

Abstract
Galactosialidosis (GS) is a lysosomal storage disease linked to deficiency of the protective protein/cathepsin A (PPCA). Similarly to GS patients, Ppca-null mice develop a systemic disease of the reticuloendothelial system, affecting most visceral organs and the nervous system. Symptoms include severe nephropathy, visceromegaly, infertility, progressive ataxia, and shortened life span. Here, we have conducted a preclinical, dose-finding study on a large cohort of GS mice injected intravenously at 1 month of age with increasing doses of a GMP-grade rAAV2/8 vector, expressing PPCA under the control of a liver-specific promoter. Treated mice, monitored for 16 weeks post-treatment, had normal physical appearance and behavior without discernable side effects. Despite the restricted expression of the transgene in the liver, immunohistochemical and biochemical analyses of other systemic organs, serum, and urine showed a dose-dependent, widespread correction of the disease phenotype, suggestive of a protein-mediated mechanism of cross-correction. A notable finding was that rAAV-treated GS mice showed high expression of PPCA in the reproductive organs, which resulted in reversal of their infertility. Together these results support the use of this rAAV-PPCA vector as a viable and safe method of gene delivery for the treatment of systemic disease in non-neuropathic GS patients.
Introduction
Galactosialidosis (GS) is a genetic disorder of lysosomal function linked to mutations in the gene encoding the serine carboxypeptidase protective protein/cathepsin A (PPCA). It is synthesized as a one-chain inactive precursor (zymogen) and is proteolytically processed in lysosomes into a two chain (32/20 kDa) mature and active enzyme. Beside its catalytic activity, PPCA has a protective function toward two glycosidases, neuraminidase-1 (NEU1) and β-galactosidase (β-GAL), with which it forms a complex.1 Association with PPCA is pivotal for the lysosomal compartmentalization, catalytic activation and stability of the two glycosidases.1,2 In GS patients, a primary defect of PPCA leads to a complete secondary deficiency of NEU1 and a partial deficiency of β-GAL.3 Given the similarities between GS and sialidosis (primary deficiency of NEU1), it is likely that the severe secondary loss of NEU1 activity in GS is the major factor contributing to disease pathogenesis.3 Three distinct clinical GS phenotypes have been described based on the age of onset and severity of the clinical symptoms: early infantile type (severe, neuropathic), late infantile type (attenuated), and juvenile/adult type (neuropathic).3 Early infantile type patients present between birth and 3 months of age with fetal hydrops, neonatal edema, proteinuria, coarse facies, inguinal hernias, telangiectasias, and death before the age of one year. Patients with late infantile GS are typically diagnosed in the first year of life with coarse facies, hepatosplenomegaly, dysostosis multiplex, retinal cherry-red spots, corneal clouding, but no or only mild neurologic involvement without mental retardation. The juvenile/adult patients comprise 70% of all GS patients and are mostly of Japanese origin. This type of GS is characterized by the late onset of the symptoms but patients develop a broad spectrum of clinical manifestations including coarse facies, spinal deformities, major neurologic symptoms with progressive mental retardation, but absence of visceromegaly.3 Autopsy material from a few reported GS cases showed severely vacuolated hepatocytes and Kupffer cells in the liver and epithelial cells in the kidney, lymphocytes, as well as vacuolar inclusions and lamellar inclusions in cultured fibroblasts.3 At present, there are no treatment options other than supportive care for individuals with GS.
To study the pathological effects of PPCA deficiency in vivo and to test various therapeutic modalities, we generated Ppca null (KO) mice, which closely mimics the systemic and neurological manifestations of early onset GS.4,5 In several proof-of-principle studies, we have treated these mice with genetically and retrovirally modified bone marrow cells overexpressing the therapeutic PPCA,5,6 as well as by enzyme replacement therapy with insect cell-produced recombinant enzyme.7 In general, the phenotype was significantly improved by these therapeutic interventions, although fertility was not specifically evaluated. Collectively these studies demonstrated the ability of the PPCA zymogen, either administered intravenously or secreted by overexpressing cells, to “cross-correct” deficient cells in multiple organs of the GS mouse model. This body of work also helped us to evaluate the relative effectiveness of different therapeutic approaches in achieving complete rescue of the systemic phenotype and in delaying the onset of the central nervous system phenotype, setting the stage for the preclinical studies presented here.
Adeno-associated viral rAAV vectors have become a promising vehicle for recombinant gene delivery because they can infect both dividing and nondividing cells, they are not integrated into the host genome, and they were shown to sustain long-term transgene expression in several murine and canine models.8,9 Such vectors have already been tested in mouse models of several lysosomal storage diseases.10,11,12,13,14,15 We have focused on AAV vectors based on serotype 8 for the treatment of hemophilia B (FIX deficiency) as the prevalence of natural immunity to this nonhuman primate isolet was thought to be lower in humans. The vector can be delivered by peripheral vein, thereby exploiting the remarkable tropism of AAV8 for the liver to facilitate efficient gene transfer to hepatocytes.16,17 Vector biodistribution after peripheral vein delivery is comparable to that achieved after delivery of an equivalent dose of vector directly into the liver. Recent biodistribution studies performed with clinical grade FIX vector in nonhuman primates demonstrated that 98% of the vector genome was present in the liver with trace amounts in other organs.18 Our liver-specific enhancer–promoter combination further confines FIX expression to the liver,19 and we predict that PPCA expression is similarly restricted. The potency of the AAV vector was enhanced by modifying the single-stranded AAV2 genome to one where the expression cassette is packaged as complementary dimers within a single virion.20 Our vector encoding human PPCA also has a self-complementary design and the liver-specific promoter; it has been designated scAACV2/8-LP1-hPPCA.
