

Abstract
Millions of individuals are prescribed platelet inhibitors, such as aspirin and clopidogrel, to reduce their risk of thrombosis-related clinical events. Unfortunately many platelet inhibitors are contraindicated in surgical settings because of their inherent bleeding risk complicating the treatment of patients who require surgery. We describe the development of a potent antiplatelet agent, an RNA aptamer-termed Ch-9.14-T10 that binds von Willebrand factor (VWF) with high affinity and inhibits thrombosis in a murine carotid artery damage model. As expected, when this potent antiplatelet agent is administered, it greatly increases bleeding from animals that are surgically challenged. To improve this antiplatelet agent's safety profile, we describe the generation of antidotes that can rapidly reverse the activity of Ch-9.14-T10 and limit blood loss from surgically challenged animals. Our work represents the first antidote controllable antiplatelet agent, which could conceivably lead to improved medical management of patients requiring antiplatelet medication who also need surgery.
Introduction
Pathological thrombosis, as manifested by cerebrovascular, cardiovascular, and peripheral vascular disease, is the leading cause of morbidity and mortality in the western world and developing countries. Accordingly, considerable research and resources have been dedicated to developing antithrombotic agents that target platelet function and these agents represent some of the most widely prescribed therapeutics in the world. Two basic strategies to antiplatelet drug design have been explored. The first approach targets surface receptors and pathways that will limit but not totally inhibit platelet function as a potentially desirable goal for chronic therapy. Two commonly used antiplatelet drugs, clopidogrel (Plavix) and aspirin, fall into this category. Both drugs inhibit specific pathways of platelet activation and/or aggregation resulting in a relatively modest antithrombotic effect. However, even modest platelet inhibition has been associated with increased surgical bleeding, and despite the potential risk for periprocedural thrombotic events these are often stopped up to 7 days in advance of an operation. The second approach to antiplatelet drug design targets surface receptors that are essential for global platelet function. Glycoprotein (GP) IIb/IIIa inhibitors, such as abciximab and eptifibatide are examples. While effectively reducing thrombotic cardiovascular events in patients with acute coronary syndrome, their overall impairment of platelet-related hemostasis creates both challenges in management and hemorrhagic risk around the time of surgery. A potent, yet controllable antiplatelet agent would facilitate the treatment of patients in surgical settings while minimizing the risk of both thrombotic and hemorrhagic periprocedural events.
Von Willebrand factor (VWF) is a protein expressed in platelets and endothelial cells. It is a critical protein for platelet function, participating in platelet adhesion, activation and aggregation through its well-defined interaction with the GP Ib-IX-V complex on the platelet surface. Damage to the blood vessel wall exposes subendothelial collagen to the circulating blood, with VWF serving as a bridge between collagen and platelets as well as between platelets themselves. This interaction also leads to the transduction of outside-in signals contributing to platelet activation. Large prospective studies have demonstrated that the risk of stroke, myocardial infarction, and death correlate with VWF levels among persons at risk.1,2 While inhibitors of VWF have been shown to limit thrombosis,3,4,5,6 one would expect their effect on hemostasis to lead to significant bleeding in the surgical setting.5,6,7 Thus a rapidly controllable VWF inhibitor would benefit patients requiring platelet inhibition in the perioperative setting.
We studied an RNA aptamer that target's VWF for its ability to inhibit platelet activity and demonstrated that it is able to prevent thrombosis following vascular injury in vivo. We observed that administration of the aptamer led to increased bleeding in surgically challenged animals; a response that was prevented with the administration of a VWF aptamer antidote which rapidly and predictably reversed the antiplatelet effect.
Results
The VWF aptamer inhibits thrombosis in vivo
Previously, we described an aptamer-termed 9.14 (Table 1) that binds and inhibits human VWF binding to human GP Ib-IX-V and prevents human platelet aggregation in vitro.8 To evaluate the ability of this aptamer to inhibit platelet function in animals, we first sought to truncate and modify aptamer 9.14 to facilitate large scale synthesis of the oligonucleotide. We created and tested truncated version of the aptamer based on progressive deletion of nucleotides from the 3′ end of the molecule (Table 1). We determined that aptamer 9.14 could be truncated from 80 nucleotides to 60 nucleotides without significantly reducing its ability to bind VWF (dissociation constant (Kd) of 44 nmol/l compared to 12 nmol/l for the full-length aptamer)8 (Figure 1a). Moreover, this truncated aptamer-termed 9.14-T10 also retained its ability to inhibit platelet function as measured in a platelet function analyzer (PFA-100) assay (Figure 1b). Truncate 9.14-T10 also tolerated a cholesterol modification to the 5′-end of the aptamer (termed Ch-9.14-T10) to increase its circulating half-life in vivo9 without altering its ability to completely inhibit platelet aggregation (Figure 1b).
