to have different roles in supporting neuron survival and mediating neuronal cell death and hence had the impacts on neurotoxicity of brain.[16] Liu et al., reported that the activation of NR2B-containing NMDA receptor resulted in excitatoxicity and an increasing neuronal apoptosis.[16] Shahib et al., showed that the increase of NR2B expression is needed at certain level otherwise it might induce apoptosis.[25]
Nerve cell damage that is caused by the rupture of brain blood vessels is the result of excessive release of glutamate from nerve cells and glial (marked by increased of the NR2B expression) and not because of hypoxic or ischemic mechanism.[33] Glutamate then activates at least three types of NMDA receptor (NR2B, Kainate, and AMPA). These receptos could increase intracellular Ca2+ level.[34] Since NMDA is paired with Ca2+ channel, activation of both will be followed by an influx of Ca2+, sodium, water, and chloride into the intracellular-space causing cellular swelling or cytotoxic-edema.[35,36] The increasing level of intracellular Ca+ will increase H2O2 (peroxide) as well. The above events will produce nitric oxide, ROS, cofactor caspases from mitochondria that activate caspase-3 as the executor caspase and eventually cause cell death.[37,38] The excessive release of glutamate induced exitatory which effect leads to a decrease in endogenous glutathione. Additional exogenous glutathione which acts as an antioxidant and anti peroxide will slow brain cell death process and prevent further damage in viable brain cells.
Conclusion
In conclusion, this study showed that glutathione administered as a single dose of 10.8 mg prevents the brain oxidative damage induced by TBI in the rats by inhibiting the expression of NR2B and caspase-3 expression. Our results suggest that glutathione must be administered shortly after brain injury since the therapeutic time window in brain injury is very narrow (between 3 to 6 hours). The outcome of this study is expected to encourage the progress of traumatic brain injury studies in Indonesia. Further studies will be needed to elucidate the adjuvant glutathione therapy mechanism in the clinical setup of TBI.
Acknowledgment
We thank Prof. Dr. med. Tri Hanggono Achmad and Prof. R. M. Padmo Santjojo for their expertise and fruitful discussion. This work has been partly supported by Universitas Padjadjaran's research grant-in-aids for Ahmad Faried.
Footnotes
Source of Support: Universitas Padjadjaran
Conflict of Interest: None declared.
References
- 1.Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Traumatic Brain Injury. 2006. [Last cited on 2011 Jun 12]. Available from: http://www.cdc.gov/ncipc/tbi/TBI.htm .
- 2.Wijanarka A, Dwiphrahasto I. Implementation of clinical governance: Indicator development of head injury clinic in emergency unit. J Health Serv Manage. 2005;8:213–20. [Google Scholar]
- 3.Arifin MZ. Doctorate Dissertation, Department of Neurosurgery. Bandung, Indonesia: Universitas Padjadjaran; 2010. Glutathione effect on glutamate receptor and cell death after traumatic brain injury. [Google Scholar]
- 4.Fakhry SM, Trask AL, Waller MA, Watts DD. Management of brain injured patients by an evidence-based medicine protocol improves outcomes and decreases hospital charges. J Trauma. 2004;56:492–500. doi: 10.1097/01.ta.0000115650.07193.66. [DOI] [PubMed] [Google Scholar]
- 5.Palmer S, Bader MK, Qureshi A, Palmer J, Shaver T, Borzatta M, et al. The impact on outcomes in a community hospital setting of using AANS Traumatic Brain Injury Guidelines. J Trauma. 2001;50:657–64. doi: 10.1097/00005373-200104000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Stein SC, Spettell CM. Delayed and progressive brain injury in children and adolescents with head trauma. Pediatr Neurosurg. 1995;23:299–304. doi: 10.1159/000120975. [DOI] [PubMed] [Google Scholar]
- 7.Gupta S. Molecular steps of death receptor and mitochondrial pathways of apoptosis. Life Sci. 2001;69:2957–64. doi: 10.1016/s0024-3205(01)01404-7. [DOI] [PubMed] [Google Scholar]
- 8.Krammer PH. CD95 (APO-1/Fas)-mediated apoptosis: Live and let die. Adv Immunol. 1999;71:163–210. doi: 10.1016/s0065-2776(08)60402-2. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz I, Kirchhoff S, Krammer PH. Regulation of death receptor-mediated apoptosis pathways. Int J Biochem Cell Biol. 2000;32:1123–36. doi: 10.1016/s1357-2725(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 10.Igney FH, Krammer PH. Death and anti-death: Tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–88. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 11.McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–60. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 12.Mutel V, Buchy D, Klingelschmidt A, Messer J, Bleuel Z, Kemp JA, et al. In vitro binding properties in rat brain of [3H]Ro25-6981, a potent and selective antagonist of NMDA receptors containing NR2B subunits. J Neurochem. 1998;70:2147–55. doi: 10.1046/j.1471-4159.1998.70052147.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim WT, Kuo MF, Mishra OP, Delivoria-Papadopoulos M. Distribution and expression of the subunits of N-methyl-D-aspartate (NMDA) receptors; NR1, NR2A and NR2B in hypoxic newborn piglet brains. Brain Res. 1998;799:49–54. doi: 10.1016/s0006-8993(98)00464-8. [DOI] [PubMed] [Google Scholar]
