

Abstract
Efficient and widespread gene transfer is required for successful treatment of Duchenne muscular dystrophy (DMD). Here, we performed the first clinical trial using a chimeric adeno-associated virus (AAV) capsid variant (designated AAV2.5) derived from a rational design strategy. AAV2.5 was generated from the AAV2 capsid with five mutations from AAV1. The novel chimeric vector combines the improved muscle transduction capacity of AAV1 with reduced antigenic crossreactivity against both parental serotypes, while keeping the AAV2 receptor binding. In a randomized double-blind placebo-controlled phase I clinical study in DMD boys, AAV2.5 vector was injected into the bicep muscle in one arm, with saline control in the contralateral arm. A subset of patients received AAV empty capsid instead of saline in an effort to distinguish an immune response to vector versus minidystrophin transgene. Recombinant AAV genomes were detected in all patients with up to 2.56 vector copies per diploid genome. There was no cellular immune response to AAV2.5 capsid. This trial established that rationally designed AAV2.5 vector was safe and well tolerated, lays the foundation of customizing AAV vectors that best suit the clinical objective (e.g., limb infusion gene delivery) and should usher in the next generation of viral delivery systems for human gene transfer.
Introduction
Duchenne muscular dystrophy (DMD) is the most common severe, life-threatening form of muscular dystrophy in childhood. DMD is associated with progressive muscle degeneration, weakness, and mortality. DMD is X-linked and is genetically inherited, caused by mutations in dystrophin, a large (427 kDa) cytoskeletal protein that is normally expressed in skeletal and cardiac muscle, as well as smooth muscle, brain, and retina in various isoforms.1 The incidence of DMD is 1 in 3,500–5,000 newborn males.2 The gene encoding dystrophin is the largest identified to date,1 and the risk of spontaneous mutation is high (1/10,000 germ cells).
Because DMD is caused by recessive and monogenic mutations in the dystrophin gene, this disease is thought to be amenable to correction or improvement by gene therapy. Gene replacement therapy for DMD is a promising strategy because in theory it could benefit all DMD patients regardless of the nature of their genetic mutations e.g., deletions and point mutations. A number of additional drugs are in development and include alternative gene therapy approaches, cell therapy, antisense oligonucleotides, and small molecule drug therapies.3 However, DMD presents with a multitude of challenges for the development of effective treatments including the largest disease gene that requires miniaturization to be compatible with gene replacement vectors, the very large mass of target tissue that is widely distributed throughout the body with layers of biological barriers, and unknown changes at the cell membrane level that may impact on vector binding and uptake. Furthermore, the immunological components of DMD pathophysiology may also impact on the development and delivery of novel therapeutics and therefore assays of immune system recognition and response must be carefully integrated into clinical trial designs.
Recombinant adeno-associated virus (rAAV) vectors carrying a miniaturized functional dystrophin gene (designated minidystrophin) have the potential to arrest or reverse muscle failure.4,5,6 Comprehensive proof of concept and preclinical testing has evaluated the effectiveness of AAV-minidystrophin gene delivery in animal models of DMD including the mdx mice and dystrophin/utrophin double knockout mice. Minidystrophin expressed after AAV delivery to dystrophin deficient models has been shown to: correctly localize to the sarcolemma, restore the missing dystrophin-associated protein complex to the cell membrane, ameliorate dystrophic pathology in mdx muscle, normalize myofiber morphology, normalize cell membrane integrity, restore missing dystrobrevin complex, partially restore α-syntrophin association with the cell membrane, partially restore nitric oxide synthase activity, reduce muscle fibrosis, reduce myofiber central nucleation, improve whole-body endurance and muscle force transduction, reduce kyphosis and limb deformation, and increase general health and lifespan.5,7,8
Genetic strategies that target the muscular dystrophies will ultimately require widespread delivery to a large volume of skeletal musculature and/or cardiac tissue. Strategies to improve transgene expression to the musculature have included the use of AAV serotypes other than AAV2 and efforts to evolve tissue specificity variants by directed capsid evolution as well as mosaic vector with a mixture of capsid from different serotypes.9,10 It is clear that no single natural AAV serotype will be useful for every clinical application, nor will directed evolution evolve all characteristics desirable for a clinical scenario simultaneously. Any given serotype may contain biological characteristics both beneficial and detrimental to the given clinical application. Instead of attempting to “fit” a known AAV serotype to a disease process or coevolve multiple traits in a single capsid, we chose to use a rational approach to identify capsid regions on alternative AAV serotypes responsible for enhanced skeletal muscle transduction and to combine these modifications into the AAV2 capsid which offers the benefits of a well-defined safety profile coupled with purification ease. The availability of capsid protein sequences from several AAV serotypes, muscle transduction profiles with different serotypes of AAV vector and antigenic epitope information for AAV2, combined with the three-dimensional structure of the AAV2 capsid11 provided us valuable information to rationally design efficient vectors for clinical trials. Through mutagenesis with insertion and substitution, a chimeric AAV2-AAV1 vector, dubbed AAV2.5, was designed to contain desirable biological properties from both parent viruses. Compared to AAV2, AAV2.5 has similar transduction efficiency in several cell lines and binds to heparin sulfate in vitro. However, AAV2.5 induces stronger transduction in skeletal muscles than AAV2 and demonstrated lower crossreactivity to AAV2 neutralizing antibodies (Nab).
The novel AAV2.5 capsid offering improved skeletal muscle gene transfer efficiency and potentially reduced immunogenicity compared with naturally occurring serotypes was next evaluated in a clinical trial for DMD utilizing the minidystrophin transgene cassette. The initial development of AAV2.5-minidystrophin clinical trial capitalized on the fact that AAV2 was the only serotype approved for clinical use, and AAV1 was the only other AAV serotype under serious consideration for clinical studies. To this end, a subset of amino acids (5aa) in AAV type 1 were constructed into type 2 capsid backbone. The engineering and testing of this chimeric capsid in clinical setting has provided a paradigm where as the investigator is no longer obligated to natural viral isolates for gene delivery and can therefore address additional clinical concerns (e.g., capsid immune response) that lie outside of primary objective of measuring therapeutic transgene expression.
Therefore the novel nature of the proposed therapeutic strategy and study population led to the inclusion of careful monitoring of both humoral and cell-mediated immune responses in this phase I clinical trial. We have recently reported elsewhere on unexpected findings related to dystrophin immunity observed in this trial.12 A subset of patients were shown post-hoc to have had pre-existing immunity to dystrophin epitopes believed to be expressed by revertant myofibers, and cellular immune responses to minidystrophin epitopes were also observed. We detail herein the immune response to the novel AAV2.5 capsid as well as other study endpoints, such as: (i) successful vector transgene delivery to all patients at each dose based on PCR analysis of biopsy sectioning, (ii) no difference in immune infiltration when comparing placebo to vector treated arms, (iii) lack of detectable immune response in empty vector only tissues, (iv) no CTL response to chimeric capsid at any dose. All and all, this trial established that rationally designed AAV capsid was safe, well tolerated and lays a foundation of customizing AAV vectors that ideally suit the clinical objective (e.g., heart tropic, liver detargeted, neuroselective, etc.).
Results
Rational design of AAV chimeric vectors
Several of the AAV serotypes characterized to date transduce mouse skeletal muscle with greater efficiency than AAV2, e.g., AAV1, AAV7, AAV8, and AAV9.13,14,15,16,17 In an effort to identify candidate amino acids responsible for enhanced transduction of skeletal muscle the VP1 amino acid sequences of these serotypes with high muscular tropism were compared to AAV2 using an alignment (Figure 1a). The criteria for selection of amino acid candidates are: (i) they must differ from AAV2 and be similar to the high muscle tropism serotypes, (ii) they must be located in a structurally variable region (VR) on the capsid surface,18,19 or, (iii) they must be located in an AAV2 antigenic region that is recognized by an antibody. Shown boxed in Figure 1a are the amino acids which met all criteria with the exception of one amino acid depicted by @ (residue 716, AAV2 Vp1 numbering) due to its close proximity to four other amino acids.
Figure 1.