The objective of our study was to perform a preclinical therapeutic trial in a large cohort of GS mice treated with a single injection of a Good Manufacturing Practices (GMP)-grade scAAV2/8-LP1-PPCA to obtain long-term, sustainable, liver-specific PPCA expression. The livers of treated GS mice should function as a factory for the corrective enzyme by secreting large quantities of the PPCA zymogen into the peripheral blood. As shown in previous studies,4,5 the circulating enzyme is internalized by resident cells in affected visceral organs resulting in increased cathepsin A (CA) activity and consequently in the rescue of the endogenous Neu1 activity. Thus, this process may afford sufficient cross-correction and amelioration of the phenotype in organs where the scAAV2/8LP1-PPCA vector is not expressed. We found a good correlation between the administered rAAV dose and the levels of PPCA expression in most organs, including in the male and female reproductive organs, with the two highest doses affording an overall more favorable outcome. Phenotypic improvements included a full restoration of normal tissue morphology and a rescue of male and female fertility.
Results
scAAV2/8LP1-PPCA-treated GS mice are similar in gross appearance to their (wild-type) WT littermates
A cohort of sixty 30-day-old male and female GS mice was injected in the tail vein with increasing doses of scAAV2/8LP1-PPCA (low dose, intermediate dose, and high dose). The doses used were 2.6 × 109 vector genomes (vg)/mouse (low dose), 2.6 × 1010 vg/mouse (intermediate dose), and 2.6 × 1011 vg/mouse (high dose). The treated mice, as well as 16 male and female untreated WT and 16 male and female KO control mice, were monitored daily for 16 weeks post-treatment. None of the treated mice died prematurely. However, the untreated GS mice exhibited symptoms that progressively worsened and included swollen, edemic, and pale paws, rough fur, puffy appearance, sluggish lethargic movement, and infertility.4 In contrast, the scAAV2/8LP1-PPCA-treated mice, regardless of the viral dose, appeared more agile and their paws showed no signs of edema. In fact, their gross appearance was indistinguishable from WT mice. By 16 weeks post-treatment, they were characterized by having smooth fur, normal movement, and significant sexual activity, as reflected by the production of normal size litters.
High levels of PPCA expression in tissues of scAAV2/8LP1-PPCA-treated GS mice
Immunohistochemical analysis of tissue sections 16 weeks post-treatment stained with polyclonal anti-PPCA antibody showed that the levels of PPCA expression in treated GS mice correlated well with the dose of virus administered (Figure 1). As anticipated, the liver showed the highest level of PPCA expression, particularly in the high-dose-treated mice. PPCA expression was also evident, though less intense, in the spleen, and in the kidney glomerular epithelial cells, Bowman's capsule, and proximal convoluted tubules (Figure 1). In the intermediate- and low-dose-treated mice, the staining intensity was overall weaker (Figure 1). In the high-dose-treated mice, strong PPCA expression was also detected in the male and female reproductive organs, testis/epididymis, and uterus/ovary, respectively (Figure 2). In the high-dose males, very strong PPCA expression was detected in the interstitial cells of both the epididymis and the testis, as well as in the Leydig cells in the seminiferous tubules (Figure 2). Similarly, in the female high-dose-treated mice, very strong PPCA expression was detected in both the corpus luteum of ovaries and the endometrial stroma of the uterus (Figure 2). PPCA was not detected in the brain and pancreas of the treated mice, regardless of the dose group (data not shown).
Figure 1.

IHC of PPCA in systemic organs of scAAV2/8LP1-PPCA-treated GS mice. Using a human PPCA-specific antibody, the enzyme was readily detected (brown punctated staining) in various tissue sections of scAAV2/8LP1-PPCA-treated GS mice and the level of expression correlated with the dose-amount (2.6 × 1011 (high), 2.6 × 1010 (intermediate), and 2.6 × 109 (low) vg/mouse) administered to the mice. PPCA, protective protein/cathepsin A.
Figure 2.
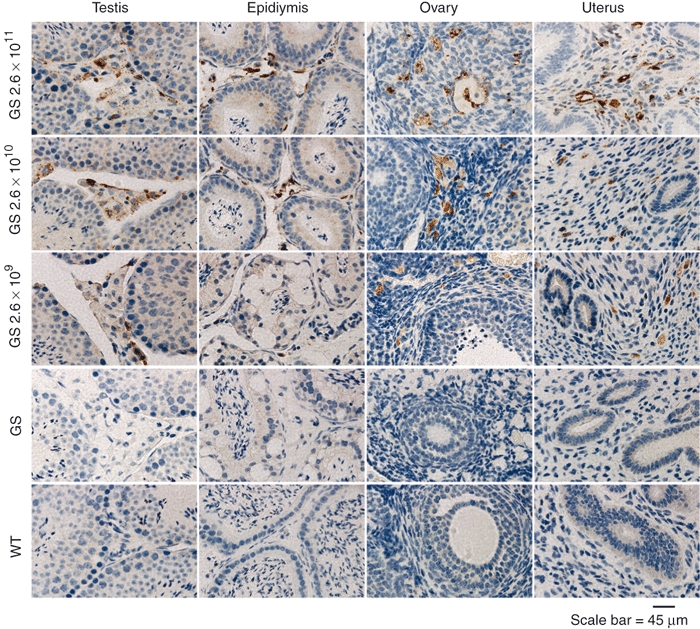
IHC of PPCA in reproductive organ tissues of scAAV2/8LP1-PPCA-treated GS mice. Using a human PPCA-specific antibody, the enzyme (brown punctated staining) was detected in the testes, epididymes, ovaries, and uteri of sections of scAAV2/8LP1-PPCA-treated GS mice and the level of expression correlated with the dose-amount (2.6 × 1011 (high), 2.6 × 1010 (intermediate), and 2.6 × 109 (low) vg/mouse) administered to the mice. PPCA, protective protein/cathepsin A.
