

Adoptive T-cell transfer is recognized as an innovative treatment strategy for various malignant diseases.1,2 To improve the efficacy and sometimes the safety of this approach, T cells can be genetically manipulated to modify their antigen specificity, to enhance their in vivo survival and trafficking to specific tissues, or to be eliminated in the event of undesired toxic effects.3 γ-retroviral vectors are frequently used to obtain robust and stable genetic modification of human T lymphocytes because these vectors can efficiently integrate within the genome and ensure that the inserted transgene is passed to the progeny of infected cells.
Although integration is the desired effect of retroviral-mediated gene transfer, it carries the risk of insertional mutagenesis, which can result in dysregulated gene expression and subsequent malignant transformation.4 The potential for insertional mutagenesis and malignant transformation may be increased in cases where replication-competent retroviruses (RCRs) are present in the vector products, as continued viral replication could result in multiple integrations within the host-cell genome. When a high-titer vector-producer packaging cell line (VPC) with a known level of RCR contamination of 103–104 virions/ml was used to transduce stem cells in 10 nonhuman primates, three animals developed virus-induced T-cell lymphoma.5 This observation led to extensive public discussions and the adoption of the current US Food and Drug Administration (FDA) guidance for RCR testing.
Almost all γ-retroviral vectors manufactured for clinical use are produced using stable VPCs derived from murine or human cell lines. The risk of RCR generation is minimized during the production of the retroviral vector supernatant by segregating vectors encoding the exogenous gene of interest from sequences encoding the viral proteins gag, pol, and env. These sequences are provided in trans by VPCs that stably express gag, pol, and env genes. Early VPCs that contained gag, pol, and env genes expressed from a single plasmid had a high incidence of RCR generation due to recombination between the vector and viral component plasmids.6 The risk of RCR generation was greatly reduced by segregating each of the viral components into separate plasmids with minimal homology, one containing gag-pol and the other env, and by minimizing homology between the vector and packaging sequences.6,7,8
Although current packaging cell line and vector designs make the generation of RCRs extremely unlikely, present FDA guidelines require extensive testing at multiple levels for the presence of RCRs, beginning with screening of packaging cell lines and vector products, followed by testing of the final gene-modified cell product, and culminating with patient monitoring after cell infusion. The extended S+L− assay—in which test material is added to a susceptible cell line that allows virus replication (amplification), after which the medium from these cells is assayed for helper virus—is commonly used to detect RCRs within packaging cell lines, viral lots, and cell products.9 Despite negative in vitro testing results for RCRs, it is further required that patients be regularly monitored postinfusion using polymerase chain reaction (PCR) or serological methods, and positive results obtained by these assays must then be confirmed by biological assays such as the S+/L− assay.
RCR testing is labor-intensive, time-consuming, and extremely costly, which severely limits the translational applicability of genetically modified cell therapies by most not-for-profit medical centers. For more than a decade, our own centers have focused on adoptive transfer of gene-modified T cells for the treatment of malignancies; other investigators, nationally and internationally, have followed similar practices. Despite the widespread and extensive use of resources to detect RCRs, there has been no systematic study examining the incidence of RCR positivity in these therapeutic products or their recipients. Here, we report a large cohort of RCR testing results from clinical trials carried out at Baylor College of Medicine, St Jude Children's Research Hospital, the National Cancer Institute, the University of Pennsylvania, and Indiana University.
Overall, 30 master cell banks (MCBs) have been generated, from which 42 viral supernatant lots have been produced (Supplementary Table S1 online). The great majority of clinical-grade MCBs (28/30) were generated using the PG13 VPC, which provides viral particles pseudotyped with the gibbon ape leukemia virus (GALV) envelope.7 Two MCBs were generated using the PA317 VPC. In most cases, the SFG (10/30) or MSGV1 (15/30) retroviral vector was used. Single-cell clones from transduced packaging cells were expanded to produce an MCB, from which clinical-grade retroviral supernatant was produced and collected. RCR screenings performed by amplification in HEK 293 cells and analyzed using the S+L− focus assay were consistently negative from all 30 MCBs and 42 viral supernatant lots.
