

Abstract
CXC chemokine receptor 4 (CXCR4) is involved in many human malignant tumors and plays an important role in tumor growth and metastasis. To explore the effects of CXCR4 expression on the malignant cells of oral squamous cell carcinoma (OSCC), Tca8113 and SCC-9 cell lines, as well as their xenograft models, of nude mice were used to detect cancer cell proliferation alteration. This study also examined the corresponding molecular mechanism after CXCR4 knockdown using a recombinant lentiviral vector expressing small interference RNA (siRNA) for CXCR4. RNA interference-mediated knockdown of CXCR4 in highly aggressive (Tca8113 and SCC-9) tumor cells significantly inhibited the proliferation of the two cell lines in vitro and in vivo. The expression levels of >1,500 genes involved in cell cycle, apoptosis, and multiple signaling pathways were also altered. These results provide new evidence of CXCR4 as a promising tumor gene therapeutic target.
Introduction
Squamous cell carcinoma is the most common malignant tumor of the oral cavity. Despite advances in chemotherapy, radiotherapy, and surgical therapy over the last two decades, the 5-year survival rate for patients with oral squamous cell carcinoma (OSCC) is still poor (~50%).1 Local and/or regional tumor recurrence develops in approximately one-third of patients, despite definitive treatment. Patients with recurrent OSCC that is refractory to chemotherapy and/or radiation therapy have a median life expectancy of several months, and the response rate to second- or third-line chemotherapeutic regimens is ~15%.2 Two-thirds of patients dying of this disease have no evidence of symptomatic distant metastases. Therefore, local and regional disease control is an urgent need for more effective therapies.
Chemokines, a superfamily of small cytokine-like proteins, can bind to and activate a family of seven transmembrane G-protein-coupled receptors, the chemokine receptors. CXC chemokine receptor 4 (CXCR4) and its ligand stromal cell–derived factor-1 (SDF-1), also known as CXCL12, have been implicated in many malignancies.3 The CXCL12/CXCR4 axis is involved in several aspects of tumor progression including angiogenesis, metastasis, and survival.4,5,6,7 Müller et al. identified that CXCR4 is commonly elevated in malignant versus normal mammary epithelial cell lines. Blocking antibody to CXCR4 inhibited metastasis in a mouse xenograft model using MDA-MB-231 human breast cancer cells.8 In addition to breast cancer, CXCR4 is also detected in malignancies of the ovary, prostate, colon, head and neck, lung, pancreas, brain, and bladder.6,9 The functional role of CXCR4 in tumor metastasis was demonstrated in multiple studies using low-molecular-weight inhibitory peptides or neutralizing antibody directed to CXCR4, which showed that inhibiting CXCR4 activity reduced tumor cell migration and metastasis in vitro and in vivo.8,10,11 Uchida et al. identified that the blockade of CXCR4 inhibited lymph node metastasis in B88 OSCC cells through shRNA and the AMD3100, a CXCR4 antagonist. After orthotopically inoculating OSCC cells into the masseter muscle of nude mice, lymph node metastases, loss in body weight, and tumor volumes were significantly inhibited in mice inoculated with shCXCR4-17 cells compared with mice inoculated with control cells.12 Aside from supporting metastasis, CXCL12 and its receptor CXCR4 directly affects the proliferation of tumor cells. Ovarian carcinoma and non-Hodgkin's lymphoma cells can grow very well in vitro in the presence of CXCL12. Moreover, this pro-proliferative effect is blocked with TN14003, a specific antagonist of CXCR4.11 Small molecular inhibitors of CXCR4, such as plerixafor, TN14003, or BKT140, are being investigated in various cancer settings. Inhibition of CXCR4 with plerixafor has shown utility by facilitating mobilization of hematopoietic stem cells (HSCs) for autologous transplant in non-Hodgkin's lymphoma and multiple myeloma.13 However, there are some potential side effects of CXCR4 antagonists. The long-term inhibition of the CXCR4-CXCL12 axis would potentially expose patients to risks of immune system and hematopoietic dysfunctions.14 The use of CXCR4 antagonists in cancer patients may also cause the mobilization of normal progenitor cells, such as hematopoietic stem cells, from their microenvironments to the blood. Mobilized hematopoietic stem cells that are normally protected in marrow niches would be exposed to the effects of cytotoxic drugs in trials where CXCR4 antagonists are administered along with cytotoxic drugs, which could result in prolonged cytopenias.15 Therefore, seeking a safe and effective therapeutic method is an urgent task for cancer therapy. In recent years, RNA interference (RNAi), a powerful gene-silencing technology with high efficiency and specificity as well as low toxicity, has been widely used for silencing malignant cellular and viral genes.16,17 This technique provides great promise in the field of cancer therapy.
This study shows that the CXCR4 expression is necessary for OSCC cells to become proliferative, and inhibiting this expression using a recombinant lentiviral vector expressing small interference RNA (siRNA) for CXCR4 can induce tumor growth inhibition in vivo. Furthermore, CXCR4 expression is identified to promote cancer cell proliferation by altering the expression of >1,500 genes involved in cell cycle, apoptosis, and multiple signaling pathways using microarray analysis technology.
Results
CXCR4 expression is increased in multiple OSCC tumors and cell lines
The CXCR4 expression was examined using real-time polymerase chain reaction (RT-PCR) in five matched normal and OSCC biopsies as well as in OSCC cell lines, including Tca8113, SCC-4, and SCC-9 cell lines. The results show that CXCR4 mRNA levels increased more than tenfold in OSCC tumors from patients 1–4 compared with their matched normal tissues. Additionally, the CXCR4 mRNA levels in Tca8113 and SCC-9 cell lines were approximately increased fourfold compared with the SCC-4 cell line. Therefore, Tca8113 and SCC-9 cell lines were selected for further studies (Figure 1a,b).
Figure 1.
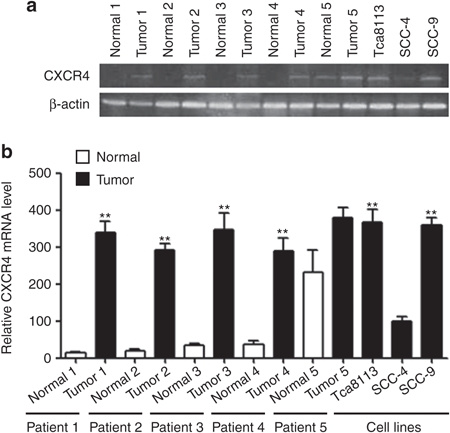
Elevated CXCR4 mRNA expression in OSCC. (a) Transcript levels of CXCR4 in five patients with OSCC and three OSCC cell lines (Tca8113, SCC-4, SCC-9). β-actin loading control is also shown. (b) Quantitative analysis of transcript levels of CXCR4, relative to β-actin, determined by qRT-PCR and compared with SCC-4 cells. Error bars indicate mean ± SD, n = 3 experiments; **P < 0.01. OSCC, oral squamous cell carcinoma; qRT-PCR, quantitative real-time polymerase chain reaction.