CA and Neu1 activities are increased in scAAV2/ 8LP1-PPCA-treated GS mice
In the livers of each of the 3 dose groups, CA activity was substantially higher than that in the livers of untreated GS and WT mice and directly correlated with the administered dose (Figure 3a). Similar CA expression profiles were detected in the spleen and kidneys of the treated mice (Figure 3a). In the gonads as well as in the uterus/epididymis of high-dose-treated mice, the levels of CA activity were similar to those measured in the corresponding WT tissues, while in the intermediate- and low-dose groups the CA levels were overall lower (Figure 3a).
Figure 3.
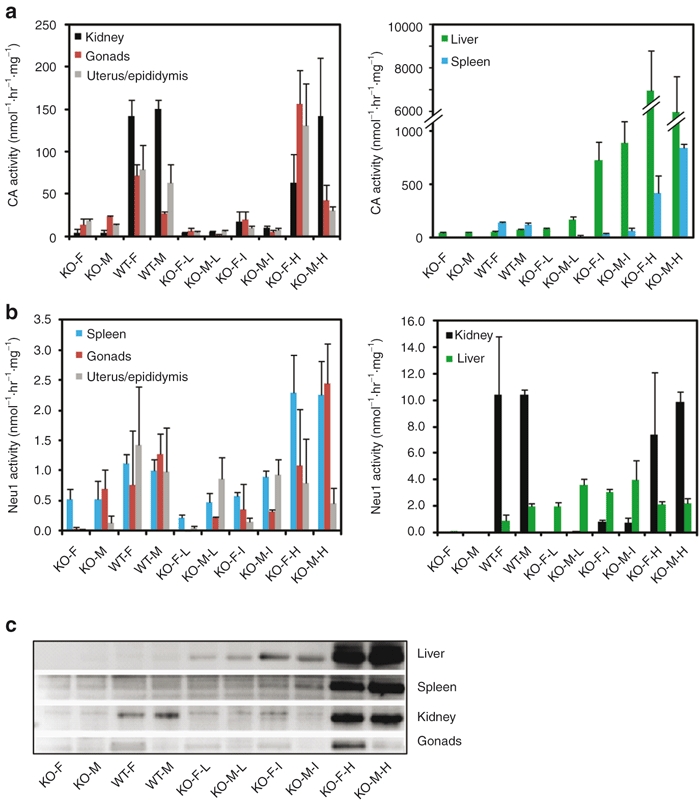
Measurements of CA and Neu1 activities in tissues of scAAV2/8LP1-PPCA-treated GS mice and immunoblot analyses. (a) CA and (b) Neu1 activities assayed in tissue homogenates of scAAV2/8LP1-PPCA-treated GS (KO) mice and untreated GS and WT controls. For each group of 10 mice, three were randomly selected for enzyme activity analysis. Error bars indicate standard deviation. (c) Immunoblot analysis of PPCA in tissues homogenates of scAAV2/8LP1-PPCA-treated KO mice and untreated GS and WT controls. Blots were incubated with a polyclonal antihuman PPCA rabbit antibody that recognizes the 32-kDa large subunit of the corrective enzyme most abundantly in the high-dose-treated GS mice and recognizes the WT mouse enzyme poorly. Untreated female GS mice (KO-F), untreated male GS mice (KO-M), untreated female WT mice (WT-F), untreated male WT mice (WT-M), low-dose-treated female GS mice (KO-F-L), low-dose-treated male GS mice (KO-M-L), intermediate-dose-treated female GS mice (KO-F-I), intermediate-dose-treated male GS mice (KO-M-I), and high-dose-treated female GS mice (KO-F-H). PPCA, protective protein/cathepsin A.
We have shown previously that restoration of PPCA by either uptake or transgenic overexpression of the enzyme rescued the endogenous Neu1 activity, both in cultured PPCA-deficient human cells and in mouse tissues; reversion of the secondary deficiency of Neu1 in multiple tissues was sufficient to attain an overall amelioration of the disease phenotype.4,6,7 Strikingly, in the liver of treated mice in each of the 3 dose groups, the Neu1 activity was restored to levels that equaled or exceeded the activities measured in WT liver (Figure 3b). In the spleen, kidneys, gonads, uterus, and epididymis of the high-dose-treated group, the levels of Neu1 activity were comparable or higher than those in WT samples (Figure 3b). In general, Neu1 activity in the tissues of treated mice was consistent with the CA activity measured in the same tissues (Figure 3a). Moreover, CA levels (Figure 3a) correlated with the amount of the 32-kDa large subunit of PPCA detected on immunoblots of liver, spleen, kidney, and gonads tissue lysates using a human PPCA-specific anti-32 antibody that poorly recognizes the murine protein (Figure 3c).
Correction of the histopathology in scAAV2/ 8LP1-PPCA-treated GS mice
Restoration of PPCA and Neu1 activities in cells of various organs resulted in a dramatic improvement of tissue morphology as demonstrated by hematoxylin-eosin (HE)-staining of tissue sections from treated GS mice. The extensive lysosomal vacuolization detected in all systemic and reproductive organs of the untreated GS mice was clearly resolved upon rAAV-PPCA treatment (Figure 4 and Supplementary Figure S1). In particular and independent of the dose group, swollen lysosomes completely disappeared from cells of the liver, spleen, kidney, lung (data not shown), heart (data not shown), testis, epididymis, ovary, and uterus except in the body of the epididymis of the low- and intermediate-dose groups (Figure 4 and Supplementary Figure S1). Unexpectedly, vacuolization of the cuboidal epithelial cells of the choroid plexus in the GS mouse brain was also fully reverted in the intermediate- and high-dose-treated GS mice and strongly reduced in low-dose-treated mice (Supplementary Figure S2).
Figure 4.