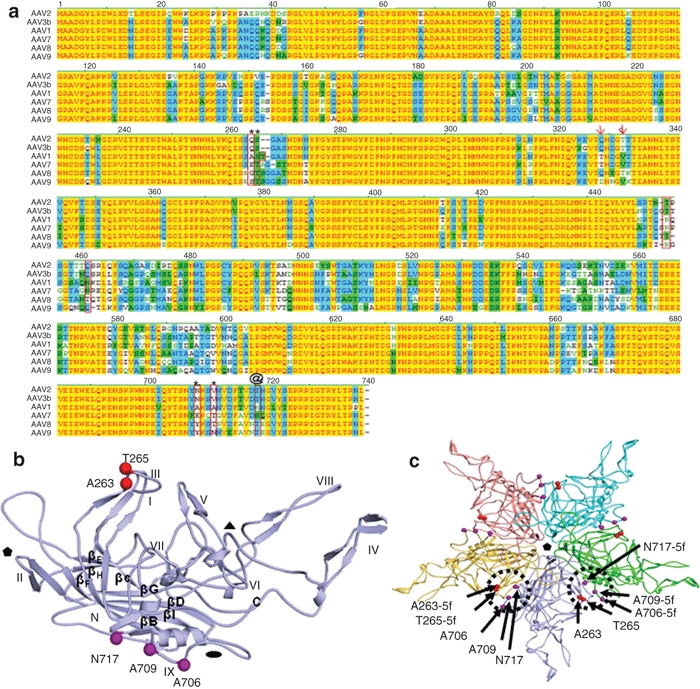
Amino acid candidates responsible for efficient skeletal muscle transduction. (a) Capsid amino acids of low skeletal muscle transducing serotypes (AAV2, AAV3) versus high skeletal muscle transducers (AAV1, AAV6, AAV7, AAV8, AAV9) were aligned using the Vector NTI program (Invitrogen). Alignments were examined for distinct amino acids of AAV2 from the others. See text for additional modeling criteria. Amino acids boxed or marked with arrows were deemed to be of interest. AAV2.5 is composed of the five amino acids indicated by * and @. (b) Location of the five amino acids on a single VP subunit which were modified in the AAV2.5 variant. Notice that the five amino acids are located on opposite positions of one subunit. (c) Location of the same five amino acids (circles and arrows) in the context of an assembled AAV capsid pentamer. Notice that the five amino acids are now in close proximity when two subunits are assembled. The five amino acid changes are located near the twofold axis of symmetry. AAV, adeno-associated virus.
Three variants were initially generated in which AAV2 was modified to resemble AAV1: AAV2.5, AAV2-Q325T/T329V, and AAV2-T450N/Q457N. In the AAV2.5 variant four residues were substituted with AAV1 amino acids (Q263A, N705A, V708A, T716N, AAV2 numbering) and one AAV1 amino acid (T265, AAV1 numbering) was inserted into the AAV2 capsid (amino acids indicated by the asterisks (*) and @ in Figure 1a and the 3D model shown in Figure 1b,c). These mutations are all on the VRs of the virion surface (VR I and VR IX, Figure 1b). Based on the AAV atomic structure, AAV2.5 was designed because residues 263 and 265 intercommunicate with residues 706, 708 and 717 (Figure 1c). The locations of all five amino acid mutations in AAV2.5 are close to the A20 antigenic epitope.18,20,21 The AAV2-Q325T/T329V variant (AAV2 numbering; depicted by arrows on Figure 1a)) partially met the set criteria, the amino acids are conserved in AAV8 but differ in AAV1, AAV7, and AAV9, and are not close to a mapped AAV2 antigenic site. The AAV2-T450N/Q457N (boxed only in Figure 1a) contains amino acids located in VR V.
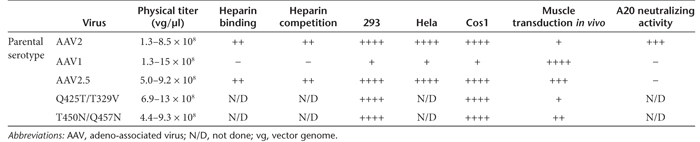
The amino acid mutations in the three AAV2 variants did not influence the ability to generate recombinant AAV vectors compared to unmodified AAV1 and AAV2 capsids containing the same transgene cassette as judged by physical particle titers (Table 1). The AAV2.5 variant resembled AAV2 with respect to its ability to bind heparin in affinity columns used for purification (Table 1). Furthermore, the transduction profiles of these variants in cultured HeLa, Cos, and 293 cells were more like that of AAV2 than AAV1 (Table 1).
Table 1. The characteristics of chimeric variants.
Genetic modification of the AAV2 capsid can improve muscle transduction
To determine whether amino acids responsible for in vivo transduction could be accurately predicted, the variants with a packaged luciferase gene (AAV2.5-luciferase, AAV2-Q325T/T329V-luciferase, and AAV2-T450N/Q457N) were evaluated for their ability to transduce skeletal muscle following injection of equivalent genome-containing particles into the gastrocnemius muscle of BALB/c mice. Luciferase expression was evaluated over time using biophotonic in vivo imaging and compared to the parental AAV1 and AAV2 (for AAV2.5) or AAV2 (for AAV2-Q325T/T329V) viruses (Figure 2a and b, respectively). In skeletal muscle, the AAV2.5 variant consistently produced higher transgene expression than AAV2 at all time points tested albeit not to the identical level as observed with AAV1 (Figure 2a). AAV1 exhibited 5–12.5-fold higher levels of light emission than AAV2; whereas AAV2.5 exhibited 1.8–5.5-fold higher light emission than AAV2. Mice injected with AAV2.5 exhibited high levels of expression up to 8.4 months postinjection. The expression level of the AAV2-Q325T/T329V variant was indistinguishable from that of AAV2 (Table 1 and Figure 2b) and was not tested further. The third variant, AAV2-T450N/Q457N exhibited a 3.5-fold enhancement in transgene expression over AAV2 (Table 1), but only at a later time point postinjection (day 42 postinjection) and was not tested any further.
Figure 2.

Evaluation of skeletal muscle transduction of AAV2 mutants. (a) Skeletal muscle transduction of AAV1, AAV2, and AAV2.5 examined over time (days 3, 7, 21, 28, 42) using in vivo biophotonic imaging. The relative light units per region of interest in each injected mouse (n = 6) are graphed over time. (b) Graphical representation of quantity of emitted light from transduction of AAV2 and AAV2-Q325T/T329V. The relative light units per region of interest in each injected mouse leg (n = 6) are graphed over time. AAV, adeno-associated virus.
Unique antigenic properties of the AAV2.5 capsid
The antigenic profile of the AAV2.5 vector was explored first by examining its recognition by the well characterized A20 anti-AAV2 monoclonal antibody.21 These studies revealed that the ability of the A20 antibody to recognize the AAV2.5 vector is extinguished compared to its strong recognition of AAV2 (Table 1). Thus this data suggest that AAV2.5 may have an immune profile distinct from AAV2.
Minimal crossreactive neutralizing antibodies exist between AAV2 and other serotypes such as AAV122 (C. Li and RJ. Samulski, unpublished results). To determine whether the neutralizing antibody profile of AAV2.5 was more like AAV2 or AAV1, sera from mice treated with AAV1-luciferase, AAV2-luciferase, or AAV2.5-luciferase vectors were analyzed for neutralizing antibody crossreactivity. Shown in Figure 3a are the dilutions of the sera from AAV2, AAV2.5, and AAV1 injected mice needed for 50% inhibition of infectivity of AAV2, AAV1, or AAV2.5 in 293 cells. The sera from AAV2-treated mice neutralized infectivity of AAV2 more efficiently than the infectivity of AAV2.5 by fivefold. A similar finding was observed in the examination of the ability of AAV1 sera to inhibit the infectivity of AAV1 and AAV2.5. The converse was found to be true for sera from AAV2.5 injected animals which was observed to be four- and eightfold more efficient at neutralizing AAV2.5 compared to AAV2 or AAV1, respectively. Therefore AAV2.5 with minimal change of 5 aa has antigenic properties that are distinct from those of the parental viruses suggesting that the engineered AAV2.5 capsid may eliminate the AAV2 or AAV1 antibody recognized epitopes or change virion three-dimension structure and such that it is less likely to be neutralized by the sera of animals pretreated with AAV2 and AAV1.
Figure 3.