Effects of CXCR4 recombinant vector and TN14003 on the protein expression of CXCR4 in Tca8113 and SCC-9 cells in vitro
First, siRNA recombinant vector from two different CXCR4 sequences (siRNA1 or siRNA2) were expressed in the highly malignant Tca8113 and SCC-9 cell lines (Figure 2a). To determine whether the recombinant vectors were transduced into the tumor cells, the green fluorescent protein (GFP) expression of Tca8113 and SCC-9 cells in the NC-LV, CXCR4-lentivirus 1 (CXCR4-LV1), and CXCR4-lentivirus 2 (CXCR4-LV2) groups was assessed using flow cytometry. The results show that the delivery efficiency of NC-LV, CXCR4-LV1, and CXCR4-LV2 was more than 95%, and no GFP expression was observed in the Tca8113 and SCC-9 groups, as expected (Supplementary Figure S1). Then, the effects of different recombinant vectors and TN14003, the antagonist of CXCR4, on CXCR4 expression were detected. After CXCR4-LV1 (multiplicity of infection (MOI) = 50), CXCR4-LV2 (MOI = 50), and NC-LV (MOI = 50) transduction for 72 hours in Tca8113 and SCC-9 cells, the expression of CXCR4 protein levels was substantially reduced by ~90% and 80% in CXCR4-LV1 expressing Tca8113 and SCC-9 cells, respectively, compared with their parental cells (Figure 2b–d). However, the expression of CXCR4 protein was substantially reduced by ~20% and 30% in CXCR4-LV2 expressing Tca8113 and SCC-9 cells, respectively (data not shown). Therefore, the CXCR4-LV1 vector was chosen for further study. Additionally, the effect of TN14003 on protein expression of CXCR4 was tested. After 50 µmol/l TN14003 treatment for 8 hours, the protein expression levels of CXCR4 was substantially reduced by ~60% in Tca8113 and SCC-9 cells (Figure 2b–d). Interestingly, TN14003 (50 µmol/l) induced a significant reduction of CXCR4 protein levels at 8 hours, but the expression of CXCR4 completely recovered at 24 hours in Tca8113 cells and 16 hours in SCC-9 cells (Supplementary Figure S2).
Figure 2.
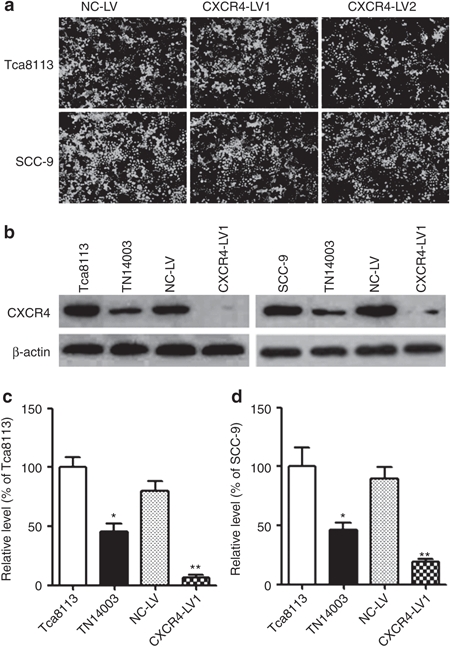
The inhibition of CXCR4 protein expression in OSCC cells treated with CXCR4-LV1 and TN14003. (a) Tca8113 and SCC-9 tongue squamous cell carcinoma cells were transduced with CXCR4-LV1 (MOI = 50), CXCR4-LV2 (MOI = 50), and NC-LV (MOI = 50) for 72 hours and were then photographed under an inverted fluorescence microscope (×100). (b) After 50 µmol/l TN14003 treatment for 8 hours and transduction with CXCR4-LV1 and NC-LV for 72 hours in Tca8113 and SCC-9 cells, CXCR4 protein levels were detected using Western blot analysis in the two cell lines. β-actin loading control is also shown. (c) Densitometric analysis of protein levels of CXCR4, relative to β-actin, determined by Western blot and compared with Tca8113 cells. (d) Densitometric analysis of protein levels of CXCR4, relative to β-actin, determined by Western blot and compared with SCC-9 cells. Error bars indicate mean ± SD; n = 3 experiments; *P < 0.05; **P < 0.01. OSCC, oral squamous cell carcinoma.
Inhibition of CXCR4 expression promotes cell cycle arrest and apoptosis of OSCC cells in vitro
To detect the effects of CXCR4 knockdown and TN14003 on OSCC cell proliferation and apoptosis, the cell cycle distribution of cells treated with CXCR4-LV1 and TN14003 in Tca8113 and SCC-9 cell lines were examined. After 50 µmol/l TN14003 treatment for 8 hours and transduction with CXCR4-LV1 (MOI = 50) and NC-LV (MOI = 50) for 72 hours in Tca8113 and SCC-9 cells, both cell lines showed decreased accumulation in S and G2/M phases and increased accumulation in the G1 as well as sub-G1 phases. After 8 hours of TN14003 treatment, the percentages of cells in the S phase decreased from ~32% to 14% in Tca8113 and from 35% to 21% in SCC-9 cells. The percentages of cells in the G2/M phase decreased from ~23% to 10% in Tca8113 and from 19% to 11% in SCC-9 cells. Conversely, the percentages of cells in sub-G1 phase increased from ~4% to 21% in Tca8113 and from 3% to 18% in SCC-9 cells. Interestingly, the percentages of cells in the G1 phase increased from ~40% to 54% in Tca8113 and from 42% to 50% in SCC-9 cells. Similarly, for the two cell lines transduced with CXCR4-LV1, the percentages of cells in the S phase decreased from ~32% to 11% in Tca8113 and from 35% to 13% in SCC-9 cells. The percentages of cells in the G2/M phase decreased from ~23% to 8% in Tca8113 and from 19% to 9% in SCC-9 cells. The percentages of cells in sub-G1 phase increased from ~4% to 32% in Tca8113 and from 3% to 28% in SCC-9 cells. The percentages of cells in G1 phase increased from ~40% to 48% in Tca8113 and from 41% to 49% in SCC-9 cells (Figure 3a–c). In addition, the cell cycle distribution of Tca8113 and SCC-9 cells at different time points of TN14003 treatment were examined. At 24 hours after TN14003 treatment, the percentages of cells in the S phase were restored to 28% and 32% in the Tca8113 and SCC-9 cells, respectively. The percentages of cells in the G2/M phase were restored to 19% and 17% in the Tca8113 and SCC-9 cells, respectively, whereas those in the sub-G1 phase were 9% and 7%, respectively. Finally, the percentages of cells in G1 phase were restored to 44% and 43% in the Tca8113 and SCC-9 cells, respectively (Supplementary Figure S3). These results suggest that CXCR4 downregulation promotes OSCC cell apoptosis.
Figure 3.
