

Abstract
Background
Acute allograft rejection is dependent on adaptive immunity, but it is unclear whether the same is true for chronic rejection. Here we asked whether innate immunity alone is sufficient for causing chronic rejection of mouse cardiac allografts.
Methods
We transplanted primarily vascularized cardiac grafts to recombinase activating gene-knockout (RAG−/−) mice that lack T and B cells but have an intact innate immune system. Recipients were left unmanipulated, received adjuvants that stimulate innate immunity, or were reconstituted with B-1 lymphocytes to generate natural IgM antibodies. In a second model, we transplanted cardiac allografts to mice that lack secondary lymphoid tissues (splenectomized aly/aly recipients) and studied the effect of NK cell inactivation on T cell-mediated chronic rejection.
Results
Acute cardiac allograft rejection was not observed in any of the recipients. Histological analysis of allografts harvested 50 to 90 days after transplantation to RAG−/− mice failed to identify chronic vascular or parenchymal changes beyond those observed in control syngeneic grafts. Chronic rejection of cardiac allografts parked in splenectomized aly/aly mice was observed only after the transfer of exogenously activated T cells. NK inactivation throughout the experiment, or during the parking period alone, reduced the severity of T cell- dependent chronic rejection.
Conclusions
The innate immune system alone is not sufficient for causing chronic rejection. NK cells predispose healed allografts to T cell-dependent chronic rejection and may contribute to chronic allograft pathology.
Keywords: innate immunity, chronic rejection, NK cells, B-1 lymphocytes
1. Introduction
Acute allograft rejection is dependent on the activation of donor-reactive host T cells [1]. T cell depletion or genetic T cell deficiency in mice prevents acute rejection and leads to long-term survival of transplanted organs [2–4]. Similarly, tolerance-induction strategies that induce T cell deletion or block T cell activation significantly prolong allograft survival [5, 6], but chronic rejection is often observed despite demonstrable donor-specific T cell unresponsiveness [5, 7]. Chronic rejection is also detected in patients presumed to be tolerant and in whom immunosuppressive drugs have been withdrawn [8]. Possible reasons for this paradox include vascular pathology caused by non-immunological factors such as hypertension, the presence of residual anti-donor T or B cell reactivity in the ‘tolerant’ host, or persistent T and B cell-independent innate immunity to the graft [8, 9].
That innate immune activation could lead to chronic allograft rejection was first suggested by a study showing that mice rendered tolerant during the neonatal period or through robust mixed hematopoietic chimerism develop chronic allograft vasculopathy [5]. The vasculopathy was mediated by natural killer (NK) cells and could be reproduced in T and B cell-deficient but NK-sufficient RAG−/− recipients. The same group later determined that NK-mediated chronic rejection did not take place unless RAG−/− hosts were concomitantly infected with lymphocytic choriomeningitis virus (LCMV) virus [10] or were reconstituted with recipient-matched CD4+ T lymphocytes [11]. Since the latter observation was made in a transplantation model in which anti-donor T cell reactivity was absent (parental hearts transplanted to F1 recipients), the authors concluded that NK cells are necessary for the development of chronic allograft vasculopathy but are not sufficient - cooperation with CD4+ T cells not responsive to donor alloantigens is required [11]. Additional support for a role of NK cells in rejection independent of T cell alloreactivity derives from studies showing that NK cells contribute to heart allograft rejection in gene knockout mice that lack a critical T cell costimulatory molecule, CD28 [12, 13]. More recently, IL-15, a cytokine that significantly expands and activates NK cells, has been shown to precipitate skin allograft rejection in RAG−/− recipients when given in large excess [14]. These studies suggest that NK cells contribute to chronic rejection but do not clearly define whether NK cells do so in the absence of concomitant factors such as viral infection or excess cytokines, or whether their contribution is indirect and requires interaction with adaptive immunity.
Here we re-addressed the question whether the innate immune system alone is sufficient for mediating chronic allograft rejection and re-examined the role of NK cells in this process. Using RAG−/− mice or mice that lack secondary lymphoid tissues as recipients of primarily vascularized cardiac grafts, we found that chronic rejection is an immunological process that does not occur in the absence of adaptive immune activation. Although NK cells were not sufficient to cause graft pathology, they predisposed healed allografts to T cell-dependent chronic rejection.
2. Original Hypothesis
We hypothesized that sufficient activation of innate immunity or the presence of natural IgM antibodies in RAG−/− recipients of primarily vascularized cardiac allografts results in chronic allograft rejection in the absence of adaptive immunity. We also speculated that host NK cell alloreactivity maintains inflammation in healed cardiac allografts thereby facilitating T cell- mediated chronic rejection.
3. Materials and Methods
3.1. Mice
BALB/c (H-2d), B10.BR (H-2k), (C57BL/6×BALB/c)F1 (H-2b/d), B6 RAG1−/− (H-2b), and BALB/c RAG1−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME). B6 and BALB/c RAG2−/− were purchased from Taconic (Germantown, NY). Alymphoplastic (aly/aly) mice were purchased from Clea Japan (Tokyo, Japan). All mice were housed in a specific pathogen-free (SPF) environment. RAG1−/− and RAG2−/− mice were used interchangeably and are referred to as RAG−/− mice throughout the manuscript.