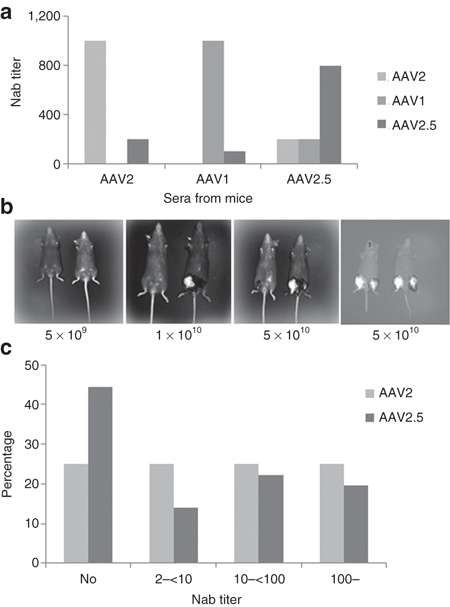
Neutralizing antibody analysis to AAV2.5. (a) Crossreactive Nab between AAV1, AAV2, and AAV2.5. C57 mice were immunized with 1 × 1010 particles of AAV/luc vectors via muscular injection. Thirty days later, sera from three mice was collected for Nab analysis. (b) The effect of AAV2 Nab on AAV2.5-induced transgene expression in vivo. Mice were immunized with AAV2/AAT viruses (left three panels) or not immunized (right panel), 2 months later, AAV2.5/luciferase (mouse right leg) and AAV2/luciferase (left leg) vectors with different dosages were applied in the same mice intramuscularly (5 × 109 particles, 1 × 1010 particles, or 5 × 1010 particles), imaging was taken 6 weeks later post-luciferase vector injection. (c) Neutralizing antibody assay for human sera. The sera from 36 human subjects were detected for Nab against AAV2 and AAV2.5. AAV, adeno-associated virus.
Genetic modification of the AAV2 capsid enables repeat administration of variant vectors
The serological data described above suggested that AAV2.5 was antigenically distinct from the parental viruses. To test whether this phenotype would make this variant refractory to pre-existing antibodies and allow readministration in subjects previously treated with AAV2 vectors, in vivo studies were performed in which mice previously injected with an AAV2-AAT vector were subsequently injected intramuscularly (i.m.) with increasing doses of either AAV2-luciferase or AAV2.5-luciferase vectors. For direct comparison, each mouse received the same dose of the two vectors in separate leg muscles. In vivo biophotonic imaging performed at 6 weeks postinjection of the luciferase expressing virus revealed no detectable expression from either vector at low doses (5 × 109 particles, Figure 3b). However, at higher doses (1 × 1010 particles (Figure 3b) and 5 × 1010 particles per leg (Figure 3b)) the AAV2.5-luciferase administered legs consistently exhibited elevated transgene expression (~10–20-fold) over its AAV2-luciferase injected counterpart (Figure 3b). The enhanced transduction by AAV2.5 is interpreted as being due to both its inherently higher skeletal muscle transduction compared to AAV2, as evidenced by its ~2–5.5-fold higher transgene expression over AAV2 observed in control mice with no previous exposure to AAV2 (Figures 2 and 3b, right panel) as well as to its ability to overcome pre-existing anti-AAV2 neutralizing antibodies. However, although it is clear that the high dose of AAV2.5 vector could escape AAV2 neutralizing antibody partially in vivo, we could not rule out the possibility that AAV2 neutralizing antibodies completely block AAV2.5 transduction after muscular injection in AAV2 pretreated mice when a much lower dose of AAV2.5 was used.
Summary of late preclinical studies
Sera from 36 individuals were screened for neutralizing antibodies and pre-existing neutralizing antibody titers of >1:2 to AAV2 were seen in 75% of individuals while titers of >1:2 to AAV2.5 were seen in only 56% of individuals (Figure 3c). Of those individuals that have AAV2 Nab, a very high Nab antibody titer (≥100) is found in 25% of these individuals; whereas, only 19.5 % of individuals with Nab to AAV2.5 exhibit high Nab titer. Generally the titer against AAV2.5 is 2–20-fold lower than that against AAV2 in the Nab positive population. The data from this human sera neutralizing antibody assay again support the conclusion that AAV2 and AAV2.5 have different immune profiles.
A penultimate preclinical study was conducted to assess possible adverse interactions between the commonly prescribed corticosteroid prednisone and AAV-minidystrophin in C57/BL10 mice. No discernable adverse interaction and influences on transgene expression were observed (data not shown, see Supplementary Materials and Methods—Preclinical AAV2.5-Minidystrophin Studies).
The AAV-minidystrophin expression cassette including the cytomegalovirus (CMV) promoter and the polyadenylation signal site in the vector plasmid was fully sequenced on both strands except the inverted terminal repeats, which were difficult to sequence. A pivotal good-laboratory-practice toxicity and biodistribution study evaluated AAV-minidystrophin delivery in C57/BL10 mice with timepoints up to 36 weeks post vector administration and doses up to 1 × 1012 vector genomes/kg, which was equivalent to 10 times the highest dose proposed in the clinical trial. Overall, the AAV-minidystrophin vector caused no significant toxicity in C57BL mice, with all animals surviving until scheduled sacrifice. Effects on clinical chemistry parameters in the toxicity study were within the normal reference ranges, and there were no differential effects observed by histopathological analyses. There were no related effects on clinical hematology parameters or gross necropsy observations. PCR biodistribution evaluations indicated that the vector remained concentrated in the injected muscle (data not shown, see Supplementary Materials and Methods—Preclinical AAV2.5-Minidystrophin Studies).
Regulatory approval process
The clinical protocol and Appendix M documents were reviewed by the Recombinant DNA Advisory Committee under the auspices of the Office of Biotechnology Activities at the National Institutes of Health (NIH). The Nationwide Children's Hospital institutional review board approved the study protocol. A Pre-IND discussion was held with FDA before initiation of the pivotal toxicology and biodistribution study, and upon conclusion of preclinical studies the IND was submitted to CBER/FDA (BB-IND 12936). The trial is registered with ClinicalTrials.gov (Reference no. NCT00428935).
Clinical study design
The experimental design was a randomized double-blind placebo-controlled study where vector was administered into one bicep and saline control in the contralateral arm. In a subset of patients, we substituted AAV empty capsid for saline in an effort to distinguish an immune response to vector versus minidystrophin transgene. The active phase of the study included a 2-week baseline screening period, a 2-day inpatient period for vector injection and acute toxicity monitoring, and a 2-year outpatient follow-up and toxicity-monitoring period. Ongoing long-term follow-up will continue out to 15 years post vector injection. (Supplementary Figure S1). Blood and urine analyses were conducted in conjunction with outpatient clinics on post administration days 8, 15, 30, 43, 53, 60, 90, and 120 and months 6, 9, 12, 18, and 24.
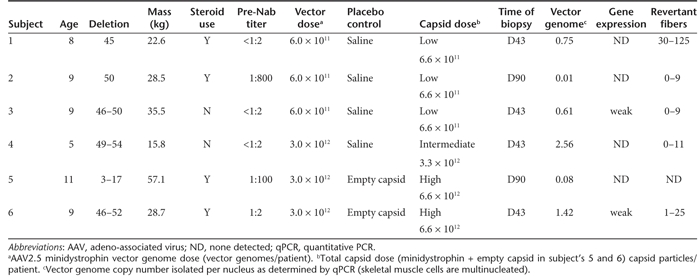
The study enrolled six DMD boys ranging in age from 5 to 11 years and ranging in mass from 16 to 57 kg, each with unique and defined dystrophin mutations. Four boys had been on corticosteroid medication schedules before vector injection (Table 2). All six patients were administered methylprednisolone (2 mg/kg, limited to <1 g total) four hours prior to vector administration, with repeat doses on the following two mornings. A MyoJect hypodermic needle (Oxford Instruments, Hawthorne, NY) was used to deliver 1.2 ml of vector in three equivalent boluses spaced 0.5 cm apart along an injection tract that was placed in a longitudinal trajectory relative to the biceps muscle orientation. Administration of AAV2.5-Minidystrophin to each subject was staggered to allow for at least 6 weeks of follow-up before the next subject receiving the vector. There were two-dose cohorts of three subjects each that received unilateral i.m. delivery of either 6 × 1011 vector genomes or 3 × 1012 vector genomes. Dosing was defined by locally administered dose as the vector was mediating production of a structural protein and not a secreted protein, and vector genomes remained localized to the region of the injection site. Internalized AAV capsid peptide competition for MHC presentation would also be more heavily influenced by the localized dosing. As a result of the inclusion of an empty capsid control vector administered to the last two subjects, there was a full log difference in the capsid dose between subjects 1–3 and 5 and 6. Biopsies were taken at 1.5 month in the majority of patients and out to 3 months in two patients (Table 2).