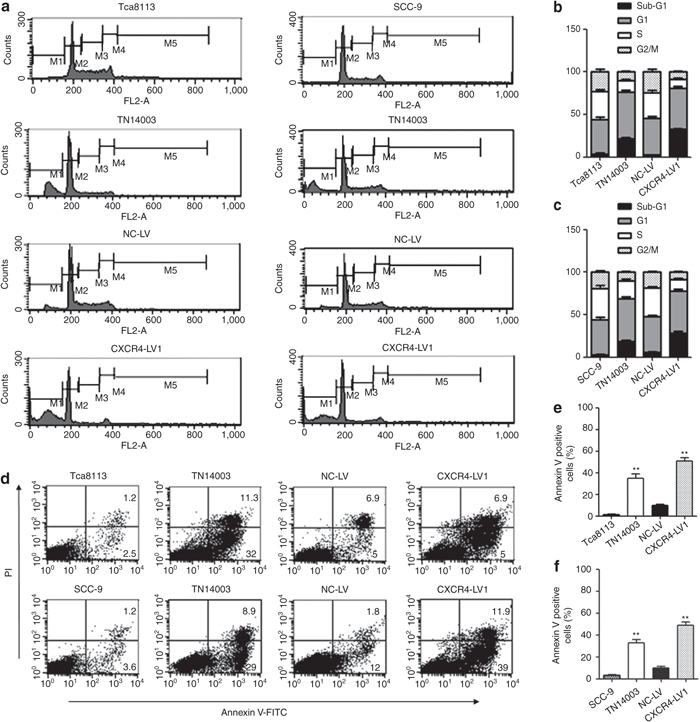
Cell cycle and apoptosis analysis of Tca8113 and SCC-9 cells which were treated with CXCR4-LV1 and TN14003. (a) After 50 µmol/l TN14003 treatment for 8 hours and transduction with CXCR4-LV1 (MOI = 50) and NC-LV (MOI = 50) in Tca8113 and SCC-9 cells for 72 hours, cell nuclei were fixed, stained with PI, and analyzed by flow cytometry. Representative histograms are shown. (b) The fractions of Tca8113 cells in sub-G1 (black), G1 (2N; gray), S (white), and G2–M (4N; speckled) are shown. (c) The fractions of SCC-9 cells in sub-G1 (black), G1 (2N; gray), S (white), and G2–M (4N; speckled) are shown. Error bars indicate mean ± SD, n = 3 experiments. (d) Apoptosis cells were stained with Annexin V-FITC and were analyzed as described in the text (see Materials and Methods). PI indicates propidium iodide. (e) Quantitative analysis of positive Annexin V Tca8113 cells in different groups. (f) Quantitative analysis of positive Annexin V SCC-9 cells in different groups. Error bars indicate mean ± SD; n = 3 experiments; **P < 0.01.
An Annexin V-FITC kit was used to determine the percentage of cells that underwent apoptosis. After 50 µmol/l TN14003 treatment for 8 hours, Annexin V/PI staining of Tca8113 and SCC-9 cells showed that ~35% and ~33% of cells were Annexin V positive, respectively. Furthermore, after transduction with CXCR4-LV1 (MOI = 50) and NC-LV (MOI = 50) in Tca8113 and SCC-9 cells for 72 hours, Tca8113 and SCC-9 cells showed that ~51% and ~48% of cells were Annexin V positive, respectively. These data suggest that the inhibitory effect on the protein expression of CXCR4 promotes Tca8113 and SCC-9 cell apoptosis (Figure 3d–f).
Inhibition of tumor cell proliferation by RNAi-mediated knockdown for CXCR4 in vitro and in vivo
CCK-8 and plate colony formation assays were used to assess the effect of CXCR4 knockdown on the proliferation of OSCC cell lines in vitro. CXCR4 knockdown significantly decreased the proliferation and colony formation of Tca8113 and SCC-9 cells (P < 0.05) (Figure 4a–e). To investigate whether shRNA targeting CXCR4 caused an inhibitory effect on preestablished tumor growth in nude mice, 36 nude mice were observed and measured in a 35-day follow-up period. On day 42, a rapid increase in the Tca8113 tumor volumes in the Mock (2,210 ± 458 mm3) and NC-LV (1,965 ± 389 mm3) groups compared with that in the CXCR4-LV1 (288 ± 85 mm3) group (P < 0.01) was observed, and the same tendency was found in the SCC-9 tumors. The volume of tumors in the Mock (2,489 ± 587 mm3) and NC-LV (2,367 ± 678 mm3) groups was significantly increased compared with the CXCR4-LV1 (323 ± 78 mm3) group (P < 0.01). However, there were no differences in tumor size and growth tendency between the Mock and the NC-LV groups either in Tca8113 or in SCC-9 tumors (Figure 5a–c). To determine whether the vectors were transduced into tumor cells, GFP expression of tumor tissues was assessed by counting 10 random fields of frozen sections under an inverted fluorescence microscope ×100 (Figure 5d). This vector independently expresses a green fluorescent protein, facilitating a direct monitoring of the delivery efficiency of the gene-silencing construct. The delivery efficiency in the NC-LV and CXCR4-LV1 groups was ~70%, and no GFP expression was observed in the Mock group, as expected. Given that the CXCR4 mRNA levels in SCC-4 cells were comparable with that of normal tissues (Figure 1), and to further confirm whether the observed effects are caused by downregulation of CXCR4 by shRNA, SCC-4 cells were also used as a control for simultaneous in vitro and in vivo experiments. CXCR4 downregulation did not inhibit SCC-4 cell proliferation in vitro and in vivo (Supplementary Figure S4). Additionally, to confirm whether the RNAi sequences have off-target effects, after transduction with CXCR4-LV1 (MOI = 50) and NC-LV (MOI = 50) in SCC-4 cells for 72 hours, the mRNA levels of multiple genes identified by microarray analysis including CCND1, MAPK3, ERBB2, MMP9, MMP13, CDKN1A, TP53, CDKN1B, BRCA1, and erythropoietin receptor in SCC-4 cells were detected. The results demonstrate that SCC-4 cells transduced by CXCR4-LV1 and NC-LV do not affect the mRNA levels of these genes (Supplementary Figure S5).
Figure 4.
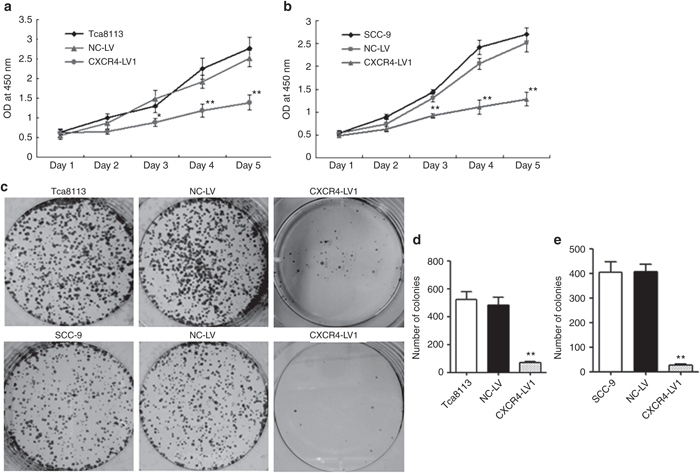
CXCR4 knockdown decreased cell proliferation of Tca8113 and SCC-9 OSCC cells in vitro. (a,b) After CXCR4-LV1 (MOI = 50) and NC-LV (MOI = 50) transduction for 72 hours in Tca8113 and SCC-9 cells, the number of viable cells was assessed by CCK-8 assay. (c) Representative photographs of plate colony formation of Tca8113 and SCC-9 cells. (d,e) Quantitative analysis of plate colony formation of Tca8113 and SCC-9 cells. Error bars indicate mean ± SD; n = 3 experiments; *P < 0.05; **P < 0.01. OSCC, oral squamous cell carcinoma.