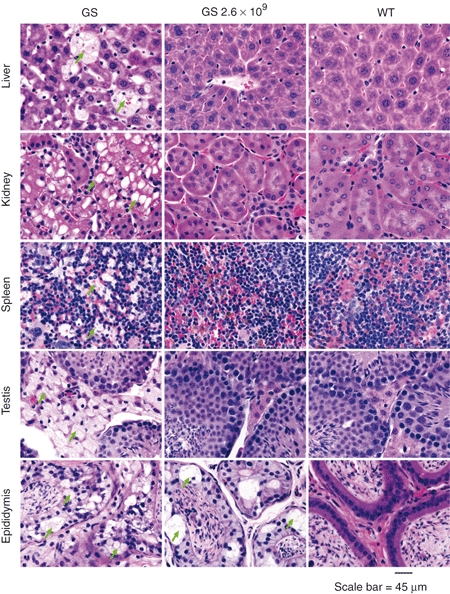
Restoration of tissue morphology in systemic and reproductive organs of low-dose scAAV2/8LP1-PPCA-treated GS mice. HE-staining shows extensive vacuolization (arrows) in the systemic and reproductive organ sections of untreated GS mice, but a complete clearance and restoration to WT morphology in the organs of low-dose (2.6 × 109 vg/mouse) scAAV2/8LP1-PPCA-treated GS mice, except in the epididymis. PPCA, protective protein/cathepsin A.
We confirmed by in situ hybridization that the scAAV2/8LP1 vector exclusively drives expression of the PPCA transgene in the liver of treated GS mice (Figure 5). Therefore, the clearance of lysosomal storage in other organs of these mice could only be accounted for by a mechanism of cross-correction mediated by a pool of PPCA zymogen present in circulation.
Figure 5.
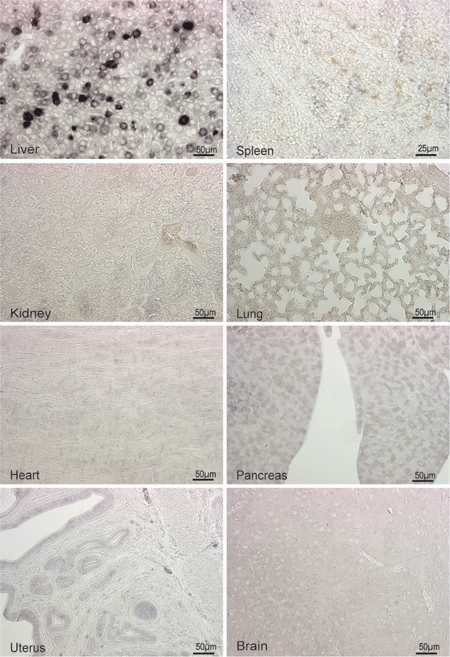
In situ cRNA hybridization of tissue sections of high-dose scAAV2/8LP1-PPCA-treated GS mice shows a high expression level of scAAV-derived PPCA transcript in the liver but a complete absence in other organs. PPCA, protective protein/cathepsin A.
Serum from high-dose scAAV2/8LP1-PPCA-treated GS mice contains exogenous PPCA
To demonstrate that liver-produced PPCA zymogen was indeed present in the peripheral blood and capable of uptake by deficient cells, we performed in vitro uptake assays on GS human fibroblasts using sera collected from mice at 16 weeks after injection. Addition to the culture medium of serum aliquots from mice treated with high-dose rAAV-PPCA increased both CA and Neu1 activities, indicating the presence of sustained levels of the PPCA zymogen in circulation (Figure 6). Similar uptake experiments done with sera from WT, nontreated GS mice, or mice treated with the low dose and intermediate dose of rAAV-PPCA were ineffective in correcting both enzyme activities (data not shown).
Figure 6.
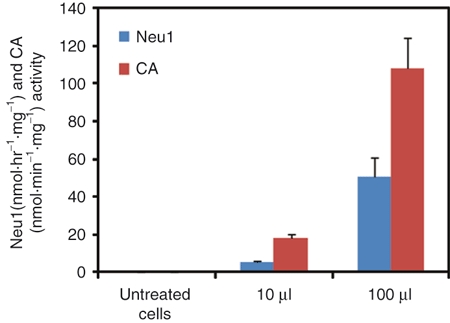
Uptake by GS fibroblasts of PPCA in serum from scAAV2/8LP1-PPCA-treated GS mice. Restoration of CA activity and rescue of endogenous Neu1 activity in GS fibroblasts cultured for 2 days in the presence of 10 or 100 µl serum isolated from the blood of high-dose scAAV2/8LP1-PPCA-treated GS mice are indicative of exogenous PPCA present in the peripheral blood of treated mice. Error bars indicate standard deviation. PPCA, protective protein/cathepsin A.
Reduction in the size of spleen and kidneys in scAAV2/8LP1-PPCA-treated GS mice
A phenotype that is characteristic of PPCA-deficient mice21 is the enlargement of their kidneys and spleens. These aberrant features could be detected by ultrasound monitoring; however, while this noninvasive method was useful to assess disease progression, it was difficult to standardize because of slight variations in the way the animals were positioned during ultrasound recording. We therefore chose to measure the weights of these organs. Spleens and kidneys of GS mice were on average significantly heavier than those of the corresponding WT mice (Figure 7). In contrast, there was no significant difference between the average weights of spleens and left/right kidneys isolated from rAAV-treated mice 16 weeks after injection and those of WT mice, regardless of the viral dose (Figure 7). These data further support the curative effect of rAAV-mediated therapy on the visceral organs of GS mice.
Figure 7.