Table 2. The clinical data in patients with AAV2.5/minidystrophin muscular delivery.
Clinical observations
Physical examinations were unremarkable with no symptoms of fever, lymphadenopathy, organomegaly, and no signs of inflammation at the injection site. During the 2-year-long active phase of the trial monitoring, no serious or mild adverse events have been observed in any subject. A few minor adverse events were observed including sore throats, rashes and nausea, but they were not considered related to the vector administration as they are commonly seen in this age group of subjects (detailed in ref. 12). Hematology and chemistry panels that included an assessment of liver function also indicated that the gene vector was well tolerated in all subjects (Supplementary Figure S2). There were no abnormal elevations in the levels of creatine kinase, alkaline phosphatase levels, or lymphocyte counts in all blood samples tested.
AAV2.5-mediated gene delivery in patients
Biopsies were undertaken in four subjects (subjects 1, 3, 4, and 6) at d43 post administration, and in two subjects (subjects 2 and 5) at d90 post administration. The biopsy procedure utilized ultrasound imaging to guide the retrieval of the injected tissue. A battery of assays were undertaken on the biopsied muscle tissue and included: PCR analysis of vector genomes, immunolabeling of minidystrophin, tissue histochemistry including CD8 T-cell infiltration.
Quantitative PCR of the vector genome in human muscle biopsies
The presence of the vector genomes was assessed in muscle biopsies from the left and right arms 1.5 and 3 months after injection. The transgene DNA was detected in 6 out of 6 patients, in only one arm with no detectable spread to the contralateral arm. The highest vector genome copy number per nucleus (diploid cell genome) detected in each patient is shown in Table 2.
Detection of minidystrophin protein in muscle tissue biopsy
Cryothin-sections of the muscle biopsies were immunofluorescently stained with antibodies recognizing the dystrophin N-terminal region (present in both minidystrophin and endogenous revertant myofibers) or the C-terminal region (absent in minidystrophin but present in revertant myofibers). Limited minidystrophin expression was observed with appropriate localization to the sarcolemma in two of six subjects. In patients 3 and 6, only a few minidystrophin positive myofibers were detected by anti-N-terminus antibody but stained negative by anti-C-terminus antibody. However, in patients 1, 2, 4 and 5, no minidystrophin positive myofibers were detected, suggesting that transgene expression was either very poor or extinguished in those patients (detailed in ref. 12).
Immunological studies
Post administration hematological testing indicated no abnormal changes in white blood cell counts, and there were no changes in markers of liver toxicity (Supplementary Figure S2). However, clinical AAV2 administration has previously been associated immune recognition of AAV capsid peptides,23 and therefore we evaluated cellular and humoral immunological recognition of AAV2.5.
Cell-mediated immunity to AAV2.5
Peripheral blood mononuclear cells (PBMCs) of the subjects were tested for recognition of potential AAV capsid and minidystrophin peptide epitopes by Elispot assay.23 Antigens used to stimulate these responses were pools of overlapping peptides spanning the AAV capsid VP1 protein. Peptides were 20 amino acids in length overlapping by 10 residues and thus would cover all epitopes of VP3 and stimulate CD4+ helper and CD8+ cytotoxic T cells. PBMCs from the subjects were cultured with the AAV capsid peptide pools for 48 hours and then developed for interferon (IFN)-γ spot formation. Subject 1 showed AAV2.5 specific responses that substantially exceeded the threshold of 50 spot-forming cells (SFC) per million PBMC collected at two timepoints, d30 and d90, but not at other timepoints including d43, d53, and d60 post vector injection. In further analyses, the d30 time-point was confirmed as positive after stimulation with peptide pools spanning the capsid protein. Cell lines generated from this sample could not be expanded to support additional characterization. We did not observe significant T cell responses to AAV2.5 capsid antigens seen at any time-point before or after vector administration in any other subjects (Figure 4), however, some samples from subjects 2, 3, and 4 were weakly positive at various time points (sporadically >50 SFC/106 PBMC).
Figure 4.
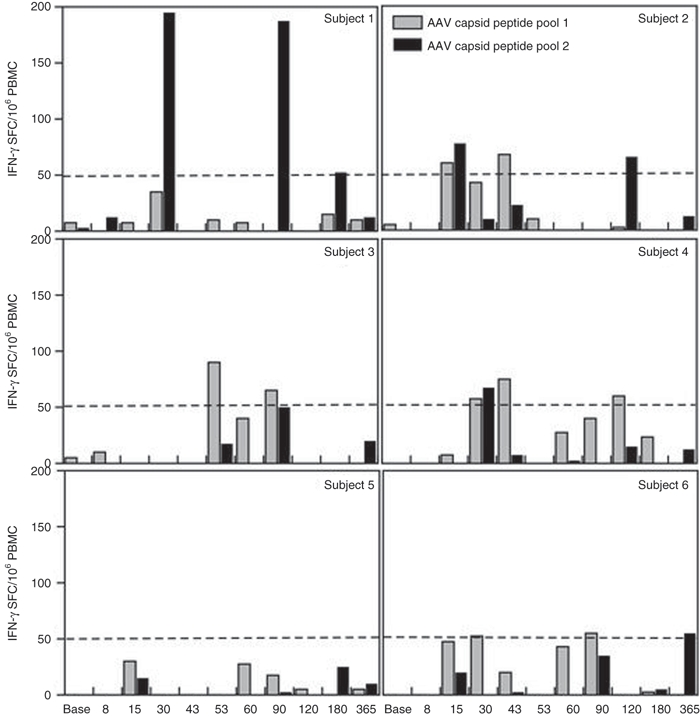
Temporal T cell response to AAV2.5 capsid. Two capsid peptide pools were comprised of peptides spanning the AAV2.5 capsid sequence. Elispot assays measured interferon (IFN)-γ release upon peptide exposure, with the threshold for peripheral blood mononuclear cell (PBMC) recognition of an epitope within the peptide pool being 50 spots/106 PBMCs. Temporal responses are shown for subjects 001–006 in separate panels for the two peptide pools. AAV, adeno-associated virus.
Lack of CD8 infiltration into injected muscle
Cellular infiltrates were quantified in a blinded examination of bicep muscle biopsies. Evaluation of muscle biopsy sections immunofluorescent stained with anti-CD8 antibody showed no statistic difference in the counts of CD8+ cells between AAV-minidystrophin injected and placebo (saline and empty capsids) injected biopsies (52.1 ± 35.2 and 41.9 ± 23.2 cells/mm3 cross-section area, respectively). These cell counts were within the range commonly observed in patients with DMD.
Humoral immunity to AAV2.5
The AAV capsid itself is a completely foreign protein and therefore a humoral immune response to vector administration was anticipated. Pre-existing humoral immunity to AAV was not an exclusion criterion in this study with the rationale that the relevance of in vitro assays of vector neutralization before i.m. administration is unknown due to a lack of previous clinical data. In order to characterize the humoral response to AAV2.5, titers of AAV neutralizing antibodies in serum were also evaluated in an assay involving in vitro transduction in the presence of sera from patients. Subject's sera were screened for neutralizing antibodies to both seroprevalent wild-type AAV2 and the synthetic AAV2.5 capsids. As detailed in Table 2, subjects 2 and 5 were observed to have significant pre-existing neutralizing antibody titers to both AAV2 and 2.5, with baseline titers of 1:800 and 1:100 to both vectors, respectively (titers are the inverse of the highest serum dilution mediating 50% neutralization to either capsids). Baseline titers were <1:2 for both AAV2 and AAV2.5 for subjects 1, 3, and 4, and Subject 6 had a baseline titer of 1:4 for AAV2 and 1:2 for AAV2.5. Classical humoral responses were observed after AAV2.5-minidystrophin administration, with increases in Nabs that peaked from weeks 2 to 6 and ranged from 50× to 1,000× baseline titers (Figure 5). Subject 2 who had high baseline titers was the only subject who did not exhibit a classical post administration strong increase in neutralizing antibody titers, with titers at all time points staying within the range of 1:800 to 1:2,000, approximately a 2.5× increase.