Figure 5.
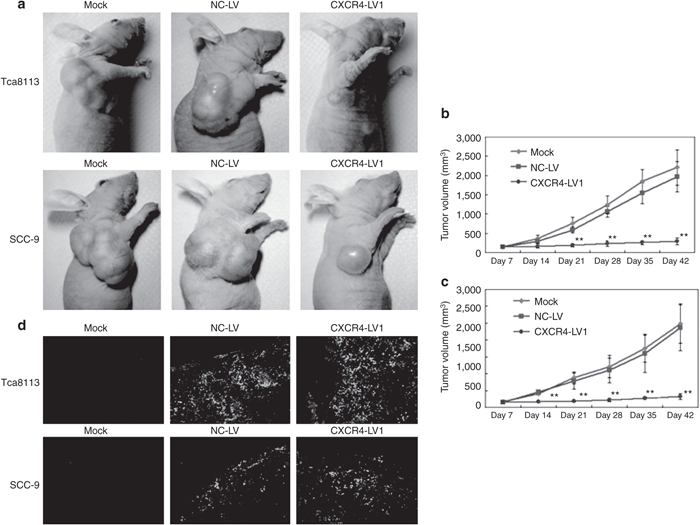
Reduced Tca8113 and SCC-9 tumor volumes in nude mice injected with CXCR4-LV1. (a) After six-time injection of saline solution (0.1 ml), NC-LV (0.1 ml), and CXCR4-LV1 (0.1 ml), on day 42, photographs of representative mice and tumors are shown. (b,c) Tumor size was measured using a vernier caliper and tumor volume was determined as described in Materials and Methods section for 42 days. Each data point is the mean value of five to six primary tumors. (d) GFP expression of tumor tissues transduced with CXCR4-LV1 and NC-LV. GFP expression of tumor tissues in different groups were observed, counted, and photographed under an inverted fluorescence microscope ×100. Scale bar = 100 µm. Error bars indicate mean ± SD; **P < 0.01.
CXCR4 knockdown promotes tumor cells apoptosis and Ki-67 protein expression in vivo
To further confirm whether CXCR4 downregulation can inhibit OSCC cell proliferation in tumor tissues, the protein expression of CXCR4 and proliferation-related gene Ki-67 were analyzed by immunohistochemistry, and the results show that CXCR4 and Ki-67 protein expression levels were weaker in the CXCR4-LV1 group than in the Mock and NC-LV groups (Figure 6a,b). The TUNEL assay was then used to evaluate apoptosis of tumor cells. The number of apoptotic cells was significantly increased in tumors treated with CXCR4-LV1 compared with those in the Mock and NC-LV groups (Figure 6c–e). These results demonstrate that CXCR4-specific shRNA can significantly inhibit tumor growth and promote tumor cell apoptosis in vivo.
Figure 6.
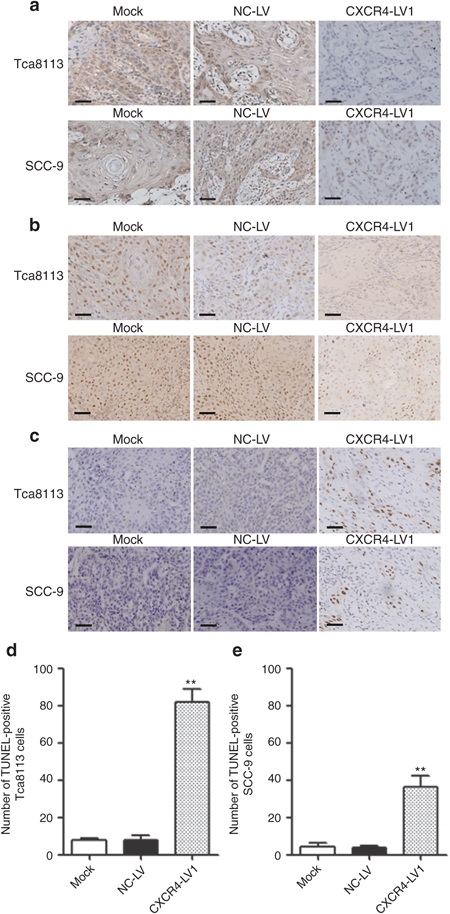
CXCR4 and Ki-67 IHC as well as TUNEL assay. (a) CXCR4-positive staining was located in the tumor nuclei and cytoplasm, and more positive cells could be seen in Mock and NC-LV cells than that in the CXCR4-LV1 Tca8113 and SCC-9 cells. (b) Ki-67-positive staining was located in the tumor nuclei, and more positive cells could be seen in the Mock and NC-LV cells than those in the CXCR4-LV1 Tca8113 and SCC-9 cells. (c) In the TUNEL assay, positive cells were located in the tumor nuclei. (d) The number of apoptotic cells significantly increased in CXCR4-LV1 Tca8113 cells compared with those in the Mock and NC-LV cells. (e) The number of apoptotic cells significantly increased in the CXCR4-LV1 SCC-9 cells compared with those in the Mock and NC-LV cells. Scale bar = 50 µm. Error bars indicate mean ± SD; **P < 0.01 IHC, immunohistochemistry.
Gene expression profile analysis
To investigate further the effects of CXCR4 knockdown in OSCC cells on downstream targets, gene expression profiling on Tca8113 and SCC-9 cells expressing either NC-LV or CXCR4-LV1 grown in culture was performed. Unsupervised clustering analysis of 1,503 genes identified two groups of genes (trees 1 and 2) that significantly changed expression levels (by >twofold) after CXCR4 downregulation in Tca8113 and SCC-9 tumor cells. Tree 1 included 467 downregulated genes and tree 2 contained 289 upregulated genes on CXCR4 downregulation (Figure 7a). Functional profiling of these genes revealed that the greatest proportion of the genes was associated with cell cycle, followed by JAK/STAT signaling, extracellular matrix (ECM) protein signaling, apoptosis, and p53 signaling. Quantitative RT-PCR was performed to confirm CXCR4-dependent expression of over 30 genes identified in the microarray analysis. Except for PLAGL1 and HOXB2 genes, the expression levels of the other 30 genes were consistent with that from the microarray analysis (Figure 7b,c).
Figure 7.
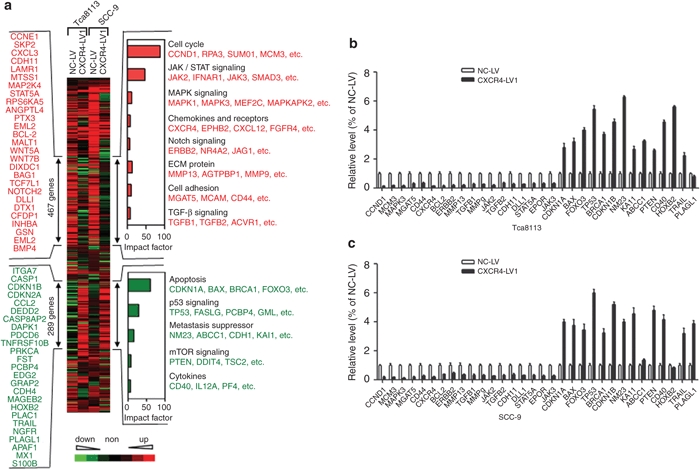
Unsupervised clustering (Cluster 3.0 software) of differentially expressed genes between control siRNA and CXCR4 siRNA1 cells from both Tca8113 and SCC-9 cells. A total of 467 CXCR4-activated genes and 289 CXCR4-repressed genes are marked by double-headed arrows. Representative CXCR4-activated (red) and repressed genes (green) are listed either vertically or under each molecular pathway. Impact factor strength of CXCR4-activated (red bars) and repressed (green bars) genes is shown. (b) Quantitative analysis of transcript levels of over 30 genes identified by microarray analysis, relative to β-actin, determined by qRT-PCR and compared with Tca8113 cells transduced with NC-LV. (c) Quantitative analysis of transcript levels of over 30 genes identified by microarray analysis, relative to β-actin, determined by qRT-PCR and compared with SCC-9 cells transduced with NC-LV. Error bars indicate mean ± SD; n = 3 experiments. qRT-PCR, quantitative real-time polymerase chain reaction.