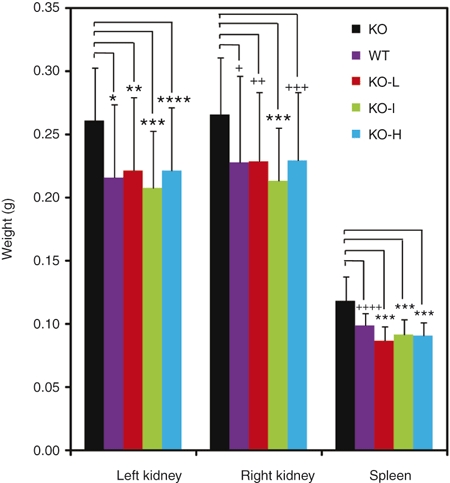
Weight of kidneys and spleen in scAAV2/8LP1-PPCA-treated GS and untreated control mice. Weights of the kidneys and spleen of euthanized mice 16-week post-treatment, females and males combined. Untreated GS mice (KO; n = 16); untreated WT (n = 16); low-dose scAAV2/8LP1-PPCA-treated GS mice (KO-L; n = 20); intermediate-dose-treated GS mice (KO-I; n = 19); high-dose-treated GS mice (KO-H; n = 21). Error bars indicate standard deviation. There are no statistically significant difference between the left/right kidney and spleen weights of WT mice and that of the GS mice in each AAV dose group; however, there are significant difference in organ weights between the untreated GS mice and the GS mice in each AAV dose group (*P = 0.0019; **P = 0.007; ***P ≤ 0.0001; ** * P = 0.003; +P = 0.0308; ++P = 0.0275; +++P = 0.0174; ++++P = 0.0004). PPCA, protective protein/cathepsin A.
Clearance of high molecular weight oligosaccharides (HMWOs) from the urine of scAAV2/8LP1-PPCA-treated GS mice
A diagnostic marker of GS in the affected children and the mouse model is the presence of aberrantly HMWOs in their urine.22 We therefore analyzed the oligosaccharide profile in urine samples of treated GS mice at 16 weeks after rAAV injection, using fluorophore-assisted carbohydrate electrophoresis.22 This FDA-approved diagnostic method for the identification of HMWOs has been employed to evaluate the efficacy of treatment and extent of correction in lysosomal storage disease (LSD) patients and in mice.22 HMWOs (>glucose-4) were readily detected in the urine of untreated GS mice, but were no longer present in urine of male and female treated mice. Only some of the low-dose-treated males still had trace amounts of HMWOs in their urine (Figure 8).
Figure 8.

Clearance of urinary oligosaccharides in scAAV2/8LP1-PPCA-treated GS mice. FACE of oligosaccharides in urine samples collected 16-week post-treatment shows an accumulation of HMWOs (>glucose-4) in two untreated male and female GS mice (KO), and complete clearance of these sugars in the urine of all scAAV2/8LP1-PPCA-treated mice with the exception of low-dose-treated male mice which have a reduction in the amount of HMWOs. KO-I, intermediate-dose-treated GS mice; KO-L, low-dose-treated GS mice; KO-H, high-dose-treated GS mice; PPCA, protective protein/cathepsin A.
Fertility is restored in scAAV2/8LP1-PPCA-treated male and female GS mice
An unexpected outcome of this therapeutic approach in GS mice was the high-level expression of PPCA detected in the reproductive organs and their overall improved histopathology. We therefore wanted to assess whether scAAV2/8LP1-PPCA treatment restored the fertility in these animals. Several pairs of male and female GS mice administered with the high dose of rAAV-PPCA were allowed to mate during the 16-week period post-treatment. All of the rAAV-treated GS mating pairs were capable of generating up to 3 litters in succession, similar to what was observed for WT mating pairs. Genotyping of the pups confirmed that all offspring were Ppca KOs. In addition, the disease phenotype of these GS pups was identical to that of KO pups born from pairs of Ppca-heterozygous mice, which indicated that the scAAV2/8LP1-PPCA had not been integrated into the genomic germline DNA of the parental mice (data not shown). Absence of scAAV2/8LP1-PPCA DNA was confirmed by PCR analysis using scAAV2/8LP1-specific primers on genomic DNA isolated from GS offspring (data not shown).
Discussion
The scope of this study was to conduct a comprehensive preclinical dose-finding trial in the mouse model of GS using a rAAV-based gene therapy approach that could potentially be translated to the clinic for the treatment of the non-neuropathic form of the disease. We report the successful, complete reversal of visceral organ histopathology, clearance of HMWOs from urine, and restoration of fertility in GS mice injected intravenously with a single dose of a liver-specific scAAV2/8LP1-PPCA vector. As expected, expression of PPCA was restricted to the liver of the treated mice. However, the enzyme was readily detected also in the visceral and reproductive organs, resulting from internalization by resident cells of circulating exogenous PPCA zymogen secreted by the hepatocytes. This was most evident in mice treated with an intermediate dose or high dose of scAAV2/8LP1-PPCA vector.
We were pleased by the extent of correction of the reproductive organs and the rescue of fertility in male and female GS mice after rAAV treatment. This outcome is noteworthy given that it results from a gene therapy approach rather than transgenic expression, as reported for two other LSD models.23,24 The cause of infertility in the female GS mice is still poorly understood. More is known about the cause of infertility in GS males.25 Previously, we have shown that Ppca deficiency is responsible for changes in the blood-epididymis barrier, which affect the luminal environment of the epididymis and result in a 70% reduction in sperm count and motility.25 After intermediate- or high-dose scAAV2/8LP1-PPCA treatment, the blood-epididymis barrier appeared normal. Although we did not perform sperm counts or test sperm motility, the number of litters and the normal size of the litters produced by the rAAV-treated GS breeding pairs suggest a full reversal of infertility in both males and females. These results have drastically improved our breeding scheme for generating Ppca null mice in larger numbers and a more timely manner.