Figure 5.
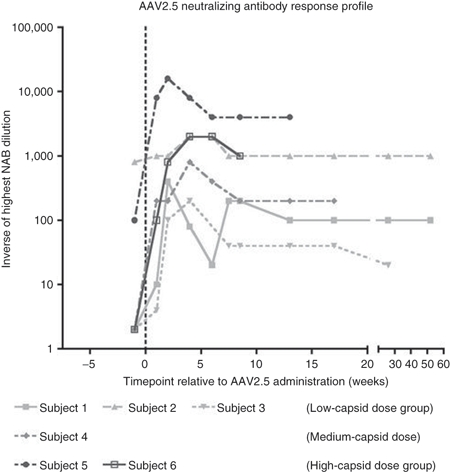
Temporal neutralizing antibody response to AAV2.5. The temporal profile of the highest dilution of serum that neutralizes AAV2.5 transduction in vitro is shown for all six subjects. Subjects 1–3 received the same dose of AAV2.5 capsid, 6.6 × 1011 capsid particles. Subject 4 received 3.3 × 1012 capsid particles. Subjects 5 and 6 received 6.6 × 1012 capsid particles due to administration of empty capsid placebo (see Table 2). AAV, adeno-associated virus.
Discussion
Our study represents the first clinical trial of a synthetic rationally designed AAV vector, AAV2.5, and provides a preliminary insight to the clinical tolerability of this first-in-class gene vector. In this study, we have utilized the atomic structure of the AAV2 virion and homologous models generated for muscle tropism serotypes and VP1 sequence alignments as a guide to rationally design an AAV2 variant (AAV2.5) with altered transduction and antigenic profile from AAV2. We have successfully identified amino acids that differ between AAV2 and muscle tropism serotypes and located in a structurally VR. Most strikingly, the genetically modified AAV2.5 vector demonstrated the enhanced muscle tropism and different antigenic properties from parents (AAV2 and AAV1). Furthermore, this study showed that i.m. administration of a laboratory-derived novel AAV2.5 gene vector encoding minidystrophin at capsid doses up to 6.6 × 1012/kg capsid particles was well tolerated by all subjects, with no vector related adverse events observed.
AAV rational design
The Q263A substitution and T265 insertion in AAV2.5 are located in one of the most VRs in the common VP3 capsid protein (structurally and at the amino acid sequence level) when AAVs are compared. They are located in VR I. A structural comparison of the loop containing these amino acids on the capsid surface of the atomic structures of AAV1 and AAV2 shows a conformational difference in the main chain (Figure 6). This surface loop region is also predicted to be different in the homologous model generated for AAV2.5 from the atomic coordinates of AAV1 (data not shown) and AAV211 (Figure 6). This is one of the three most structurally divergent surface loops in main chain conformation when the structure of AAV8, another muscle tropism serotype, was compared to that of AAV2.24 The Q263A substitution and T265 insertion were also among the nine residues identified as contributing to AAV1 skeletal muscle transduction by the AAV1/AAV2 domain swap study by Hauck et al.25 Like the AAV2.5 vector, their AAV-221-IV vector approached but did not quite reach the transduction profile of AAV1, strongly suggesting other capsid motif may be required.
Figure 6.
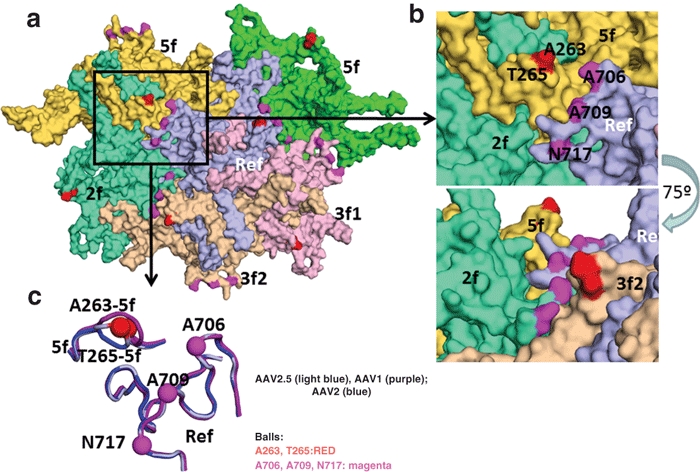
Topology of AAV2.5 virion. Surface topology view of the five VP monomers (in different colors) immediately surrounding the reference monomer in light blue. The symmetry operation that brings the monomer into contact is given in the labels. The positions of the AAV2.5 residue mutations are colored as in Figure 1b,c. (b) Top panel is a close up of the boxed region in the a. The bottom panel shows the same image rotated by ~75° and shows that the region containing the 263/265 amino acids are raised on the capsid surface. (c) Image showing a surface view (same as in Figure 1) of the loop containing the 263/265 region, VR III and VR IX (close to the 706, 709, 717 mutations) for AAV1 (purple), AAV2 (blue), and AAV2.5 (light blue). The positions of the AAV2.5 mutants are shown in the balls and labeled. AAV, adeno-associated virus.
Gene expression and immunological recognition
AAV2.5-mediated minidystrophin delivery to skeletal muscle was confirmed by vector DNA quantitative PCR (qPCR). Retrieved genome copy numbers (up to 2.56 copies/diploid genome in subject 4) were within the range of that observed in previous trials of i.m. administration of AAV vectors (Supplementary Table S1). However, the overall minidystrophin transgene expression detected in this clinical trial was low. Limited minidystrophin positive myofibers were detected in patients 3 and 6, but not in other patients. However, patients 2 and 5 had elevated T cell Elispot responses to dystrophin epitopes from endogenous revertants and/or transgene product after vector delivery, suggesting a transient transgene expression.12 As an alternative to the hypothesis that minidystrophin-stimulated transient recognition by T cell PBMCs resulted in loss of expression before biopsy, the low levels of transgene expression could be partially explained by numerous factors, e.g., pre-existing anti-AAV neutralizing antibodies (seen in patients 2 and 5), pre-existing T cell responses to endogenous revertant dystrophin epitopes that are also present on minidystrophin (seen in patients 2 and 4),12 silencing of the CMV promoter in human dystrophic muscles, disease-associated inflammation, and low tropism of AAV2.5 to human muscle tissue.
With respect to these possibilities, we observed significantly higher vector genome copy numbers in muscle from patients who both lacked pre-existing Nab and who were also biopsied at earlier time-point. Subjects 4 and 6, who received the high vector dose, also had the highest vector genome copy numbers detected (2.56 and 1.42 vg/dg, respectively), consistent with animal studies. Although subjects 4 and 6 retained the highest genome copy number, they showed no detectable (subject 4) or very low levels of minidystrophin expression (subject 6), a strong indication of CMV promoter silencing in the injected muscle. Lower levels of vector DNA were detected in subjects 1 and 3 (0.75 and 0.61 vg/dg, respectively), but only subject was found to have minidystrophin positive myofibers. Subjects 2 and 5 had high levels of pre-existing neutralizing antibodies (1:800 and 1:100) to AAV2.5. They also had elevated T cells recognizing dystrophin. The humoral and T-cell immunity could have jointly contributed to the very low vector genome copy numbers (0.01 and 0.08 vg/dg, respectively) and the lack of detectable minidystrophin expression, again as predicted from animal studies (see Figure 3). Unfortunately due to the small size of this safety study, the variability in subject characteristics and the two biopsy timepoints, it is premature to draw definitive conclusions regarding the association between pre-existing immunities and gene transfer and expression efficiency, but mark a key observation that has been noted by more recent AAV studies (>50 exclusion of patients with pre-existing Nab in AAV1 CUPID trial).