Discussion
In this study, RNAi-mediated CXCR4 silencing could significantly downregulate the protein levels of CXCR4 in OSCC cell lines and their xenograft model of nude mice. More importantly, CXCR4 knockdown significantly promoted cell cycle arrest and apoptosis of tumor cells and inhibited tumor growth, resulting in the alteration of expression of many cell cycle-, apoptosis-, and signaling-related genes. This phenomenon suggests that CXCR4 is a promising gene-targeting therapy for oral cancer. Quantitative RT-PCR results show that CXCR4 is overexpressed in several different OSCC tumors and cell lines, suggesting the potential roles of this protein in OSCC progression. To confirm the role of CXCR4 in OSCC, CXCR4 recombinant lentiviral vector was used and a specific inhibitor of CXCR4, TN14003, was recruited to inhibit CXCR4 expression in Tca8113 and SCC-9 cell lines. Cell cycle plays an important role in cell proliferation, differentiation, and tumor progression, regulated by intracellular and extracellular signal transduction pathway.18 In this study, cells of phase G1 significantly increased, and cells of S and G2/M phases significantly decreased in cells treated with TN14003 and CXCR4-LV1, suggesting that CXCR4 downregulation could induce phase G1 arrest and apoptosis as well as inhibit tumor cell proliferation. To confirm the effects of CXCR4 expression on cell proliferation, cancer cell proliferation and colony formation potentials were investigated. The finding that CXCR4 knockdown led to a significant decrease in the Tca8113 and SCC-9 cell proliferation and colony formation was consistent with many previous reports, which showed that high levels of CXCR4 expression promoted OSCC cell proliferation.19,20 After constructing the xenograft tumor models using Tca8113 and SCC-9 cells in nude mice, the tumor volume in the CXCR4-LV1 treatment group was significantly lower than those in the Mock and NC-LV groups on day 42. To detect the effects of CXCR4 expression on OSCC tumor growth in vivo, the apoptosis of Tca8113 and SCC-9 cells with TUNEL assay and the Ki-67 protein expression with immunohistochemistry were examined. The results show that the number of apoptotic cells significantly increased and Ki-67 protein expression significantly decreased in tumors treated with CXCR4-LV1 compared with those in the Mock and NC-LV groups. These data indicate that RNAi-mediated CXCR4 gene silencing could inhibit tumor growth and induce apoptosis of OSCC cells both in vitro and in vivo.
Finally, to identify the potential molecular mechanism of effects of CXCR4 downregulation on tumor growth inhibition, microarray analysis technology was used to detect the gene expression profile of Tca8113 and SCC-9 cells treated with CXCR4 recombinant lentiviral vector and control lentiviral vector. A total of 1,503 genes with altered expression levels (by >twofold) were identified. A total of 467 genes were downregulated in Tca8113 and SCC-9 cells treated with CXCR4-LV1 compared with Tca8113 and SCC-9 cells treated with NC-LV1, which were mainly involved in cell cycle regulation, JAK/STAT signaling, and ECM protein regulation. In addition, 289 genes were upregulated in Tca8113 and SCC-9 cells treated with CXCR4-LV1 compared with those treated with NC-LV1, which were mainly involved in apoptosis, p53 signaling, and metastasis suppressor. Subsequently, RT-PCR was performed to confirm the CXCR4-dependent expression of over 30 genes identified in the microarray analysis. The expression of many genes is known to have important role in tumor progression. CCND1, also known as cyclin D1, is overexpressed in many human tumors21,22,23,24 that are induced by growth factors and occur at multiple levels including increased transcription, translation, and protein stability. Cyclin D1, a kinase-independent function, plays important role for G1 phase transition, by sequestering cyclin-dependent kinases (Cdks) inhibitors such as CDKN1A (p21) and CDKN1B (p27) for efficient activation of CDK2-containing complexes.25,26 Based on the study of p53 influence on apoptosis and cell cycle, the expression of the key molecule of p53 passage, p21, was further confirmed in an attempt to explore the associated molecular mechanism of CXCR4 on OSCC cells. p21 mediates p53-dependent G1 growth arrest.27 Earlier studies reported that p21 suppresses tumor growth by promoting cell cycle arrest in response to various stimuli. Substantial evidence from biochemical and genetic studies has indicated that p21 acts as a master effector of multiple tumor suppressor pathways for promoting antiproliferative activities that were independent of the classical p53 tumor suppressor pathway.28 p27 is an atypical tumor suppressor that regulates G0 to S phase transitions by binding to and regulating the activity of Cdks.26,29 In G0 and early G1 phases, p27 translation and protein stability are maximal, and p27 binds, as well as inhibits, cyclin E-CDK2. The progressive decrease in p27 during G1 phase allows cyclin E-CDK2 and cyclin A-CDK2 to activate the transcription of genes that are required for the G1–S transition and participate in the initiation of DNA replication.30 Several retrospective multivariate studies in head and neck cancers have shown that reduced p27 is associated with an adverse outcome. Reduced p27 correlated strongly with advanced disease stage and increased tumor size.31,32 Bcl-2 family functions as a “life/death switch” that integrates diverse inter- and intracellular cues to determine whether the stress apoptosis pathway should be activated.33 p53 gene could precisely regulate cell apoptosis via activating the apoptosis promoter of Bcl-2 family, such as bax and bid, or inhibiting the antiapoptosis molecules, such as Bcl-2.34 Additionally, the following genes that have roles in tumor progression were also analyzed: β1,6 N-acetylglucosaminyltransferase V (MGAT5) and CD44,35,36 cell adhesion and promote tumor invasion; transforming growth factors β1 and β2 (TGFB1, TGFB2),37,38 stimulate invasion; matrix metalloproteases 9 and 13 (MMP9, MMP13),9,39,40 degrade ECM and promote metastasis; and delta-like 1 (DLL1) and ERBB2 (HER-2 or NEU),41,42 Notch signaling and tumor growth. Notably, CXCR4 upregulated genes involved in JAK/STAT signaling, such as erythropoietin receptor, as well as Janus kinases 2 and 3 (JAK2, JAK3), signal transducer and activator of transcriptions 5A and 5B (STAT5A, STAT5B). Previous studies have suggested a role for erythropoietin/erythropoietin receptor signaling in the proliferation and survival of cancer cells.43 The overexpression of erythropoietin can recruit JAK2 and activate STAT factors, STAT5A and STAT5B, leading to distinct transcriptional regulation events to promote tumor acquisition of aggressive phenotypes.44,45 Additionally, CXCR4 expression also repressed the metastasis suppressor genes NM23 and KAI1.46 Taken together, in this study, CXCR4 knockdown significantly decreased the tumor growth of OSCC through regulating downstream cell cycle, apoptosis, and multiple signaling-related genes. This study may provide new evidence for CXCR4 as a promising gene therapeutic target for cancer.