Table 1. Sequences and binding properties of VWF aptamer truncates.
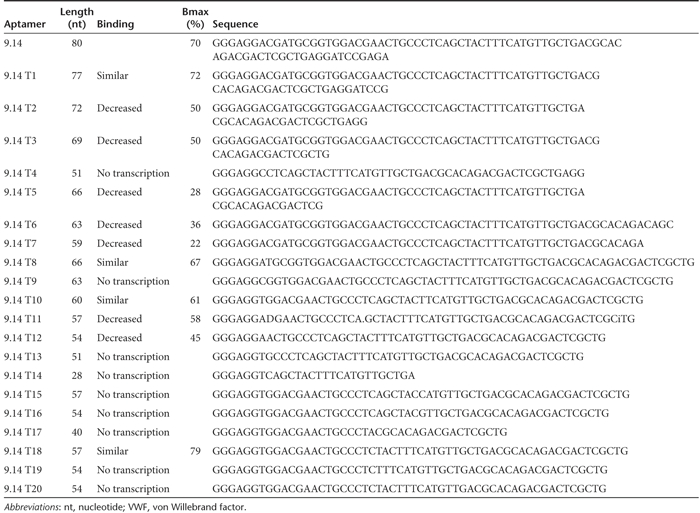
Figure 1.
Analysis of truncated aptamer. (a) von Willebrand factor (VWF) aptamer truncate 9.14-T10 binds with similar affinity compared to full-length aptamer. Binding was performed using a nitrocellulose-filter binding. assay. Filled circles represent full-length aptamer VWF 9.14. Filled squares represent aptamer VWF 9.14-T10. The x-axis represents VWF concentration and the y-axis represents the fraction of RNA bound to the protein. (b) VWF aptamer 9.14 and derivatives inhibit platelet activity in a platelet function analyzer (PFA)-100 assay. The inhibitory activities of VWF aptamers 9.14, 9.14-T10, and Ch-9.14-T10 were measured at a 50 nmol/l concentration in a PFA-100 assay. Error bars represent the mean ± SEM. Each data point was performed in duplicate.
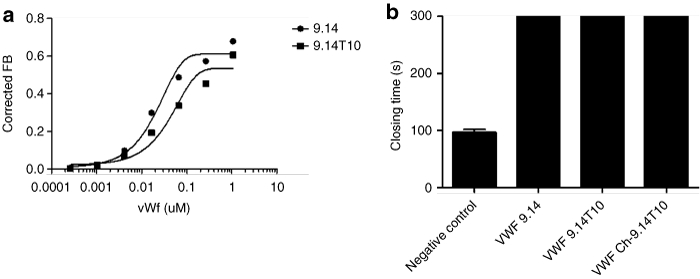
To determine whether the aptamer could inhibit platelet function in vivo, we evaluated its ability to limit thrombosis in a ferric chloride-induced damage model of the common carotid artery in mice. After intubation, cannulation of the left jugular vein and placement of a flow probe around the right common carotid artery, each animal received an intravenous bolus injection of aptamer Ch-VWF 9.14 T10 (3 mg/kg, n = 11) or phosphate-buffered saline (n = 11). Next, we placed Watmann paper (1 mm2) soaked in 10% ferric chloride (370 mol/l) on the carotid artery proximal to the flow probe and left it on for 5 minutes to induce endothelial damage before being removed.10 The average time to thrombosis of the common carotid artery in the negative control group was ~10 minutes. By contrast the carotid arteries of all aptamer Ch-9.14 T10-treated mice remained patent until the end of the experiment (60 minutes) (P < 0.0001 compared to the negative control group) (Figure 2a). Moreover, no significant change in blood flow was observed in aptamer-treated animals from the beginning of the experiment and for the entire 60 minutes of the experiment when the procedure was electively terminated (Figure 2a).
Figure 2.
VWF aptamer is a potent antithrombotic agent in vivo. (a) von Willebrand factor (VWF) Aptamer Ch-9.14-T10-treated animals maintained carotid. artery patency following ferric chloride-induced damage of murine carotid arteries. Transit time or blood flow (ml/minute) through the damaged carotid artery did not decline in animals treated with the aptamer compared to control animals treated with phosphate-buffered saline (PBS) whose carotid blood flow ceased during the course of the experiment (n = 11 per group). Filled squares represent aptamer Ch-9.14-T10-treated animals. Open circles represent PBS-treated animals. No significant change was observed in the blood flow in the aptamer-treated group during the course of the experiment. However, a significant difference in blood flow between the aptamer Ch-9.14-T10-treated mice and mice that did not receive the aptamer was observed (P < 0.0001). x-Axis represents time in minutes over which the experiment took place. y-Axis represents transit time or blood flow in ml/minute. A transit time of 0 indicates that the carotid artery is completely occluded. Error bars represent mean ± SEM. (b) Carotid arteries of aptamer Ch-9,14-T10-treated animals maintained 100% patency while all animals that did not receive the (c) aptamer had thrombi that completely occluded their damaged carotid arteries (P < 0.0001) (n = 11/group). Histopathological sections were stained with hematoxylin and eosin.