With the caveat that many experimental differences exist between trials, it remains interesting to compare the current results with previous clinical studies. While minidystrophin expression was observed only at low levels in one subject from each dose cohort, there are similarities with clinical observations in other trials using similar doses of AAV via i.m. injections in the AAV2-FIX, AAV2-AAT, and AAV1-AAT skeletal muscle clinical trials (Supplementary Table S2). In this present study, we observed T cell recognition of nonself-epitopes encoded by the minidystrophin in subject 5.12 Unpredicted T cell responses against self-epitopes encoded by the minidystrophin and the endogenous revertant dystrophin were also observed in subjects 2 and 4, apparently as a result of priming by rare revertant myofibers. In contrast to this and previous studies, in the recently reported LGMD 2D trial with AAV1 α-sarcoglycan,26,27 no T-cell immunity against either the endogenous or the transgene-encoded α-sarcoglycan was detected. In fact, 5 out of 6 patients had efficient and long-term transgene expression. The starkly different outcomes between the DMD and LGMD 2D clinical trials could be attributed to different complexity and underlying pathology of the diseases, choosing of patients (deletion versus point mutations) and the use of different AAV serotype vectors (AAV2.5 versus AAV1), promoters (nonspecific CMV versus muscle-specific tMCK) and the transgenes (minidystrophin versus α-sarcoglycan). The ongoing oligo exon skipping studies for DMD should provide supportive evidence for “revertant myofiber” hypothesis or simply note that peripheral T-cell recognition is not indicative of cytotoxic T-cell response.
In the present study, cell-mediated responses to the AAV capsid peptides were absent at baseline in all subjects. This absence of T cell response to AAV capsid epitopes in subjects 2 and 5 who had high levels of pre-existing AAV Nab was somewhat unexpected. Interestingly, we did not observe any T cell response to AAV capsid in subjects 5 and 6 who received a 1 log higher AAV2.5 capsid dose than Cohort 1 due to the fivefold dose escalation and the additional equal dose of empty capsid vector in the contralateral arm. In a minority of subjects, Elispot IFN-γ release approached and sometimes exceeded the threshold values considered positive, but there was no consistent pattern or obvious relationship to the timing of vector administration.
Significance of immunological data
This trial was the first to use a laboratory-derived novel AAV capsid to mediate gene delivery. The vector was well tolerated and there were no vector-related adverse events. These data are similar to a meta-analysis of all adverse events reported in 12 AAV trials enrolling >400 subjects overall, in which no AAV vector related adverse event pattern was observed. The safety profile observed in this trial supports further clinical evaluation of the AAV-minidystrophin. However, T cell-mediated immune recognition of vector derived neoantigens must continue to be monitored in future trials to determine whether there are dose thresholds or specific response stimuli that can be avoided. Notwithstanding, the relationship remains elusive between T cell recognition and a response against transduced cells. Manno et al. reported contemporaneous increases in PBMCs that recognized the AAV2 capsid with an increase in transaminase levels and reduction in circulating FIX concentrations.23 In a recent study of AAV1-α-antitrypsin (AAT) gene delivery to muscle,28,29 T cell recognition of AAV capsid epitopes was also observed. In contrast to the FIX trial, quantitative examination of blood AAT levels suggested that the PBMC T cell recognition of AAV capsid did not have an impact on gene expression as it was stable over an extended duration.28,29 A recent publication by the Walker group suggests that in some scenarios peripheral PBMCs may become apoptotic in muscle tissue and a functional immune response targeting an antigen may be muted or curtailed.30 The impact of immunological recognition is likely to be multifactorial. For example, Vandenberghe described the generation of high levels of T cells against capsid of serotypes capable of binding heparin or heparan sulfate proteoglycans.31 Although AAV2.5 contains heparin binding capabilities we observed weak or minimal T cell activation to the AAV2.5 capsid using ELISPOT, suggesting that besides vector serotype and tropism, the route of administration, capsid particle dose, choice of promoter, and underlying disease pathology are likely to be critical determinants of any immune effector response.
Future directions
Pre-existing humoral immunity will continue to be a critical factor to be considered in the selection of study populations and in the determination of enrollment criteria. The general population is exposed predominantly to AAV2 in the first decades of life.32 Patients with pre-existing high-titer Nab should be avoided in future trial designs. Effort is needed to continue the development of novel chimeric AAV vectors that evade the common pre-existing Nab.33,34 Diminished seroprevalence of pre-existing crossreactive neutralizing antibodies is a goal of novel capsid development. The preliminary evaluation of AAV2.5 in vivo and preliminary study of humoral responses showed reduced crossreactivity to AAV2.5 in animal and human sera. The pre-existing humoral immunity to AAV2.5, we observed is attributable to AAV2 crossreactivity, with lower titers of anti-AAV2.5 Nab being observed compared with titers of anti-AAV2. These results support the predictive utility of the assays and methods used to develop the AAV2.5 capsid. However, we did observe less of a difference in crossreactivity between AAV2 and AAV2.5 Nab titers in the current study than in the preclinical evaluations, which could be due to the small sample sizes of both studies. As was seen in our studies, this ability to develop a new generation of more tissue-selective gene vectors must be tempered with our limited abilities to predict human immunological recognition/responses with the available preclinical models. Recent findings suggest that pre-existing anti-AAV8 neutralizing titers of 1:10 or greater may result in altered biodistribution patterns in nonhuman primates.35 This observation is of limited relevance to the current study that involved direct i.m. administration. However, future trials involving delivery via the vasculature may be impacted by this finding.
Immunological recognition of both capsid and transgene-encoded epitopes should be carefully monitored in any ongoing or future clinical study that targets DMD, in particular, potentially neoantigens not previously presented to the immune system, as well as antigens generated by revertant myofibers (see ref. 12).
The route of administration is known to impact upon the host immune response,36,37 and future trials of isolated limb delivery (Fan et al., this issue) and eventual systemic delivery will need to continue to monitor for immune responses. Intravascular AAV delivery results in some level of gene transfer to the liver, thymus, and spleen, which has been associated with transgene product tolerance in preclinical studies.38 Our preliminary preclinical limb perfusion study in the DMD dog model using AAV2i8, AAV8, or AAV9 mediated long-term minidystrophin expression (data not shown). Although the immunological mechanisms behind these observations are unclear, either immune tolerance or T-cell apoptosis or both could favor blood-vessel-mediated AAV vector delivery to the muscle, which has a more uniform vector and antigen distribution than local i.m. injection. Given that regional limb delivery and eventual body wide delivery of the AAV vectors require proportionally higher vector doses of up to 1015 vg per patient, this will be associated with unprecedented foreign antigen load. The success of future clinical development of AAV based gene transfer for DMD will continue to require careful evaluation of the relation between vector doses, cell-mediated immune recognition and T cell immune response at all levels of preclinical and clinical development. To this end we have initiated the next generation AAV capsid for efficient muscle delivery via vascular route that detargets liver but efficiently transduces muscle and cardiac tissue.39,40
Materials and Methods
Plasmid and DNA mutagenesis. The starting plasmid for these experiments was the packaging plasmid pXR2.41 All plasmid mutagenesis was performed using either the Quick Change Multi Site Mutagenesis or the Quick Change Site Directed Mutagenesis kits (both from Stratagene, Santa Clara, CA). When possible, mutagenesis of multiple amino acids was done concurrently. When concurrent mutagenesis was not possible because distance between nucleotides was not optimal, mutagenesis was performed sequentially within inclusion of previous nucleotide changes within the new primer to prevent reversion. Accuracy of the nucleotide changes was verified by DNA sequence analyses followed by DNA subcloning to eliminate any unwanted artifacts generated by the mutagenesis.
Recombinant virus production (preclinical studies). All recombinant AAV viruses were generated using the standard triple transfection method using the XX6-80 adenoviral helper plasmid with a packaging plasmid (either AAV1, AAV2 or a modified packaging plasmids) and an inverted terminal repeat plasmid [containing either green fluorescent protein, luciferase, or hAAT].42
rAAV was purified using standard methodology and the physical titer of the different viral preparations was evaluated using dot blot hybridization.42 For direct comparison of in vivo transduction efficiency, the vectors being compared within one experiment were evaluated on the same dot blot.
In vitro vector characterization. Batch binding of rAAV to heparin agarose was performed as described previously.43 The ability of the virus encoding the firefly luciferase complementary DNA to transduce cultured cells was also evaluated. Briefly, 293, Cos1, or HeLa cells were infected with 1,000 particles of rAAV/cell and Ad dl309 at a multiplicity of infection of 10. Cells lysates were then assessed via luminometry 24 hours post-transduction.
Animals. C57BL and BALB/c mice were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were maintained and treated in accordance with the Animal Care and Use Committee of UNC Chapel Hill. All care and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals (DHHS Publication No. [NIH] 85-23), and all procedures received prior approval by the University of North Carolina Institutional Animal Care and Usage Committee.