Materials and Methods
Cell lines and reagents. SCC-9, SCC-4, and Tca8113 human tongue squamous cell carcinoma cell lines were obtained from ATCC and State Key Laboratory of Oral Diseases at Sichuan University. The primary and secondary antibodies to human CXCR4, Ki-67, and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Cell proliferation and cytotoxicity assay kits were from Dojindo Laboratories (Tokyo, Japan). Annexin V-FITC/PI Apoptosis kit was purchased from KeyGEN BioTECH (Nanjing, China).
Construction of lentiviral vector for knockdown of human CXCR4 expression. Two siRNAs were designed based on the CXCR4 sequence (NM_001008540): siRNA 1: 5′-GGTGGTCTATGTTGGCGTCTG-3′ or siRNA 2: 5′-GGCAGTCCATGTCATCTAC-3′. Oligonucleotides with a sequence predicted to induce efficient RNAi of CXCR4 (containing sense and antisense sequences) were synthesized (siRNA 1 sense: 5′-GATCCCGGGTGGTCTATGTTGGCGTCTGGAA GCTTGCAGACGCCAACATAGACCACCTTTTTT-3′, antisense: 5′-CTAGAAAAAAGGTG GTCTATGTTGGCGTCT GCAAGCTTCCAGACGCC AACATAGACCA CCCGG-3′; siRNA 2 sense: 5′-GATCCCGGGCAGTCCATGTC ATCT ACGAAGCTTGGTAGATGACATGGACTGCCTTTTTT-3′, antisense: 5′-CTAGAAAAAAGGCAGTCCATGTCATCTACCAA GCTTCGTAGATGACATGGACTGCCCGG-3′). These oligonucleotides were annealed in sodium chloride-tris-EDTA buffer at 94 °C for 5 minutes and cooled gradually. The double-stranded products were cloned downstream to the human U6 promoter of the pRNAT/U6 vector. The products were designated as CXCR4-shRNA1 and CXCR4-shRNA2. A control vector (NC-shRNA) was constructed (5′-GAAGCAGCACGACTTCTTC-3′) with no significant homology to any mammalian gene sequence and, therefore, served as a nonsilencing control. Different shRNAs were screened by cotransfection with a human CXCR4 cDNA plasmid into 293 T cells with Lipofectamine LTX and plus (Invitrogen, Carlsbad, CA). The sequence of shRNA was then cloned into plasmid pGCL-GFP, which encodes an HIV-derived lentiviral vector containing multiple cloning sites for insertion of shRNA constructs to be driven by an upstream U6 promoter and a downstream cytomegalovirus promoter-GFP fluorescent protein (marker gene) cassette flanked by loxP sites. The resulting lentiviral vector contained two human CXCR4 shRNAs, CXCR4-LV1 and CXCR4-LV2. A negative control lentiviral vector containing NC-shRNA was constructed by a similar process (NC-LV). The recombinant lentivirus of siRNA targeting CXCR4, as well as the control lentivirus, was prepared and titered to 109 transfection units/ml. Tca8113, SCC-4, and SCC-9 cells were infected by the addition of lentivirus into the cell culture at an MOI of ~50.
Cell culture and treatment. The cells were cultured at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (Gibco, Rockville, MD), 2 mmol/l glutamine, 1 mmol/l sodium pyruvate, 10 mmol/l 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 100 U/ml penicillin G, 100 mg/ml streptomycin, and 50 ng/ml SDF-1 (Sigma, St. Louis, MO). The cell lines were treated with TN14003 (50 µmol/l) for 4, 8, 16, and 24 hours. For transduction assay, the cells were seeded onto 6-well plate with a concentration of 2 × 105 per well. After 24 hours, the cells were transduced in Opti-MEM I medium with lentiviral vectors in accordance with the manufacturer's instructions. Cultured for another 72 hours, fluorescence-activated cell sorting was performed for quantitation of infection efficiency.
Patients. Five patients with OSCC who received surgical treatment in the West China Hospital of Stomatology, Sichuan University, were randomly chosen for this study. All patients had surgery as their first treatment and did not undergo preoperative radiotherapy and chemotherapy.
Quantitative RT-PCR. Total RNA was isolated from frozen tumor tissue or 1 × 106 cells with the TRIzol reagent (Invitrogen, Rockville, MD). Total RNA was subsequently reverse transcribed to cDNA using the SuperScript First-Strand Synthesis System (Invitrogen, Rockville, MD). Quantitative RT-PCR was performed using the SYBR premix Ex Taq II Kit (Takara, Kyoto, Japan). Specific primers are listed in Supplementary Table S1. The comparative threshold cycle method was used to calculate amplification fold. β-actin gene was used as a reference control gene to normalize the expression value of target genes. Triple replicates were performed for each gene, and the average expression value was computed for subsequent analysis. The relative expression level of the genes was calculated using the 2−ΔΔCt method.47
Western blot. The cells were lysed in lysis buffer (phosphate-buffered saline containing 1% Triton X-100, protease inhibitor cocktail, and 1 mmol/l phenylmethylsulfonyl fluoride) at 4 °C for 30 minutes. Equal quantities of protein were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After the transfer to immobilon-P transfer membrane, successive incubations with anti-CXCR4 and β-actin antibody, as well as horseradish peroxidase-conjugated secondary antibody, were conducted. Immunoreactive proteins were then detected using the enhanced chemoluminescence (ECL) system. Bands were scanned using a densitometer (GS-700; Bio-Rad, Hercules, CA), and quantification was performed using Quantity One 4.6.3 software.
Cell cycle assay. Cells (1 × 106) were obtained from the appropriate samples, and nuclei were stained with propidium iodide. Flow cytometry was performed using an FACSCalibur (BD Biosciences, San Jose, CA), and the fraction of cells in each phase of cell cycle was determined using the cell cycle analysis platform in the FlowJo software (Tree Star, Ashland, OR).
Apoptosis assay. Cells (1 × 106) were collected, washed, and resuspended in phosphate-buffered saline, Annexin V-fluorescein isothiocyanate (FITC; 5 µl/ml). Then, propidium iodide (KeyGEN, Nanjing, China) was added and the cells were incubated for 20 minutes at 4 °C. Cells were analyzed using FACScan flow cytometer (Becton Dickinson, San Jose, CA) with FlowJo software. The TUNEL assay was performed using In Situ Cell Death Detection Kit, POD (Roche Applied Science, Basel, Switzerland), according to the manufacturer's protocol. The sections were observed in a bright-field microscope, and the number of apoptotic cells in the tumor tissue in each section was counted in 10 different microscopic fields.