Histological analysis of the carotid arteries of all the mice confirmed that vessels in the aptamer Ch-9.14-T10-treated animals were patent and devoid of thrombi (Figure 2b). This observation was in stark contrast to all PBS-treated control mice, which had thrombi that had completely occluded their arteries (P < 0.0001 of the Ch-9.14-T10-treated compared to PBS-treated controls) (Figure 2c).
The VWF aptamer increases bleeding from surgically challenged animals
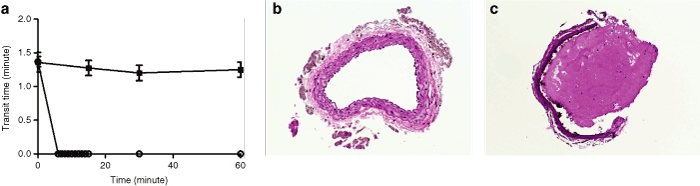
Once we determined that aptamer Ch-9.14-T10 was a potent antithrombotic agent in vivo, we evaluated its potential safety profile. Therefore, we surgically challenged animals that had received the aptamer to determine the degree of bleeding. We employed a murine tail-transection bleeding model in which aptamer Ch-9.14-T10 was administered and 5 minutes later, the animal's tails were transected and the volume of blood lost over the next 15 minutes was measured. As anticipated from the bleeding diathesis described in VWF-knockout mice,11,12 mice treated with varying doses of aptamer Ch-9.14-T10 (10, 5, 3, and 1 mg/kg, n = 5 for each dose) exhibited significantly enhanced bleeding as compared with control animals (Figure 3) (P < 0.0001 comparing aptamer-treated mice at each dose to control animals). Moreover, this effect was dose-dependent and most of the aptamer-treated animals did not stop bleeding for the duration of the experiment, whereas all of the PBS-treated animals formed a platelet plug at the tail transaction site and stopped bleeding within 15 minutes. These results demonstrate that, as expected for a potent platelet inhibitor, aptamer Ch-9.14-T10 can lead to significant blood loss in surgically challenged animals. By contrast as shown in Supplementary Figure S1, no evidence of bleeding was observed in the brains of normal, adult mice that had been treated with aptamer Ch-9.14-T10 (3 mg/kg) for 1 hour and not surgically challenged. These results taken together suggest that the aptamer does not cause spontaneous bleeding, but can dramatically facilitate it at sites of vascular/tissue injury.
Figure 3.
von Willebrand factor (VWF) aptamer Ch-9.14-T10 inhibits platelet plug formation in mice. (n = 5). Mice (18–24 g) were treated with 10, 3, 1, and 0.3 mg/kg of aptamer, then the distal 2 mm of their tails were clipped and blood loss measured. Animals treated with all doses of aptamer had significant blood loss when surgically challenged compared to animals not given the aptamer (P < 0.0001). y-Axis represented blood loss in µl. Error bars represent the mean ± SEM.
Therefore, after determining that aptamer Ch-9.14-T10 can prevent thrombosis but can also enhance bleeding, we sought to develop antidote molecules that could be used to rapidly reverse the activity of aptamer Ch-9.14-T10 if needed.
Antidote oligonucleotides and CDP both neutralize VWF aptamer activity in vitro
We designed complementary antidote oligonucleotides (AOs) based on the sequence of aptamer Ch-9.14-T10 and base-pairing rules (Figure 4a). Initially, we tested the antidotes for their ability to reverse the aptamer's activity in a platelet function assay (PFA-100) (Figure 4b). In comparison to the other AOs, antidote oligonucleotide 1 (AO1) was the most potent reversal agent and it completely reversed the activity of aptamer Ch-9.14-T10 in 2 minutes (P = 0.01 compared to other AO). We then tested AO1 at varying concentrations and found that the lowest concentration of AO1 (fivefold molar excess of AO1 over aptamer Ch-9.14-T10) completely reversed aptamer activity in a PFA-100 assay (Figure 4c). These results demonstrate that VWF aptamer Ch-R9.14-T10 can be completely reversed by AO1 in vitro.
Figure 4.