Representative data from 29 clinical studies using gene-modified T-cell products manufactured with γ-retroviral vectors from the tested MCBs are shown in Table 1. These studies utilized either polyclonal activated T cells (ATCs); cytotoxic T lymphocytes (CTLs) specific for antigens encoded by Epstein–Barr virus (EBV), cytomegalovirus, or adenovirus; or tumor-infiltrating lymphocytes (TILs). As shown, a variety of transgenes were expressed from marker proteins (Neo, ref. 10) to tumor-antigen receptors (chimeric antigen receptors (ref. 11)) or transgenic αβ-T-cell receptors (ref. 12) to cytokines (interleukin-2, ref. 13) and suicide genes (iCaspase9, ref. 14). S+L− focus-forming assays or PG-4 plaque assays were performed to detect RCR generation within 297 T-cell products. RCRs were absent in all T-cell products thus far tested, with 15 results still pending. Three additional T-cell products were generated but not tested for RCRs, as the cells were maintained in culture for less than 4 days after retroviral transduction. Current FDA recommendations do not require RCR testing when cells are cultured under these short-term conditions.
Table 1. RCR results from T-cell products and patient-monitoring samples.
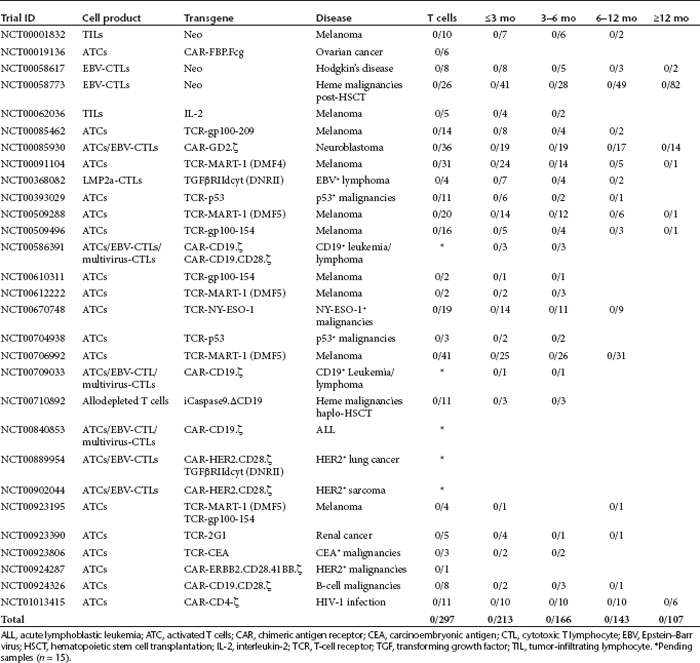
FDA guidelines require patient follow-up analysis for RCRs to be performed at 3, 6, and 12 months and yearly thereafter postinfusion. RCR screening was performed by PCR analysis of peripheral blood mononuclear cells to detect the presence of the GALV, amphotropic envelope, or gag-pol. A total of 629 follow-up samples have been analyzed, ranging from 1 month to 8 years after infusion (Table 1). In most studies occurring since 2001, yearly samples collected from 1 to 15 years postinfusion were banked but not analyzed for RCRs. Thus far, we have not detected RCRs in any patient sample.
Although the results of RCR testing have consistently been negative, the costs of the procedure are considerable. Certain tests were performed in-house or through the National Gene Vector Biorepository, which defrayed a significant part of the trial cost. Because estimating true cost to academic institutions can be difficult, we utilized prices provided by two separate commercial sources to estimate the costs incurred by RCR testing. As indicated in Supplementary Table S2, the commercial cost of testing 30 MCBs plus 42 retroviral supernatant lots ranges between $1,497,036 and $1,593,726. The total cost of testing 312 T-cell products ranges between $3,088,800 and $3,092,856, and the total in-house cost of testing 629 patient samples is around $314,500.
We believe that the data reported herein have significant implications for the conduct of clinical trials using T cells genetically manipulated with γ-retroviral vectors. FDA requirements for RCR screening have changed little over the past 20 years since this novel therapy was first introduced into the clinic. Our analysis indicates that modern VPCs such as PG13, which are characterized by independent gag-pol and env components and pseudotyped viral particles, significantly reduce the risk of RCR generation. In addition to screening the MCBs, investigators must screen 5% of the final vector product and 108 end-of-production cells of each vector lot. These requirements were set somewhat arbitrarily, balancing the need to provide rigorous screening with the technical and practical implications of testing a large percentage of a vector lot. In an effort to ensure that the RCR assays and sample sizes adequately screened for RCRs, the FDA further required testing of ex vivo manipulated cell products and patient samples. Our data demonstrating the lack of RCRs in ex vivo γ-retroviral-transduced T-cell products and patient samples over 29 trials, including HIV-infected patients, suggest that the current FDA requirements for screening MCBs and viral lots are adequate and appropriate.