Although we were not able to detect immunoreactivity for PPCA and CA activity in total brain tissue or in specific regions of the brain at 16-week post-treatment, rAAV-treated GS mice showed complete correction of the extensive lysosomal vacuolization characteristic of the choroid plexus of untreated GS mice. It is known that levels of lysosomal enzyme activity as low as 2%–10% those of the WT enzyme can be sufficient to prevent, reverse, or ameliorate disease pathology in some LSD patients or mouse models of LSDs.26 Therefore, the most likely explanation for the resolved choroid plexus pathology in treated GS mice is that circulating, exogenous PPCA, albeit clearly below the detection limit, was internalized by epithelial cells of the highly vascular choroid plexi in all four ventricles of the brain. Because GS affects mainly the reticuloendothelial system, this could also explain the remarkable overall improvement of the brain cyto-architecture in treated GS mice. These initial data suggest that it will be valuable to evaluate central nervous system correction in more detail in the future.
Potential variations in the curative efficacy between mice treated at young versus older age, or gender-based differences are critically important when considering clinical applications for the treatment of LSD patients. It has been reported that AAV1-mediated α-galactosidase expression in a mouse model of Fabry disease was greatly influenced by the age and gender of the treated mice.27 The AAV treatment of neonatal Fabry mice resulted in a greater efficacy than similar treatment of adult Fabry mice, regardless of the gender of the recipients. Moreover, adult female mice showed poorer efficacy than male recipients likely as a result of androgen dependence of AAV vector transduction.28 Another factor that may influence the efficacy of AAV treatment is the generation of neutralizing antibodies against the therapeutic enzyme. Problems with androgen dependence and the generation of neutralizing antibodies were circumvented in adult Pompe and Fabry mice using an AAV2-8 serotype vector and a liver-specific regulatory cassette to drive expression of the corrective enzymes,29,30 similar to the vector used in our study. Preliminary data suggest that older GS mice with a full-blown disease may be more difficult to correct than younger mice, although further studies are needed to thoroughly evaluate the effect of age on phenotypic correction.
The potential use of this gene therapy approach for the treatment of GS patients is strongly supported by the results of our ongoing trial for hemophilia B using an analogous vector, scAAV2/8-LP1-scFIXco. We have observed a dose-dependent increase in FIX, which, in the higher dose cohort, was between 5% and 10%, which is well into the therapeutic range. The two participants in our trial who received the highest vector dose developed evidence of immune hepatitis and were treated with oral corticosteroids for 4–6 weeks. Fortunately, FIX production has been sustained after steroid withdrawal. Our experience with the FIX vector in mice, nonhuman primates, and humans suggests that a 400-fold higher dose than the minimum effective dose per mouse will be required in GS patients. Our initial proposed clinical dose is 1 × 1012 vg/kg, just below the dose of 2 × 1012 vg/kg, which triggered the immune response in our hemophilia trial. Accordingly, we will be prepared to initiate corticosteroid treatment in the context of our planned trial for GS. In conclusion, the results of this study set the stage for the potential translation of this therapeutic modality to the clinic for the treatment of non-neuropathic GS patients.
Materials and Methods
Animal studies. All experiments were performed on Ppca−/− (GS or KO) mice and WT littermate controls4 in the FVB background. The handling of and experiments on the mice were approved by the St. Jude Children's Research Hospital Animal Care and Use Committee.
scAAV2/8-LP1-PPCA vector production and titration. The scAAV2/8LP1-PPCA construct contains a liver-specific regulatory element (LP1) that consists of the core domains from the human apolipoprotein hepatic control region and the human α-1–antitrypsin gene promoter17 that drives the hepatocyte-exclusive expression of the 1.44-kb human PPCA cDNA.31 The scAAV vector particles were made using a Good Manufacturing Practice (GMP) process-comparable transient transfection procedure in the Children's GMP, LLC facility on the St. Jude campus, as described for our hemophilia B vector.32 As qPCR under reports vector genome titers for self-complementary vectors (Fagone P and Gray JT., unpublished results), scAAV2/8LP1-PPCA vector genome titers were determined by direct loading and electrophoresis of detergent-treated vector particles on native agarose gels, staining with fluorescent dye, and quantitation of signal relative to known mass standards.
Experimental design, injection of AAV, and tissue and urine collection. In total, 93 mice were used in this study. Three doses of scAAV2/8LP1-PPCA, i.e., 2.6 × 109 (low), 2.6 × 1010 (intermediate), or 2.6 × 1011 (high) vg, were injected into the tail veins of three groups of GS mice (10 males and 10 females for the low dose, 10 males and 10 females for the intermediate dose, and 11 males and 10 females for the high dose group, respectively) at age of 30 days (±5). A total of 16 male and female WT mice, and 16 male and female KO mice were not subjected to rAAV injection. The mice were monitored for 16 weeks after injection, after which they were euthanized and dissected. Serum was collected from all treated mice and stored at −70 °C. Urine was collected prior to treatment, and again just before euthanasia. Nine different tissues (liver, kidney, spleen, lung, heart, brain, pancreas, testis, epididymis, uterus, and ovary) were collected at time of euthanasia, and the weight and size of the spleen and kidneys were measured. Tissues were either fixed in 10% neutralized formalin and paraffin-embedded for histology and immunohistochemical analyses or frozen in liquid nitrogen for biochemical analyses.
CA and Neu1 activity assays and western blot analysis. Frozen liver, spleen, kidney, brain, testis, epididymis, uterus, and ovary of three mice of each of the virus dose groups as well as of untreated WT and KO controls were homogenized on ice in 3 volumes of water (wt/vol) using the Omni-Prep Multi-Sample Homogenizer and soft-tissue disposable grinders (Omni, Kennesaw, GA). Neu1 catalytic activity was measured against synthetic 4-methylumbelliferyl-α-D-N-acetylneuraminic acid (Sigma, St. Louis, MO),33 and CA activity was measured against de di-peptide Z-Phe-Ala.34 Tissue homogenate samples (10–30 µg) were resolved on 4%–12% gradient polyacrylamide gels under denaturing and reducing conditions, transferred onto Hybond-PVDF membranes (Amersham, Piscataway, NJ), and incubated with affinity-purified anti-PPCA antibody6 and peroxidase-conjugated goat–anti-rabbit secondary antibody (Jackson Immuno Research, West Grove, PA). The enzyme was visualized using Super Signal West Femto Chemiluminescence reagent (Pierce, Thermo Scientific, Rockford, IL) and the Chemi-Doc imaging system (Bio-Rad, Hercules, CA).