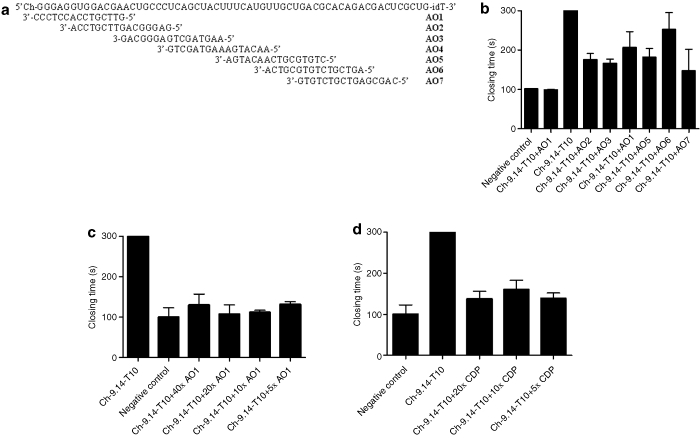
Antidotes for von Willebrand factor (VWF) aptamer Ch-9.14-T10. (a) Antidote oligonucleotide design based upon Watson–Crick base-pairing to aptamer VWF Ch-9.14-T10 (top). (b) Antidote oligonucleotide 1 (AO1) completely reverses the activity of aptamer Ch-9.14-T10 (50 nmol/l) as measured in the platelet function analyzer (PFA)-100 assay. Each antidote oligonucleotide (AO) was added in 50-fold molar excess over aptamer Ch-9.14-T10. y-Axis represent closing time in seconds. Error bars represent mean and SEM. All measurements were done in duplicate. (c) AO1 and (d) the Universal antidote β-cyclodextrin-containing polycation (CDP) reverse that activity of aptamer Ch-9.14-T10 in a PFA-100 assay at tenfold molar excess over aptamer. AO1 and CDP were added in a 5–20-fold molar excess of 50 nmol/l aptamer Ch-9.14-T10. Negative control samples that did not receive an antidote were treated with phosphate-buffered saline. y-Axis represent closing time in seconds. Error bars represent mean and SEM. All measurements were done in duplicate.
β-Cyclodextrin-containing polycation (CDP) is a polymer that can bind to nucleic acid aptamers and inhibit their activity.13 We tested the ability of CDP to reverse the activity of Ch-9.14-T10 in the PFA-100. As shown in Figure 4d, we observed that CDP could reverse the activity of the aptamer when added at a modest excess (more than fivefold molar excess) over the aptamer.
After establishing that AO1 and CDP both inhibit the activity of Ch-9.14-T10 in vitro, we tested the activity of the aptamer and antidotes in vivo.
AOs and universal antidotes can counteract the activity of the VWF aptamer in mice and thereby limit blood loss in surgically challenged animals
We evaluated the ability of the AO1, and the universal antidote, CDP, for their respective abilities to reverse aptamer Ch-R9.14-T10 activity in vivo. We once again used a murine tail transaction-bleeding model. Animals received aptamer Ch-R9.14-T10 (3 mg/kg) by intraperitoneal injection. Five minutes after aptamer administration, we injected PBS, AO1, or CDP into the tail vein at tenfold molar excess over aptamer. The animals were then surgically challenged by tail transection and blood loss was monitored. Administration of AO1 or CDP completely reversed the enhanced bleeding provoked by aptamer Ch-9.14-T10 within 2 minutes (P < 0.0001 between animals given or not given the antidote). Blood loss from the surgically challenged animals given the aptamer but not given the antidote was 135 ± 34 µl. By contrast, blood loss from animals treated with the aptamer and then given AO1 or CDP was reduced to 23 ± 12 and 17 ± 10 µl, respectively. This amount of blood loss is not significantly different from animals surgically challenged but not administered the aptamer where blood loss was 22 ± 16 µl (P > 0.95 between no aptamer and aptamer plus AO1- and aptamer plus CDP-treated animals) (Figure 5a).
Figure 5.