In vivo bioluminescence imaging of luciferase. 1 × 1010 viral genome-containing particles (vg) were injected into the gastrocnemius male BALB/c mice. A total of six limbs were injected for each vector type using 25 µl of virus. The mice were anesthetized by intraperitoneal administration of 2.5% averdin. A solution of the luciferase substrate luciferin (150 mg/kg; Invitrogen, Carlsbad, CA) was injected intraperitoneally. A grey scale reference image of animals was generated and mice were imaged for 5 minutes using a NightOwl cabinet with charge couple device camera (Berthold Technologies, Bad Wildbad, Germany). Total photon emission from selected and defined areas within the images of each mouse was quantified with the WinLight32 software (Berthold Technologies). The photon signal was presented as a pseudocolor image representing light intensity (red most and blue least intense). This image was superimposed on the reference image for orientation.
Detection of neutralizing anti-AAV antibodies. For comparison of humoral immune response among AAV capsids in mice, 100 µl phosphate-buffered solution containing 1 × 1010 particles of rAAV/GFP virus for each capsid type was intraperitoneally injected into 6–10-week-old mice at day 0, and boosted at day 14. Blood sera were collected from mice via retro-orbital plexus at indicated time points or from human via peripheral vein. Nab titer was assayed using methodology described by Moskalenko et al. with slight modifications.44 Briefly, 293 cells were seeded in a 48-well plate at a density of 105 cells/well in 200 µl DMEM containing 10% fetal bovine serum. The cells were cultured for 3–4 hours at 37 °C and allowed to adhere to the well. AAV-GFP (1 × 108 particles) was incubated with mice sera at serial dilution with phosphate-buffered solution for 2 hours at 4 °C in a total volume of 25 µl. The mixture was added to cells in a final volume of 200 µl which contained 4 × 10 6 particles of adenovirus dl309 and incubated 24 hours or 48 hours at 37 °C. GFP-expressing cells were counted under a fluorescent microscope. The neutralizing antibody titer was calculated using the highest dilution where the percentage of GFP expressed cells were 50% less than control without sera.
Readministration. For readministration application of AAV vectors in vivo, mice were immunized with AAV/AAT vectors by muscular injection, and then challenged with AAV/luciferase vectors 2 months later. The luciferase transgene expression was measured 6 weeks following last vector injection.
Human subjects (preclinical). The sera from adult volunteers were screened for neutralizing antibodies to AAV. Individuals were researchers at the University of North Carolina at Chapel Hill (Chapel Hill, NC). The study was approved by the institutional review board of the University of North Carolina at Chapel Hill.
Homologous model building for AAV2.5. The available atomic coordinates for AAV2 (ref. 11; PDB accession No.1LP3) and AAV1 (L. Govindasamy and M. Agbandje-McKenna, unpublished results) were used to generate a homologous model for the AAV2.5 variant to enable comparison with the parental viruses. The model was generated by submitting the VP3 sequence of AAV2.5 to the SWISS-MOLEL model building program with the coordinates of structures of the AAV1 and AAV2 VP3 supplied as template.45 The model of AAV2.5 VP3 was then superimposed onto the AAV1 and AAV2 structures using the SSM option of the COOT program for comparative analysis.46,47 To visualize the location of the mutated residues in the context of an assembled capsid in the predicted structure of AAV2.5 icosahedral symmetry operators were applied to the VP3 model coordinates by matrix multiplication using the program O and the amino acids were highlighted in the context of the symmetry related VPs.48 Figures showing ribbon drawings and surface representations were generated using the program PyMOL.
Regulatory approval process. The clinical protocol and Appendix M documents were reviewed by the Recombinant DNA Advisory Committee under the auspices of the Office of Biotechnology Activities at the NIH. The Nationwide Children's Hospital institutional review board approved the study protocol. A Pre-IND discussion was held with FDA prior to initiation of the pivotal toxicology and biodistribution study, and upon conclusion of preclinical studies the IND was submitted to CBER/FDA (BB-IND 12936). The trial is registered with ClinicalTrials.gov (Reference no. NCT00428935).
Study design. The experimental design was a randomized double-blind placebo-controlled study where vector was administered into one bicep and saline control in the contralateral arm. In a subset of patients, we substituted AAV empty capsid for saline in an effort to distinguish immune response to vector versus minidystrophin transgene. The active phase of the study included a 2-week baseline screening period, a 2-day inpatient period for vector injection and acute toxicity monitoring, and a 2-year outpatient follow-up and toxicity-monitoring period. Ongoing long-term follow-up will continue out to 15 years post vector injection (Supplementary Figure S1). Blood and urine analyses were conducted in conjunction with outpatient clinics on postadministration days 8, 15, 30, 43, 53, 60, 90, and 120 and months 6, 9, 12, 18, and 24.
The trial was comprised by two-dose cohorts, with each patient receiving bilateral injections, with double-blinded randomization of AAV2.5-minidystrophin vector to one bicep and placebo to the contralateral bicep. Four patients received a saline placebo injection into the contralateral bicep, and two patients in dose cohort II received an empty capsids placebo injection into the contralateral bicep with an identical dose as of the minidystrophin-containing full capsids.
qPCR analysis of AAV2.5 vector genomes. DNA was isolated from frozen muscle biopsies using a Qiagen DNeasy Tissue kit. 0.5% of the recovered DNA from each sample was used as template in duplicate qPCR using three primer sets targeting the minidystrophin transgene and two primers targeting human genomic DNA reference sites at the laminB2 and globin loci (Supplementary Table S3). The minidystrophin primers crossed exon splicing junctions, so they specifically amplify the complementary DNA transgene but not the endogenous dystrophin gene. qPCR was performed.
Minidystrophin vector. The vector genome packaged in AAV2.5 encoded the aminoterminal actin-binding domain, five rod repeat domains (R1, R2, R22, R23, and R24), three hinge domains (H1, H3, and H4), and the cysteine-rich domain of the human dystrophin gene. The CMV immediate early promoter regulated transgene expression, along with a bovine growth hormone derived polyadenylation signal.
Both the test vector and the empty capsid reagents were generated by transient transfection of mammalian HEK293 cell cultures, undertaken at the Human Applications Laboratory, University of North Carolina.
With Dot–Blot tittering, the total lot yield was calculated as 1.72 × 1013 vector genomes at a concentration of 4.4 × 1012 vector genomes/ml. This final product was then further diluted in sterile 1× phosphate-buffered solution 5% sorbitol to generate the clinical dose concentrations (see Table 2) for additional characterization and lot release testing. The clinical dose is determined by the vector genome dose, which is 6 × 1011 vector genomes/subject in the first low dose cohort and 3 × 1012 vector genomes/subject in the second high dose cohort.
Apart from two critical differences, the manufacturing procedures used in the production of the empty capsid lot were identical to those used in the production of rAAV2.5 minidystrophin. The first difference is merely the absence of the AAV-minidystrophin-containing plasmid in the cell transfection stage. The second difference is the use of a dot blot western to titer the empty capsids (in units of capsid particles) in the absence of a single-stranded DNA insert. The AAV2.5 CMV-empty capsid lot underwent nearly identical release testing (titering assays differed) to that performed for AAV2.5 CMV-3978, and in addition was released with a CoA using the same procedures.
Estimation of capsid particle titer. Transmission electron microscopy was undertaken to evaluate the ratio of genome-containing particles to the total number of genome-containing (full) or empty capsid particles, with the ratio estimated as being 0.907. The total dose of AAV2.5 capsid particles was then estimated from this ratio and the genomic particle titer generated by the dot blot assay.
Evaluation of T-cell reactivity to capsid derived peptides. Peripheral blood T cell responses to the novel AAV capsid were quantified by IFN-γ ELISpot assay, as described before.12 Briefly PBMC isolated on Ficoll hypaque gradients were cultured with synthetic peptides (20 amino acids in length, overlapping by 10 residues) that spanned the VP1 capsid protein. To identify individual peptides within a pool that elicited IFN-γ activity, so that each peptide was present in two of the intersecting mapping subpools. After incubation at 37 °C for 36 hours. IFN-γ SFC were counted. Fewer than 10 SFC/well were observed with peptides from a control pool (enhanced GFP). Responses were considered positive when SFC exceeded 50/106 PBMC in duplicate wells.
SUPPLEMENTARY MATERIAL Figure S1. Trial procedures timeline. Figure S2. Temporal profiles of blood chemistry, hematology, and urinalyses. Table S1. Genome detection in subject biopsies. Table S2. Comparison of AAV.Minidys dose–response-related expression. Table S3. Quantitative PCR primers. Materials and Methods.