- 14.Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, et al. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: Correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38:611–23. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 15.Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–82. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–57. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kantos HA, Povlishok JT. Oxygen radicals in brain injury. Cent Nerv Sys Trauma. 1986;3:257–63. doi: 10.1089/cns.1986.3.257. [DOI] [PubMed] [Google Scholar]
- 18.Braugler JM, Hall ED. Central nervous system trauma and stroke.Biochemical concentrations for oxygen radical formation and lipid peroxidation. Free Rad Biol Med. 1989;6:289–301. doi: 10.1016/0891-5849(89)90056-7. [DOI] [PubMed] [Google Scholar]
- 19.Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. Neurotrauma. 2000;17:871–90. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- 20.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–71. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 21.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, et al. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–9. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikawa S, Kinouchi H, Kamii H, Gobbel GT, Chen SF, Carlson E, et al. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J Neurosurg. 1996;85:885–91. doi: 10.3171/jns.1996.85.5.0885. [DOI] [PubMed] [Google Scholar]
- 23.Fan P, Yamauchi T, Noble LJ, Ferriero DM. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma. 2003;20:437–45. doi: 10.1089/089771503765355513. [DOI] [PubMed] [Google Scholar]
- 24.Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quinn PJ, et al. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J Neurochem. 2000;7:2178–89. doi: 10.1046/j.1471-4159.2000.0752178.x. [DOI] [PubMed] [Google Scholar]
- 25.Shahib MN, Syamsunarno MR, Faried A, Yuliana D, Anggraeni D, Yuniarti L, et al. The effect of glycine max extract diets on changes in NR2B gene expression, cognitive vitality and neurotoxicity in high concentrate consumption. Kitakanto Med J. 2010;60:41–7. [Google Scholar]
- 26.Faried A, Kimura H, Faried LS, Usman N, Miyazaki T, Kato H, et al. Expression of carbohydrate antigens in human esophageal squamous cell carcinoma: Prognostic application and its diagnostic implication. Ann Surg Oncol. 2006;14:960–7. doi: 10.1245/s10434-006-9200-z. [DOI] [PubMed] [Google Scholar]
- 27.Feuerstein GZ, Wang X, Barone FC. Inflammatory gene expression in cerebral ischemia and trauma.Potential new therapeutic targets. Ann N Y Acad Sci. 1997;825:179–93. doi: 10.1111/j.1749-6632.1997.tb48428.x. [DOI] [PubMed] [Google Scholar]
- 28.Shohami E, Beit-Yannai E, Horowitz M, Kohen R. Oxidative stress in closed-head injury: Brain antioxidant capacity as an indicator of functional outcome. J Cereb Blood Flow Metab. 1997;17:1007–19. doi: 10.1097/00004647-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez C, Mayo JC, Sainz RM, Antolín I, Herrera F, Martín V, et al. Regulation of antioxidant enzyme: A significant role for melatonin. J Pineal Res. 2004;36:1–9. doi: 10.1046/j.1600-079x.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Cheng E, Brooke S, Chang P, Sapolsky R. Over-expression of antioxidant enzymes protects cultured hippocampal and cortical neurons from necrotic insults. J Neurochem. 2003;87:1527–34. doi: 10.1046/j.1471-4159.2003.02123.x. [DOI] [PubMed] [Google Scholar]
- 31.Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol. 1987;394:501–27. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–87. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 33.Ascher P, Nowak L. The role of divalent cations in the NMDA response of mouse central neurons in culture. J Physiol. 1988;399:247–66. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogura A, Miyamoto M, Kudo Y. Neural death in vitro: Parallelism between survivability of hyppocampal neurons and sustained elevation of cytosolic Ca2+ after exposure to glutamate receptor agonist. Exp Brain Res. 1988;73:447–58. doi: 10.1007/BF00406601. [DOI] [PubMed] [Google Scholar]
- 35.Verma A. Opportunities for neuroprotection in traumatic brain injury. J Head Trauma Rehabil. 2000;15:1149–61. doi: 10.1097/00001199-200010000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Murthy TV, Bhatia P, Sandhu K, Prabhakar T, Gogna RL. Secondary brain injury: Prevention and intensive care management. Indian J Neurotrauma. 2005;2:7–12. [Google Scholar]
- 37.Bigford GE, Alonso OF, Dietrich D, Keane RW. A novel protein complex in membrane Rafts lingking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J Neurotrauma. 2009;26:703–20. doi: 10.1089/neu.2008.0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: Bioenergic function in cell life and death. J Cereb Blood Flow Metab. 1999;19:231–45. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]