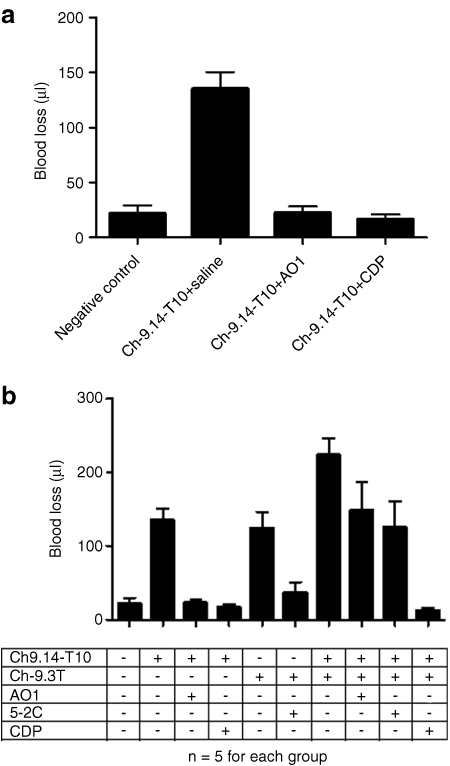
Antidote control of von Willebrand factor (VWF) aptamer activity in vivo. (a) Antidote oligonucleotide 1 (AO1) or β-cyclodextrin-containing polycation (CDP) can reverse the activity of aptamer Ch-9.14-T10 in surgically challenged animals. Mice (n = 5 per group) were treated with aptamer Ch-9.14-T10 (3 mg/kg) by intraperitoneal injection. AO1 or CDP were administered at a tenfold molar excess over aptamer via tail vein injection, then the animals were surgically challenged by clipping their tails and blood loss was measured. Animals that did not receive an antidote experienced significant blood loss compared to those given either or the antidotes (AO1- and CDP compared to no antidote, P < 0.0001). However, no significant difference was observed in blood loss from animals given the aptamer and either of the antidotes (AO1-treated or CDP-treated) and animals not given the aptamer (P = 0.74). (b) CDP reverses the activity of the anti-FIXa aptamer Ch9.3T aptamer and the VWF aptamer Ch-9.14-T10 simultaneously. Mice (n = 5/group) were treated with the VWF aptamer (3 mg/kg) alone, the anti-FIXa aptamer (10 mg/kg) alone or both aptamers. Animals were then given antidote oligonucleotides AO1 (for VWF aptamer) or 5-2C (for FIXa aptamer), the universal antidote CDP or no antidote (PBS vehicle control) and surgically challenged. Administration of antidote oligonucleotides AO1 and 5-2C to animals that received both aptamers decreased blood loss significantly compared to the negative control antidote (P = 0.03) but this was well above blood loss levels from animals that did not receive either aptamer. Blood loss from animals that received both aptamers and the universal antidote (CDP) was similar to that from mice that did not receive either aptamer (untreated, P = 0.27) (y-axis shows blood loss in µl). Error bars represent the mean ± SEM.
Universal antidotes can counteract the activity of multiple antithrombotic aptamers in vivo while AOs only control the activities of individual aptamers
Since both an AO1 and a universal antidote (CDP) were able to rapidly reverse the activity of the VWF aptamer and prevent aptamer-dependent bleeding from surgically challenged animals, we investigated how these control agents may differ. We have previously shown that an antifactor IXa (FIXa) aptamer-termed Ch9.3T inhibited coagulation FIXa activity in mice and caused increased bleeding and that enhanced bleeding could be controlled by administration of an AO-termed 5-2C.9 In this experiment, we wanted to determine whether a universal antidote could simultaneously neutralize both an anticoagulant and an antiplatelet aptamer. This question was of particular interest because in many clinical settings such as percutaneous coronary intervention where both anticoagulants and antiplatelet agents are employed in combination. Therefore, we administered the anti-FIXa aptamer Ch9.3T (10 mg/kg) and the VWF aptamer Ch-R9.14-T10 (3 mg/kg) to animals. Five minutes later, we administered PBS or a tenfold molar excess of an AO (AO1 or 5-2C) or the universal antidote CDP. Then animals were surgically challenged by tail transection as previously described. Animals treated with the anticoagulant and antiplatelet aptamer combination, surgically challenged and not given an antidote (PBS control group) lost large amounts of blood (223 ± 53 µl). As expected this amount of blood loss was significantly greater than animals treated with the antiplatelet aptamer, Ch-R9.14-T10 alone (135 ± 34 µl, P = 0.014) (Figure 5a,b). Administration of AO1 or 5-2C decreased blood loss to a similar level [148 ± 88 and 125 ± 79 µl, respectively (P = 0.72)], but remained elevated compared to animals that did not receive either aptamer [22 ± 17 µl, (P = 0.03)].
Administration of the universal antidote CDP to animals that had received both the anticoagulant and the antiplatelet aptamer, significantly reduced blood loss from surgically challenged animals (12 ± 8 µl), a level markedly reduced compared to animals treated with both aptamers and given one of the matched AOs (P = 0.009 and P = 0.01 compared to AO1 and 5-2C treated animals, respectively). Moreover this amount of blood loss was not significantly different from mice that had not received an aptamer (P = 0.27) (Figure 5b).
Discussion
These experiments demonstrate that the VWF aptamer Ch-9.14 T10 is a potent antiplatelet agent that can block thrombosis in vivo. As anticipated, administration of this potent platelet inhibitor enhances bleeding from animals that are surgically challenged. We also demonstrate that a matched antidote oligonucleotide as well as a universal antidote can rapidly reverse the aptamer's antiplatelet activity and thereby limit surgically induced bleeding.