Immunohistochemical analysis and in situ hybridization of mouse tissue sections. Six-µm-thick paraffin-embedded tissue sections were subjected to deparaffinization and antigen retrieval using standard histology methods. After blocking with 0.1% bovine serum albumin and 0.5% Tween-20, the sections were incubated overnight at room temperature with anti-PPCA antibody.6 The sections were washed and incubated with biotinylated secondary goat anti-rabbit antibody (Jackson ImmunoResearch Laboratory, West Grove, PA) for 1 hour. Endogenous peroxidase was removed by incubating the sections with 0.1% hydrogen peroxidase for 30 minutes. Antibody detection was performed using the ABC Kit and diaminobenzidine substrate (Vector Laboratories, Burlingame, CA), and sections were counterstained with hematoxylin according to standard method.
For histopathological examination, paraffin-embedded tissues from all 96 mice were cut into 6-µm-thick slices and stained with a standard HE method.
In situ hybridization of tissue sections with a digoxigenin-labelled PPCA cRNA probe was performed according to the method described previously.35
FACE analysis. Urine samples (10 µl) were tested for the presence of undegraded high molecular weight oligosaccharides using the FACE urinary carbohydrate analysis kit according to the manufacturer's protocol (Glyko, Novato, CA).
In vitro uptake assay by GS fibroblasts of exogenous PPCA present in the serum of scAAV2/8LP1-PPCA-treated mice. GS fibroblasts from an early infantile patient were seeded to near-confluency in 6-well plates as previously described.4 Ten- and 100-µl aliquots of serum of low-, intermediate-, and high-dose scAAV2/8LP1-PPCA-treated mice were added to the culture medium of Ppca-null GS fibroblasts and incubated for 48 hours, after which the cells were harvested and assayed for Neu1 and CA activity.
Statistical analysis. The weights of the dissected left and right kidneys and spleen were measured after euthanasia. Two-way ANOVA model was used to analyze the data. The model included group effect and gender effect. Tukey–Kramer method was used to adjust for multiple pairwise comparisons between different groups. The same analysis was applied to the left kidney, right kidney, and spleen data separately. All analyses were performed using SAS software (SAS Institute, Cary, NC), Windows version 9.2.
SUPPLEMENTARY MATERIAL Figure S1. Restoration of tissue morphology in systemic and reproductive organs of intermediate- and high-dose scAAV2/8LP1-PPCA-treated GS mice. Figure S2. Clearance of vacuolization in the brain of scAAV2/8LP1-PPCA-treated GS mice.
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK52025, GM60905), the Assisi Foundation of Memphis, and the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children's Research Hospital. A. d'A. holds an endowed chair in Genetics and Gene Therapy from the Jewelry Charity Fund. The authors wish to thank Pat Streich for her assistance in preparing the manuscript.
Supplementary Material
Restoration of tissue morphology in systemic and reproductive organs of intermediate- and high-dose scAAV2/8LP1-PPCA-treated GS mice.
Clearance of vacuolization in the brain of scAAV2/8LP1-PPCA-treated GS mice.
REFERENCES
- Bonten EJ, Arts WF, Beck M, Covanis A, Donati MA, Parini R.et al. (2000Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis Hum Mol Genet 92715–2725. [DOI] [PubMed] [Google Scholar]
- van der Spoel A, Bonten E., and, d'Azzo A. Transport of human lysosomal neuraminidase to mature lysosomes requires protective protein/cathepsin A. EMBO J. 1998;17:1588–1597. doi: 10.1093/emboj/17.6.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Azzo, A, Andria G, Strisciuglio P., and, Galjaard H.2001Galactosialidosis Scriver C, Beaudet A, Sly W., and, Valle D.eds). The Metabolic and Molecular Bases of Inherited Disease8th edn.vol. 3McGraw-Hill: New York; pp. 3811–3826. [Google Scholar]
- Zhou XY, Morreau H, Rottier R, Davis D, Bonten E, Gillemans N.et al. (1995Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells Genes Dev 92623–2634. [DOI] [PubMed] [Google Scholar]
- Leimig T, Mann L, Martin Mdel P, Bonten E, Persons D, Knowles J.et al. (2002Functional amelioration of murine galactosialidosis by genetically modified bone marrow hematopoietic progenitor cells Blood 993169–3178. [DOI] [PubMed] [Google Scholar]
- Hahn CN, del Pilar Martin M, Zhou XY, Mann LW., and, d'Azzo A. Correction of murine galactosialidosis by bone marrow-derived macrophages overexpressing human protective protein/cathepsin A under control of the colony-stimulating factor-1 receptor promoter. Proc Natl Acad Sci USA. 1998;95:14880–14885. doi: 10.1073/pnas.95.25.14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonten EJ, Wang D, Toy JN, Mann L, Mignardot A, Yogalingam G.et al. (2004Targeting macrophages with baculovirus-produced lysosomal enzymes: implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis FASEB J 18971–973. [DOI] [PubMed] [Google Scholar]
- Harding CO, Gillingham MB, Hamman K, Clark H, Goebel-Daghighi E, Bird A.et al. (2006Complete correction of hyperphenylalaninemia following liver-directed, recombinant AAV2/8 vector-mediated gene therapy in murine phenylketonuria Gene Ther 13457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- Sun B, Zhang H, Franco LM, Young SP, Schneider A, Bird A.et al. (2005Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II Mol Ther 1157–65. [DOI] [PubMed] [Google Scholar]