Acknowledgments
The participation and cooperation of the trial participants and their guardians was greatly appreciated. The authors would like to acknowledge the following individuals for providing expert assistance on this project, including trial planning and conduct, data collection and analysis, and reviewing this manuscript: Jerry Mendell, Chris Walker, Chris Shilling, Kristine Campbell, L. Radino-Klapac, Z. Sahenk, S. Lewis, G. Galloway, V. Malik, B. Coley, R. Clark, D. McCarty, S. Hesterlee, J. Larkindale, S. Moore, V. Alekseeva, Aravind Asokan. D.E.B., R.J.S., and X.X. hold stock in Asklepios BioPharmaceutical Inc. C.L., S.J.G., R.J.S., and X.X. have received research funding from Asklepios BioPharmaceutical Inc. This work was funded by a Translational Corporate Grant to Asklepios BioPharmaceutical Inc. from the Muscular Dystrophy Association USA. This study was also supported by NIH research grants 1R01AI080726 and 5R01DK084033 (to C.L. and R.J.S.), 5U54AR056953 (to R.J.S.) and R01 AI072176 (R.J.S. and M.A.-M).
Supplementary Material
Trial procedures timeline.
Temporal profiles of blood chemistry, hematology, and urinalyses.
Genome detection in subject biopsies.
Comparison of AAV.Minidys dose–response-related expression.
Quantitative PCR primers.
REFERENCES
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C., and, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, Takeda S.et al. (2011Current status of pharmaceutical and genetic therapeutic approaches to treat DMD Mol Ther 19830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L.et al. (2006rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice Nat Med 12787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li J, Fu FH., and, Xiao X. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthop Res. 2009;27:421–426. doi: 10.1002/jor.20781. [DOI] [PubMed] [Google Scholar]
- Wang B, Li J., and, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li J, Fu FH, Chen C, Zhu X, Zhou L.et al. (2008Construction and analysis of compact muscle-specific promoters for AAV vectors Gene Ther 151489–1499. [DOI] [PubMed] [Google Scholar]
- Wang B, Li J, Qiao C, Chen C, Hu P, Zhu X.et al. (2008A canine minidystrophin is functional and therapeutic in mdx mice Gene Ther 151099–1106. [DOI] [PubMed] [Google Scholar]
- Koerber JT, Jang JH., and, Schaffer DV. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol Ther. 2008;16:1703–1709. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang L, Johnson JS, Zhijian W, Grieger JC, Ping-Jie X.et al. (2009Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium Mol Ther 172067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A.et al. (2002The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy Proc Natl Acad Sci USA 9910405–10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S.et al. (2010Dystrophin immunity in Duchenne's muscular dystrophy N Engl J Med 3631429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda VR, Schuettrumpf J, Herzog RW, Nichols TC, Robinson N, Lotfi Y.et al. (2004Safety and efficacy of factor IX gene transfer to skeletal muscle in murine and canine hemophilia B models by adeno-associated viral vector serotype 1 Blood 10385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H, Monahan PE, Liu Y, Samulski RJ., and, Walsh CE. Sustained and complete phenotype correction of hemophilia B mice following intramuscular injection of AAV1 serotype vectors. Mol Ther. 2001;4:217–222. doi: 10.1006/mthe.2001.0449. [DOI] [PubMed] [Google Scholar]
- Wu Z, Asokan A., and, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Louboutin JP, Wang L., and, Wilson JM. Gene transfer into skeletal muscle using novel AAV serotypes. J Gene Med. 2005;7:442–451. doi: 10.1002/jgm.686. [DOI] [PubMed] [Google Scholar]
- Xiao W, Chirmule N, Berta SC, McCullough B, Gao G., and, Wilson JM. Gene therapy vectors based on adeno-associated virus type 1. J Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA.et al. (2006Structurally mapping the diverse phenotype of adeno-associated virus serotype 4 J Virol 8011556–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padron E, Bowman V, Kaludov N, Govindasamy L, Levy H, Nick P.et al. (2005Structure of adeno-associated virus type 4 J Virol 795047–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R.et al. (2006Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization J Virol 80821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Hügle-Dörr B, Girod A, Petersen G, Hallek M., and, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X.et al. (2004Clades of Adeno-associated viruses are widely disseminated in human tissues J Virol 786381–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ.et al. (2006Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response Nat Med 12342–347. [DOI] [PubMed] [Google Scholar]
- Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E.et al. (2007Structure of adeno-associated virus serotype 8, a gene therapy vector J Virol 8112260–12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck B, Xu RR, Xie J, Wu W, Ding Q, Sipler M.et al. (2006Efficient AAV1-AAV2 hybrid vector for gene therapy of hemophilia Hum Gene Ther 1746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S.et al. (2010Sustained α-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D Ann Neurol 68629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G.et al. (2009Limb-girdle muscular dystrophy type 2D gene therapy restores α-sarcoglycan and associated proteins Ann Neurol 66290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT.et al. (2009Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy Proc Natl Acad Sci USA 10616363–16368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotte, TR, Trapnell, BC, Humphries, M, Carey, B, Calcedo, R, Rouhani, F.et al. (2011Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha(1)-Antitrypsin: interim results Hum Gene Therepub ahead of print). [DOI] [PMC free article] [PubMed]
- Velazquez VM, Bowen DG., and, Walker CM. Silencing of T lymphocytes by antigen-driven programmed death in recombinant adeno-associated virus vector-mediated gene therapy. Blood. 2009;113:538–545. doi: 10.1182/blood-2008-01-131375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe LH, Wang L, Somanathan S, Zhi Y, Figueredo J, Calcedo R.et al. (2006Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid Nat Med 12967–971. [DOI] [PubMed] [Google Scholar]
- Calcedo R, Vandenberghe LH, Gao G, Lin J., and, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS.et al. (2008Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles Mol Ther 161252–1260. [DOI] [PubMed] [Google Scholar]
- Maheshri N, Koerber JT, Kaspar BK., and, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- Wang L, Calcedo R, Bell P, Lin J, Grant RL, Siegel DL.et al. (2011Impact of Pre-Existing Immunity on Gene Transfer to Nonhuman Primate Liver with Adeno-Associated Virus 8 Vectors Hum Gene Therin press). [DOI] [PMC free article] [PubMed]
- Toromanoff A, Adjali O, Larcher T, Hill M, Guigand L, Chenuaud P.et al. (2010Lack of immunotoxicity after regional intravenous (RI) delivery of rAAV to nonhuman primate skeletal muscle Mol Ther 18151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toromanoff A, Chérel Y, Guilbaud M, Penaud-Budloo M, Snyder RO, Haskins ME.et al. (2008Safety and efficacy of regional intravenous (r.i.) versus intramuscular (i.m.) delivery of rAAV1 and rAAV8 to nonhuman primate skeletal muscle Mol Ther 161291–1299. [DOI] [PubMed] [Google Scholar]
- Herzog RW. Hepatic AAV gene transfer and the immune system: friends or foes. Mol Ther. 2010;18:1063–1066. doi: 10.1038/mt.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R.et al. (2010Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle Nat Biotechnol 2879–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulicherla N, Shen S, Yadav S, Debbink K, Govindasamy L, Agbandje-McKenna M.et al. (2011Engineering Liver-detargeted AAV9 Vectors for Cardiac and Musculoskeletal Gene Transfer Mol Ther 191070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X.et al. (2002Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity J Virol 76791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieger JC, Choi VW., and, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- Rabinowitz JE, Bowles DE, Faust SM, Ledford JG, Cunningham SE., and, Samulski RJ. Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol. 2004;78:4421–4432. doi: 10.1128/JVI.78.9.4421-4432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalenko M, Chen L, van Roey M, Donahue BA, Snyder RO, McArthur JG.et al. (2000Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure J Virol 741761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J., and, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Krissinel E., and, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60 Pt 12 Pt 1:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Emsley P., and, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60 Pt 12 Pt 1:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW., and, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr, A, Found Crystallogr. 1991;47 (Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial procedures timeline.
Temporal profiles of blood chemistry, hematology, and urinalyses.
Genome detection in subject biopsies.
Comparison of AAV.Minidys dose–response-related expression.
Quantitative PCR primers.