Aptamer Ch-9.14-T10 was able to maintain vessel patency for >1 hour in a murine FeCl3 vascular injury thrombosis model and histopathologic analysis of the damaged carotid artery showed minimal evidence of platelet accumulation. We have previously determined that Ch-9.14-T10 binds to the A1 region of VWF and inhibits its interaction with platelet GP Ib,8 interfering with platelet adhesion to subendothelial collagen and impeding a key signal transduction pathway which subsequently leads to platelet aggregation through GP IIb/IIIa.14,15 Thus, we believe that this mechanism of VWF inhibition is responsible for the potent antiplatelet effect observed in vivo.
While aptamer Ch-9.14-T10 exhibited potent in vivo antiplatelet activity, several antiplatelet agents have now been described with this ability.7,16 However, this is the first description of an antiplatelet agent that can be neutralized rapidly and effectively by administration of an antidote molecule in vivo. Parenteral platelet inhibitors are used extensively by interventional cardiologists and radiologists and are now being used by neurosurgeons to aid in the surgical management of stroke and stent-assisted coiling of aneurysms.17,18,19,20,21,22 Both interventionalists and surgeons across vascular specialties are enthusiastic to use these types of drugs more extensively but are reluctant to do so because of concern for bleeding complications. Attenuating their pharmacodynamic activity in the event of bleeding requires blood product transfusion or recombinant clotting protease administration. Unfortunately, such procedures yield protracted and unknown quantitative inhibitory effects, are potentially prothrombotic and may be ineffective.
A number of agents targeting VWF, including monoclonal antibodies and aptamers, have been shown to inhibit its activity and in turn, platelet function.3,4,5,6,7,23,24,25 However, antidotes have not been described for any of these drugs, limiting their overall clinical used in surgical settings. The VWF aptamer and its antidotes, including universal antidotes such as CDP represent the first controllable antiplatelet agent and may provide clinicians with much needed options in surgical settings where thrombotic and hemorrhagic risk coexist.
Materials and Methods
Synthesis of aptamer truncates and AOs. The AOs and primers used in the truncation of the aptamer were synthesized and purified by IDT (Coralville, IN). Software predicting RNA secondary structure (Mfold by M. Zuker) was used to aid in the design of truncates. Briefly, primers were designed to make progressively shorter DNA templates for aptamer molecules. T7 RNA polymerase was then used to transcribe RNA aptamers and tested each of these in binding assays.
Binding assay. Dissociation constants (Kd) of each truncate were determined using double-filter nitrocellulose-filter binding assays.12 All binding studies were performed in binding buffer F (20 mmol/l HEPES, pH 7.4, 150 mmol/l NaCl, 2 mmol/l CaCl2, and 0.1% bovine serum albumin) at 37 °C. Human purified VWF (with factor VIII and factor VIII free) was purchased from Hematologic Technologies (Essex Junction, VT) and used in the double-filter nitrocellulose-filter binding assay to determine the Kd of individual clones. Briefly, RNA was dephosphorylated using bacterial alkaline phosphatase (Gibco BRL, Gaithersburg, MD) and end-labeled at the 5′ end with T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and [γ32P] ATP (Amersham Pharmacia Biotech, Piscataway, NJ). Direct binding was performed by incubating 32P-RNA with VWF in physiological buffer + 1 mg/ml bovine serum albumin at 37 ºC for 5 minutes. The fraction of the nucleic acid–protein complex which bound to the nitrocellulose membrane was quantified with a phosphoimager (Molecular Dynamics, Sunnyvale, CA).
Nonspecific binding of the radiolabeled nucleic acid was subtracted out of the binding such that only specific binding remained.26
Platelet function analysis. The PFA-100 (Dade Behring, Deerfield, IL), is a whole blood assay that measures platelet function in terms of clot formation time.27 It is highly sensitive to VWF levels. Briefly, whole blood (840 µl) was mixed with VWF aptamer in PBS with magnesium and calcium (Sigma-Aldrich, St Louis, MO) and incubated for 5 minutes at room temperature. This mixture was then added to a collagen/adenosine diphosphate cartridge and tested for its closing time. The cartridge contains a microscopic aperture cut into a biologically active membrane at the end of a capillary. The whole blood is drawn through the aperture and the membrane is coated with collagen and adenosine diphosphate or collagen and epinephrine which activate platelets. The activated platelet form a plug which occludes the aperture and stops blood flow. The time it takes for this to occur represents the closing time. The maximum closing time that the PFA-100 machine records is 300 seconds. The effect of the AOs on the activity of the aptamer was measured by mixing whole blood with aptamer, incubating for 5 minutes followed by addition of antidote or negative control, and testing the mixture in the PFA-100.