Cell proliferation assay. Cell proliferation assays were performed using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Cells were plated in 96-well plates at 1 × 104 cells per well and incubated for 5 days. Ten microliters of Cell Counting Assay Kit-8 solution was added to each well daily. The cells were incubated for another 2 hours. The value of optical density was measured at a wavelength of 450 nm using a microplate reader (VARI OSKAN FLASH 3001; Thermo, Marietta, OH). The amount of the formazan dye generated by the activities of dehydrogenases in cells is directly proportional to the number of living cells. Additionally, the plate colony formation assay was performed to evaluate the colony formation ability of tumor cells. The tumor cells were cultured at 1,000 cells/5 ml with DMEM medium and 10% fetal bovine serum in a 6-well plate. After 10 days in culture, the cells were fixed with methanol for 10 minutes and stained with 1% crystal violet solution for 20 minutes to visualize colonies for counting.
Tumor xenografts model and CXCR4 recombinant lentiviral vector treatment. The construction of the tumor xenografts model was conducted at the Laboratory Animal Center of Sichuan University (Chengdu, China). The animal experiment was authorized by West China Hospital Ethics Committees. Fifty-four 4-week-old nude mice were bred in an aseptic condition. The animals were kept at a constant humidity and temperature (25–28 °C) with a 12-hour light/dark cycle. After 1 week of acclimatization, 0.1 ml of Tca8113, SCC-4, and SCC-9 cell suspension at a concentration of 2 × 106 cells/ml was subcutaneously injected into the right lateral of axilla region. Tumor xenografts were staged for 7 days and reached ~150 mm3 in size. Then, same volumes (0.1 ml) of saline solution (Mock), control vector (NC-LV), and CXCR4-specific lentiviral vector (CXCR4-LV1) were injected into multiple sites intratumorally in three different groups, respectively, every 7 days. The size of tumors was measured with vernier calipers, and the volume of tumor was determined using the simplified formula of a rotational ellipsoid (L × W2 × 0.5). On day 42, mice were euthanized and the tumors were removed for further study.
Immunohistochemistry. Five-micrometer sections of formalin-fixed, paraffin-embedded tissue were cut onto silanized glass slides, deparaffinized in xylene, rehydrated in graded ethanol concentrations (100%, 95%, 70%, and 50%), and finally submerged in phosphate-buffered saline. The sections were blocked for endogenous peroxidase with 3% hydrogen peroxide solution for 15 minutes and placed in an autoclave with 0.01 mol/l sodium citrate solution at 121 °C for 3 minutes for antigen retrieval. Sections were incubated with the CXCR4 and Ki-67 primary antibody overnight at 4 °C and then incubated with biotin-labeled secondary antibody at room temperature for 1 hour. Negative controls were performed by replacing the primary antibody with phosphate-buffered saline. Diaminobenzidine tetrahydrochloride was used as a chromogen. All sections were then counterstained with hematoxylin and then observed in a bright-field microscope.
Microarray analysis. The 44K Human Genome Oligo Microarray, including 44,000 60-mer oligonucleotide probes representing 41,000 unique genes and transcripts, was purchased from Agilent Technologies (Palo Alto, CA). In brief, total RNA from Tca8113 and SCC-9 cells transduced with NC-LV and CXCR4-LV1, respectively, were harvested using TRIzol and the RNeasy kit (Qiagen, Germany). The amplification and labeling of 500 ng of total RNA were performed according to the Agilent Quick Amp labeling kit's protocol using Cy3. Hybridization was performed for 16 hours at 50 °C. After hybridization and washing, the processed slides were scanned with the Agilent DNA microarray scanner (part number G2505B; Agilent, Santa Clara, CA). The resulting text files extracted from Agilent Feature Extraction Software (version 10.5.1.1) were imported into the Agilent GeneSpring GX software (version 10.0) for further analysis. To identify the differentially expressed genes, a fold-change screening between the two groups obtained from the experiment was performed. The threshold used to screen up- or downregulate genes is fold change >2. Hierarchical clustering and tree diagram were generated by Cluster 3.0 software.
Statistical analysis. Data are expressed as mean ± SD (standard deviation), when normally distributed. The statistical significance of differences was determined by Student's two-tailed t-test in two groups and one way ANOVA in multiple groups. Differences at probability of less than 0.05 were considered statistically significant. All data were analyzed with SPSS 15.0 software.
SUPPLEMENTARY MATERIAL Figure S1. GFP expression of Tca8113 and SCC-9 cells transduced by recombinant retroviral vector. Figure S2. The CXCR4 protein expression in Tca8113 and SCC-9 cells treated with TN14003. Figure S3. Cell cycle analysis of Tca8113 and SCC-9 cells which were treated with CXCR4 antagonist TN14003. Figure S4. Inhibition of CXCR4 protein expression in SCC-4 cells treated with CXCR4-LV1. Figure S5. Quantitative real-time polymerase chain reaction analysis. Table S1. Genes and Primer Sequences for the quantitative Real-Time RT-PCR.
Acknowledgments
We thank Xiaoyu Li, Yurong Liu, and Min Zhou from State Key Laboratory of Oral Diseases, Sichuan University for their technical assistance. This project was supported by National Natural Science Foundation of China (Grant 30973345) and Doctoral Fund of Ministry of Education of China (Grant 20090181110082). The authors declare no conflict of interest.
Supplementary Material
GFP expression of Tca8113 and SCC-9 cells transduced by recombinant retroviral vector.
The CXCR4 protein expression in Tca8113 and SCC-9 cells treated with TN14003.
Cell cycle analysis of Tca8113 and SCC-9 cells which were treated with CXCR4 antagonist TN14003.
Inhibition of CXCR4 protein expression in SCC-4 cells treated with CXCR4-LV1.
Quantitative real-time polymerase chain reaction analysis.
Genes and Primer Sequences for the quantitative Real-Time RT-PCR.