Our data also question the need for continued testing of T-cell products and patient samples for RCRs. This testing does not appear to provide additional safety assurances above those obtained by the screening of vector products. Given the lack of apparent benefit, one must also consider the associated high costs of testing. Because many of these therapies have already produced well-documented, complete, and long-term responses in otherwise lethal disorders,12,15 the significant expense of testing will impede translation of this therapy into the clinic.
We make the following proposals for future T-cell gene therapy protocols. We believe it is important that the MCB, end-of-production cells, and viral lots continue to be rigorously analyzed to exclude the presence of RCRs before they are released for clinical use; the current FDA recommendation for screening γ-retrovirus lots for RCRs should continue unchanged. Final T-cell products that incorporate γ-retroviral vectors, together with samples from treated patients, should be collected and archived for retrospective studies, but need not be actively screened for RCR contamination. It should be noted that the majority of retroviral vectors used in this analysis were generated using the PG13 cell line. Currently, we do not know whether these data can be extrapolated to other packaging cell lines, other methods of vector production, or other vector systems (e.g., lentiviral vectors). Such virus-production methods will need to be tested experimentally. Future efforts may reasonably be dedicated to the development of rapid and inexpensive assays for the detection of RCR contamination of manipulated T-cell products, but for the present, our data show that the changes we propose can be introduced without measurably affecting patient safety and, by reducing the cost of each protocol, will allow further effective treatments to be safely developed.
SUPPLEMENTARY MATERIAL Table S1. RCR results from packaging cell lines and viral supernatant lots. Table S2. Cost analysis of RCR testing.
Acknowledgments
Kenneth Cornetta is the founder of Rimedion Inc. Gwendolyn Binder-Scholl participated in this work while at the University of Pennsylvania. As of the writing of this paper, she is an employee of Adaptimmune Ltd.
This work was supported in part by grants from the Leukemia and Lymphoma Society Specialized Center of Research (SCOR; grant 7018), the National Institutes of Health (NIH) (3P50CA126752, P01CA094237, and 5U54HL081007), and the National Center for Research Resources/NIH National Gene Vector Biorepository program (P40 RR024928). The authors declared no conflict of interest.
Supplementary Material
RCR results from packaging cell lines and viral supernatant lots.
Cost analysis of RCR testing.
References
- Rosenberg SA., and, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner MK., and, Heslop HE. Adoptive T cell therapy of cancer. Curr Opin Immunol. 2010;22:251–257. doi: 10.1016/j.coi.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera JF, Brenner MK., and, Dotti G. Immunotherapy of human cancers using gene modified T lymphocytes. Curr Gene Ther. 2009;9:396–408. doi: 10.2174/156652309789753338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, von Kalle KC, Schmidt M, Le Deist F, Wulffraat N, McIntyre E.et al. (2003A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency N Engl J Med 348255–256. [DOI] [PubMed] [Google Scholar]
- Donahue RE, Kessler SW, Bodine D, McDonagh K, Dunbar C, Goodman S.et al. (1992Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer J Exp Med 1761125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CM., and, Miller AD. Production of high-titer helper virus-free retroviral vectors by cocultivation of packaging cells with different host ranges. J Virol. 1991;65:3887–3890. doi: 10.1128/jvi.65.7.3887-3890.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C., and, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65:2220–2224. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD., and, Chen F. Retrovirus packaging cells based on 10A1 murine leukemia virus for production of vectors that use multiple receptors for cell entry. J Virol. 1996;70:5564–5571. doi: 10.1128/jvi.70.8.5564-5571.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Reeves L., and, Cornetta K. Safety testing for replication-competent retrovirus associated with gibbon ape leukemia virus–pseudotyped retroviral vectors. Hum Gene Ther. 2001;12:61–70. doi: 10.1089/104303401450979. [DOI] [PubMed] [Google Scholar]
- Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA.et al. (2010Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients Blood 115925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G.et al. (2008Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma Nat Med 141264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM.et al. (2006Cancer regression in patients after transfer of genetically engineered lymphocytes Science 314126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S.et al. (2008Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2 Hum Gene Ther 19496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C.et al. (2011Inducible apoptosis as a safety switch for adoptive cell therapy N Engl J Med 3651673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A., and, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RCR results from packaging cell lines and viral supernatant lots.
Cost analysis of RCR testing.