Murine in vivo studies. All in vivo experiments were approved by the Duke University Institutional Animal Care and Use Committee. All experiments were completed with the operator and the neuropathologist blinded to the treatment group.
Carotid injury. Adult C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) (18–24 g) were intubated and the left jugular vein was cannulated. Next, the right common carotid artery was exposed and a transonic flow probe (Transonic Systems Incorporated, Ithaca, NY) was placed around the vessel. The blood flow was measured for 5 minutes to achieve a stable baseline followed by intravenous injection of negative control or aptamer Ch-9.14-T10. Carotid artery thrombosis was activated by 10% ferric chloride-soaked Whatmann paper as previously described.28 The blood flow was then measured for 60 minutes. The time to occlusion was recorded. The animals were then sacrificed. The brain and common carotid arteries were harvested for analysis. The brain and carotid arteries were prepared on slides and stained with hematoxylin and eosin stains (Duke University Department of Pathology, Durham, NC) and reviewed by a neuropathologist (T.J.C.), who was blinded to the treatment groups.
Tail transaction. Adult mice (18–24 g) received Ch9.14T10 or saline by intraperitoneal injection. After 5 minutes, the mice were injected with saline or tenfold molar excess of AO1. After 2 minutes, 2 mm of the distal tail was amputated and blood was collected for 15 minutes in 1 ml of PBS at 37 °C. Blood loss was determined by measuring the absorbance of saline at 550 nm and comparing the result to a standard curve constructed from known volumes of mouse blood as previously described.29
Data analysis. All data is expressed as mean ± SD. All data was inputted into Graphpad Prism (Graphpad Software, La Jolla, CA). All statistical analysis was performed using Graphpad Prism or Graphpad Instat (Graphpad Software).
SUPPLEMENTARY MATERIAL Figure S1. Brain histology of aptamer-treated mice.
Acknowledgments
This work was supported in part by grants from the US National Institutes of Health (HL065222 to B.A.S.), Duke University's CTSA grant (1 UL1 RR024128-01 from NCRR/NIH to B.A.S. and R.C.B.) and the Neurosurgery Research Education Foundation (NREF Research Fellowship to SMN). B.A.S. is the scientific founder of Regado Biosciences which is focused upon developing antithrombotic aptamers.
Supplementary Material
Brain histology of aptamer-treated mice.
REFERENCES
- Conway DS, Pearce LA, Chin BS, Hart RG., and, Lip GY. Prognostic value of plasma von Willebrand factor and soluble P-selectin as indices of endothelial damage and platelet activation in 994 patients with nonvalvular atrial fibrillation. Circulation. 2003;107:3141–3145. doi: 10.1161/01.CIR.0000077912.12202.FC. [DOI] [PubMed] [Google Scholar]
- Kamphuisen PW, Eikenboom JC., and, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21:731–738. doi: 10.1161/01.atv.21.5.731. [DOI] [PubMed] [Google Scholar]
- Cosmi B. ARC-1779, a PEGylated aptamer antagonist of von Willebrand factor for potential use as an anticoagulant or antithrombotic agent. Curr Opin Mol Ther. 2009;11:322–328. [PubMed] [Google Scholar]
- Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R.et al. (2009Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779 J Thromb Haemost 71155–1162. [DOI] [PubMed] [Google Scholar]
- Wu D, Vanhoorelbeke K, Cauwenberghs N, Meiring M, Depraetere H, Kotze HF.et al. (2002Inhibition of the von Willebrand (VWF)-collagen interaction by an antihuman VWF monoclonal antibody results in abolition of in vivo arterial platelet thrombus formation in baboons Blood 993623–3628. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Vreys I, Stassen JM, Yoshimoto R, Vermylen J., and, Hoylaerts MF. Antagonism of vWF inhibits both injury induced arterial and venous thrombosis in the hamster. Thromb Haemost. 1998;79:202–210. [PubMed] [Google Scholar]
- Cadroy Y, Hanson SR, Kelly AB, Marzec UM, Evatt BL, Kunicki TJ.et al. (1994Relative antithrombotic effects of monoclonal antibodies targeting different platelet glycoprotein-adhesive molecule interactions in nonhuman primates Blood 833218–3224. [PubMed] [Google Scholar]
- Oney S, Nimjee SM, Layzer J, Que-Gewirth N, Ginsburg D, Becker RC.et al. (2007Antidote-controlled platelet inhibition targeting von Willebrand factor with aptamers Oligonucleotides 17265–274. [DOI] [PubMed] [Google Scholar]
- Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, Quick G., Jret al. (2004Antidote-mediated control of an anticoagulant aptamer in vivo Nat Biotechnol 221423–1428. [DOI] [PubMed] [Google Scholar]
- Westrick RJ, Winn ME., and, Eitzman DT. Murine models of vascular thrombosis (Eitzman series) Arterioscler Thromb Vasc Biol. 2007;27:2079–2093. doi: 10.1161/ATVBAHA.107.142810. [DOI] [PubMed] [Google Scholar]
- Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Culleré M, Hynes RO.et al. (1998A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis Proc Natl Acad Sci USA 959524–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergolizzi RG, Jin G, Chan D, Pierre L, Bussel J, Ferris B.et al. (2006Correction of a murine model of von Willebrand disease by gene transfer Blood 108862–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oney S, Lam RT, Bompiani KM, Blake CM, Quick G, Heidel JD.et al. (2009Development of universal antidotes to control aptamer activity Nat Med 151224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri ZM. Von Willebrand factor. Curr Opin Hematol. 2003;10:142–149. doi: 10.1097/00062752-200303000-00008. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM, Dent JA., and, Saldívar E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood. 1999;94:172–178. [PubMed] [Google Scholar]
- Jackson SP., and, Schoenwaelder SM. Antiplatelet therapy: in search of the 'magic bullet'. Nat Rev Drug Discov. 2003;2:775–789. doi: 10.1038/nrd1198. [DOI] [PubMed] [Google Scholar]
- Gandhi CD, Johnson DM., and, Patel AB. The endovascular management of intracranial vascular disease including the MERCI device. Curr Treat Options Cardiovasc Med. 2007;9:99–108. doi: 10.1007/s11936-007-0003-8. [DOI] [PubMed] [Google Scholar]
- Nelson PK, Sahlein D, Shapiro M, Becske T, Fitzsimmons BF, Huang P.et al. (2006Recent steps toward a reconstructive endovascular solution for the orphaned, complex-neck aneurysm Neurosurgery 595 Suppl 3S77–92; discussion S3. [DOI] [PubMed] [Google Scholar]
- Bendok BR, Padalino DJ, Levy EI, Qureshi AI, Guterman LR., and, Hopkins LN. Intravenous abciximab for parent vessel thrombus during basilar apex aneurysm coil embolization: case report and literature review. Surg Neurol. 2004;62:304–311. doi: 10.1016/j.surneu.2003.10.045. [DOI] [PubMed] [Google Scholar]
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet glycoprotein IIb/IIIa in unstable angina: receptor suppression using integrilin therapy. N Engl J Med. 1998;339:436–443. doi: 10.1056/NEJM199808133390704. [DOI] [PubMed] [Google Scholar]
- Scarborough RM, Kleiman NS., and, Phillips DR. Platelet glycoprotein IIb/IIIa antagonists. What are the relevant issues concerning their pharmacology and clinical use. Circulation. 1999;100:437–444. doi: 10.1161/01.cir.100.4.437. [DOI] [PubMed] [Google Scholar]
- Topol EJ, Byzova TV., and, Plow EF. Platelet GPIIb-IIIa blockers. Lancet. 1999;353:227–231. doi: 10.1016/S0140-6736(98)11086-3. [DOI] [PubMed] [Google Scholar]
- Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN.et al. (2007First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers Circulation 1162678–2686. [DOI] [PubMed] [Google Scholar]
- Mayr FB, Knöbl P, Jilma B, Siller-Matula JM, Wagner PG, Schaub RG.et al. (2010The aptamer ARC1779 blocks von Willebrand factor-dependent platelet function in patients with thrombotic thrombocytopenic purpura ex vivo Transfusion 501079–1087. [DOI] [PubMed] [Google Scholar]
- Spiel AO, Mayr FB, Ladani N, Wagner PG, Schaub RG, Gilbert JC.et al. (2009The aptamer ARC1779 is a potent and specific inhibitor of von Willebrand Factor mediated ex vivo platelet function in acute myocardial infarction Platelets 20334–340. [DOI] [PubMed] [Google Scholar]
- Wong I., and, Lohman TM. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc Natl Acad Sci USA. 1993;90:5428–5432. doi: 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P. Platelet function analysis. Blood Rev. 2005;19:111–123. doi: 10.1016/j.blre.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Konstantinides S, Schäfer K, Thinnes T., and, Loskutoff DJ. Plasminogen activator inhibitor-1 and its cofactor vitronectin stabilize arterial thrombi after vascular injury in mice. Circulation. 2001;103:576–583. doi: 10.1161/01.cir.103.4.576. [DOI] [PubMed] [Google Scholar]
- Fay WP, Parker AC, Ansari MN, Zheng X., and, Ginsburg D. Vitronectin inhibits the thrombotic response to arterial injury in mice. Blood. 1999;93:1825–1830. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Brain histology of aptamer-treated mice.