REFERENCES
- Yu T, Wang XY, Gong RG, Li A, Yang S, Cao YT.et al. (2009The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster J Exp Clin Cancer Res 2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi S., and, Grandis JR. Gene therapy for the treatment of oral squamous cell carcinoma. J Dent Res. 2003;82:11–16. doi: 10.1177/154405910308200104. [DOI] [PubMed] [Google Scholar]
- Teicher BA., and, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- Barbieri F, Bajetto A, Stumm R, Pattarozzi A, Porcile C, Zona G.et al. (2008Overexpression of stromal cell-derived factor 1 and its receptor CXCR4 induces autocrine/paracrine cell proliferation in human pituitary adenomas Clin Cancer Res 145022–5032. [DOI] [PubMed] [Google Scholar]
- Gassmann P, Haier J, Schlüter K, Domikowsky B, Wendel C, Wiesner U.et al. (2009CXCR4 regulates the early extravasation of metastatic tumor cells in vivo Neoplasia 11651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung B, Jhurani S, Ahn KS, Mastuo Y, Yi T, Guha S.et al. (2008Zerumbone down-regulates chemokine receptor CXCR4 expression leading to inhibition of CXCL12-induced invasion of breast and pancreatic tumor cells Cancer Res 688938–8944. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME.et al. (2001Involvement of chemokine receptors in breast cancer metastasis Nature 41050–56. [DOI] [PubMed] [Google Scholar]
- Yu T, Wu Y, Helman JI, Wen Y, Wang C., and, Li L. CXCR4 promotes oral squamous cell carcinoma migration and invasion through inducing expression of MMP-9 and MMP-13 via the ERK signaling pathway. Mol Cancer Res. 2011;9:161–172. doi: 10.1158/1541-7786.MCR-10-0386. [DOI] [PubMed] [Google Scholar]
- Tamamura H, Hori A, Kanzaki N, Hiramatsu K, Mizumoto M, Nakashima H.et al. (2003T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer FEBS Lett 55079–83. [DOI] [PubMed] [Google Scholar]
- Bertolini F, Dell'Agnola C, Mancuso P, Rabascio C, Burlini A, Monestiroli S.et al. (2002CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin's lymphoma Cancer Res 623106–3112. [PubMed] [Google Scholar]
- Uchida D, Onoue T, Kuribayashi N, Tomizuka Y, Tamatani T, Nagai H.et al. (2011Blockade of CXCR4 in oral squamous cell carcinoma inhibits lymph node metastases Eur J Cancer 47452–459. [DOI] [PubMed] [Google Scholar]
- Burger JA., and, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N.et al. (2004Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5 Nat Immunol 5943–952. [DOI] [PubMed] [Google Scholar]
- Burger JA., and, Stewart DJ. CXCR4 chemokine receptor antagonists: perspectives in SCLC. Expert Opin Investig Drugs. 2009;18:481–490. doi: 10.1517/13543780902804249. [DOI] [PubMed] [Google Scholar]
- Novina CD., and, Sharp PA. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- Masiero M, Nardo G, Indraccolo S., and, Favaro E. RNA interference: implications for cancer treatment. Mol Aspects Med. 2007;28:143–166. doi: 10.1016/j.mam.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Peter M., and, Herskowitz I. Joining the complex: cyclin-dependent kinase inhibitory proteins and the cell cycle. Cell. 1994;79:181–184. doi: 10.1016/0092-8674(94)90186-4. [DOI] [PubMed] [Google Scholar]
- Hong JS, Pai HK, Hong KO, Kim MA, Kim JH, Lee JI.et al. (2009CXCR-4 knockdown by small interfering RNA inhibits cell proliferation and invasion of oral squamous cell carcinoma cells J Oral Pathol Med 38214–219. [DOI] [PubMed] [Google Scholar]
- Meng X, Wuyi L, Yuhong X., and, Xinming C. Expression of CXCR4 in oral squamous cell carcinoma: correlations with clinicopathology and pivotal role of proliferation. J Oral Pathol Med. 2010;39:63–68. doi: 10.1111/j.1600-0714.2009.00801.x. [DOI] [PubMed] [Google Scholar]
- Jares P, Colomer D., and, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7:750–762. doi: 10.1038/nrc2230. [DOI] [PubMed] [Google Scholar]
- Jin M, Inoue S, Umemura T, Moriya J, Arakawa M, Nagashima K.et al. (2001Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II Lung Cancer 34207–218. [DOI] [PubMed] [Google Scholar]
- Barnes DM., and, Gillett CE. Cyclin D1 in breast cancer. Breast Cancer Res Treat. 1998;52:1–15. doi: 10.1023/a:1006103831990. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Müller H, Strauss M, Gusterson B., and, Bartek J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995;55:949–956. [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM.et al. (1994p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest Genes Dev 89–22. [DOI] [PubMed] [Google Scholar]
- Sherr CJ., and, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ., and, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Abbas T., and, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst L., and, Reed SI. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol. 1998;227:25–41. doi: 10.1007/978-3-642-71941-7_2. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Targets of cyclin-dependent protein kinases. Curr Opin Cell Biol. 1993;5:187–193. doi: 10.1016/0955-0674(93)90101-u. [DOI] [PubMed] [Google Scholar]
- Pruneri G, Pignataro L, Carboni N, Buffa R, Di Finizio D, Cesana BM.et al. (1999Clinical relevance of expression of the CIP/KIP cell-cycle inhibitors p21 and p27 in laryngeal cancer J Clin Oncol 173150–3159. [DOI] [PubMed] [Google Scholar]
- Fujieda S, Inuzuka M, Tanaka N, Sunaga H, Fan GK, Ito T.et al. (1999Expression of p27 is associated with Bax expression and spontaneous apoptosis in oral and oropharyngeal carcinoma Int J Cancer 84315–320. [DOI] [PubMed] [Google Scholar]
- Adams JM., and, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z., and, Haupt Y. Apoptosis – the p53 network. J Cell Sci. 2003;116 Pt 20:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- Lagana A, Goetz JG, Cheung P, Raz A, Dennis JW., and, Nabi IR. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol Cell Biol. 2006;26:3181–3193. doi: 10.1128/MCB.26.8.3181-3193.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai T, Maruyama Y, Mio K, Nishiyama H, Suga M., and, Sato C. Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. J Biol Chem. 2011;286:1999–2007. doi: 10.1074/jbc.M110.184010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Hu Z, Wen J, Wang K., and, Liu Y. TGF-beta promotes invasion and metastasis of gastric cancer cells by increasing fascin1 expression via ERK and JNK signal pathways. Acta Biochim Biophys Sin (Shanghai) 2009;41:648–656. doi: 10.1093/abbs/gmp053. [DOI] [PubMed] [Google Scholar]
- Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ.et al. (2009Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2 Neuro-oncology 11368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J.et al. (2011KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9 Oncogene 301901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P, Döppler H, Copland JA, Simpson KJ., and, Toker A. FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol Cell Biol. 2009;29:4906–4917. doi: 10.1128/MCB.00077-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayzafoon M, Abdulkadir SA., and, McDonald JM. Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines. J Biol Chem. 2004;279:3662–3670. doi: 10.1074/jbc.M308158200. [DOI] [PubMed] [Google Scholar]
- Hirose H, Ishii H, Mimori K, Ohta D, Ohkuma M, Tsujii H.et al. (2010Notch pathway as candidate therapeutic target in Her2/Neu/ErbB2 receptor-negative breast tumors Oncol Rep 2335–43. [PubMed] [Google Scholar]
- Acs G, Acs P, Beckwith SM, Pitts RL, Clements E, Wong K.et al. (2001Erythropoietin and erythropoietin receptor expression in human cancer Cancer Res 613561–3565. [PubMed] [Google Scholar]
- Lai SY, Childs EE, Xi S, Coppelli FM, Gooding WE, Wells A.et al. (2005Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma Oncogene 244442–4449. [DOI] [PubMed] [Google Scholar]
- Hwa V, Little B, Kofoed EM., and, Rosenfeld RG. Transcriptional regulation of insulin-like growth factor-I by interferon-gamma requires STAT-5b. J Biol Chem. 2004;279:2728–2736. doi: 10.1074/jbc.M310495200. [DOI] [PubMed] [Google Scholar]
- Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- Livak KJ., and, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GFP expression of Tca8113 and SCC-9 cells transduced by recombinant retroviral vector.
The CXCR4 protein expression in Tca8113 and SCC-9 cells treated with TN14003.
Cell cycle analysis of Tca8113 and SCC-9 cells which were treated with CXCR4 antagonist TN14003.
Inhibition of CXCR4 protein expression in SCC-4 cells treated with CXCR4-LV1.
Quantitative real-time polymerase chain reaction analysis.
Genes and Primer Sequences for the quantitative Real-Time RT-PCR.