- McEachern KA, Nietupski JB, Chuang WL, Armentano D, Johnson J, Hutto E.et al. (2006AAV8-mediated expression of glucocerebrosidase ameliorates the storage pathology in the visceral organs of a mouse model of Gaucher disease J Gene Med 8719–729. [DOI] [PubMed] [Google Scholar]
- Passini MA, Macauley SL, Huff MR, Taksir TV, Bu J, Wu IH.et al. (2005AAV vector-mediated correction of brain pathology in a mouse model of Niemann-Pick A disease Mol Ther 11754–762. [DOI] [PubMed] [Google Scholar]
- Cardone M, Polito VA, Pepe S, Mann L, D'Azzo A, Auricchio A.et al. (2006Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery Hum Mol Genet 151225–1236. [DOI] [PubMed] [Google Scholar]
- Jung SC, Han IP, Limaye A, Xu R, Gelderman MP, Zerfas P.et al. (2001Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice Proc Natl Acad Sci USA 982676–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman ML, Baek RC, Comer LA, Fernandez JL, Seyfried TN., and, Sena-Esteves M. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol Ther. 2007;15:30–37. doi: 10.1038/sj.mt.6300004. [DOI] [PubMed] [Google Scholar]
- Davidoff AM, Gray JT, Ng CY, Zhang Y, Zhou J, Spence Y.et al. (2005Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models Mol Ther 11875–888. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN.et al. (2006Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver Blood 1072653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Rosales C, McIntosh J, Rastegarlari G, Nathwani D, Raj D.et al. (2011Long-term Safety and Efficacy Following Systemic Administration of a Self-complementary AAV Vector Encoding Human FIX Pseudotyped With Serotype 5 and 8 Capsid Proteins Mol Ther 19876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, McIntosh J, Ng CY, Zhou J, Spence Y.et al. (2007Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates Blood 1091414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman E, Evans V, Graham IR, Athanasopoulos T, McIntosh J, Nathwani AC.et al. (2009Preliminary evaluation of a self-complementary AAV2/8 vector for hepatic gene transfer of human apoE3 to inhibit atherosclerotic lesion development in apoE-deficient mice Atherosclerosis 204121–126. [DOI] [PubMed] [Google Scholar]
- de Geest N, Bonten E, Mann L, de Sousa-Hitzler J, Hahn C., and, d'Azzo A. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum Mol Genet. 2002;11:1455–1464. doi: 10.1093/hmg/11.12.1455. [DOI] [PubMed] [Google Scholar]
- Klock JC., and, Starr CM. The different faces of disease. FACE diagnosis of disease. Adv Exp Med Biol. 1995;376:13–25. doi: 10.1007/978-1-4615-1885-3_2. [DOI] [PubMed] [Google Scholar]
- De Gasperi R, Friedrich VL, Perez GM, Senturk E, Wen PH, Kelley K.et al. (2004Transgenic rescue of Krabbe disease in the twitcher mouse Gene Ther 111188–1194. [DOI] [PubMed] [Google Scholar]
- Donohue C, Marion S., and, Erickson RP. Expression of Npc1 in glial cells corrects sterility in Npc1(-/-) mice. J Appl Genet. 2009;50:385–390. doi: 10.1007/BF03195698. [DOI] [PubMed] [Google Scholar]
- Hermo L, Korah N, Gregory M, Liu LY, Cyr DG, D'Azzo A.et al. (2007Structural alterations of epididymal epithelial cells in cathepsin A-deficient mice affect the blood-epididymal barrier and lead to altered sperm motility J Androl 28784–797. [DOI] [PubMed] [Google Scholar]
- Desnick RJ., and, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3:954–966. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Hirai Y, Ishizaki M, Takahashi H, Hanawa H, Fukunaga Y.et al. (2009Long-term inhibition of glycosphingolipid accumulation in Fabry model mice by a single systemic injection of AAV1 vector in the neonatal period Mol Genet Metab 9691–96. [DOI] [PubMed] [Google Scholar]
- Koeberl DD. Age-related efficacy with an AAV vector in Fabry disease mice. Mol Genet Metab. 2009;96:83–84. doi: 10.1016/j.ymgme.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Sun B, Bird A, Young SP, Kishnani PS, Chen YT., and, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler RJ, Cherry M, Barbon CM, Li C, Bercury SD, Armentano D.et al. (2007Correction of the biochemical and functional deficits in fabry mice following AAV8-mediated hepatic expression of alpha-galactosidase A Mol Ther 15492–500. [DOI] [PubMed] [Google Scholar]
- Galjart NJ, Gillemans N, Harris A, van der Horst GT, Verheijen FW, Galjaard H.et al. (1988Expression of cDNA encoding the human “protective protein” associated with lysosomal beta-galactosidase and neuraminidase: homology to yeast proteases Cell 54755–764. [DOI] [PubMed] [Google Scholar]
- Allay JA, Sleep SE, Long S, Tillman DM, Clark R, Carney G.et al.(2011GMP production of self-complementary serotype 8 AAV vector for a hemophilia B clinical trial Hum Gene Therepub ahead of print). [DOI] [PMC free article] [PubMed]
- Galjaard, H. Genetic Metabolic Disease: Diagnosis and Prenatal Analysis. Elsevier Science Publishers B.V.: Amsterdam; 1980. [Google Scholar]
- Galjart NJ, Morreau H, Willemsen R, Gillemans N, Bonten EJ., and, d'Azzo A. Human lysosomal protective protein has cathepsin A-like activity distinct from its protective function. J Biol Chem. 1991;266:14754–14762. [PubMed] [Google Scholar]
- Schaeren-Wiemers N., and, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Restoration of tissue morphology in systemic and reproductive organs of intermediate- and high-dose scAAV2/8LP1-PPCA-treated GS mice.
Clearance of vacuolization in the brain of scAAV2/8LP1-PPCA-treated GS mice.