3.2. Mouse procedures
All heart donors and recipients were age- (6 – 8 weeks) and sex-matched. Heterotopic transplantation of primarily vascularized heart allografts was performed by a single microsurgeon (QL). In this model, the pulmonary artery and ascending aorta of the heart graft are anastomosed to the recipient’s inferior vena cava and abdominal aorta, respectively [15]. Grafts were monitored daily by palpation for the first week and then thrice weekly. Clinical rejection was defined as cessation of palpable heartbeat, at which time the abdomen was opened to confirm rejection and the graft was harvested for histological analysis. Splenectomy was performed in aly/aly mice at least one week prior to heart transplantation according to an established procedure [16]. NK depletion of recipients, where specified, was accomplished by injecting 250 μg of anti-NK1.1 mAb (clone PK136, BioXCell, West Lebanon, NH) i.p. on days −1, 0, and +1 relative to heart transplantation and weekly thereafter until day 50. Control animals received isotype-control IgG. NK depletion was confirmed by flow cytometric analysis of peripheral blood cells using a polyclonal PE-conjugated anti-NKp46 mAb (R&D Systems, Minneapolis, MN) [17]. Complete Freund’s Adjuvant (CFA, Sigma, St. Louis, MO), poly I:C, and CpG-1826 (both from InvivoGen, San Diego, CA) were administered as indicated in Table 1. All procedures were performed in accordance with the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Table 1.
Absence of acute and chronic cardiac allograft rejection in RAG−/− recipients
| Donor | Recipient | Recipient Treatment | Harvest Time (Days) | n | ARa | CRb |
|---|---|---|---|---|---|---|
| B6 RAG−/− | B6 RAG−/− | None | 72 – 75 | 6 | 0 | 0 |
| BALB/c RAG−/− | B6 RAG−/− | 1×107 B6 T cells i.v.c | 11–14d | 3 | 3 | 0 |
| B10.BR | B6 RAG−/− | None | 79 – 82 | 7 | 0 | 0 |
| B6 RAG−/− | BALB/c RAG−/− | CFAe | 77 – 85 | 7 | 0 | 0 |
| B6 RAG−/− | BALB/c RAG−/− | Poly I:Cf | 60 – 75 | 4 | 0 | 0 |
| B6 RAG−/− | BALB/c RAG−/− | CpGf | 70 – 75 | 4 | 0 | 0 |
| B6 RAG−/− | BALB/c RAG−/− | B-1 reconstitution | 50 (n = 2) 90 (n = 1) |
3 | 0 | 0 |
| B6 RAG−/− | BALB/c RAG−/− | Alloimmunizationg | 49 – 57 | |||
| B6 RAG−/− | BALB/c RAG−/− | Alloimmunization + CFA | 48 – 57 | 7 | 0 | 0 |
AR = Acute Rejection
. CR = Chronic Rejection.
T cells were injected 50 days after transplantation.
Hearts were harvested upon cessation of palpable heart beat (median graft survival = 11.5 days after T cell injection) and rejection was confirmed histologically.
CFA (200 μl) was administered i.p. 7 days prior to transplantation.
Poly I:C (100 μg) and CpG-1826 (25 μg) were administered i.v. on the day of transplantation and once or twice weekly thereafter.
Alloimmunization was performed by injecting 2 × 107 B6 RAG−/− splenocytes i.p. 7 days prior to transplantation.
3.3. Histological analysis
Heart tissue was frozen in Tissue-Tech O.C.T. compound or fixed in aqueous-buffered zinc formalin and paraffin-embedded. Fixed sections were stained with Hematoxylin & Eosin (H&E), Masson-Trichrome (MT), and Verhoeff Van Gieson (VVG) elastin stain, and analyzed in a blinded fashion by a pathologist (FKB or AJD). Acute and chronic rejection were scored based on type and severity of cellular infiltrates, vascular pathology (including obliterative arteriopathy), myocyte dropout, and interstitial fibrosis. Obliterative arteriopathy was quantitated by calculating the average ratio of intima to total vessel wall thickness in at least 10 intra-parenchymal ateries per graft. Subepicardial vessels and those present close to suture line were excluded from evaluation.
3.4. Analysis of graft-infiltrating cells
To quantitate NK cells that infiltrated heart grafts parked in RAG−/− or splenectomized aly/aly recipients, mice were sacrificed 50 days after transplantation and perfused with 20 ml PBS + 0.5% heparin solution via the left ventricle of the native heart. Heart grafts were removed and homogenized, RBCs were lysed in hypotonic buffer, and tissue was digested at 37°C with Collagenase IV (350 U/ml) and DNAse I (20 ng/ml) (Sigma). Infiltrating cells were isolated by centrifugation on a Lympholyte M gradient (CedarLane Labs, NC). NK cells were quantitated by flow cytometry by gating on NK1.1+ cells after live/dead cell discrimination and exclusion of lineage-positive (B220+, CD4+, CD8+) cells.
3.5. Adoptive cell transfer
Spleens and peripheral lymph nodes were harvested 7 days after immunizing B6 mice with allogeneic BALB/c or (C57BL/6×BALB/c)F1 splenocytes. T cells were enriched by negative selection using magnetic cell separation (AutoMACS, Miltenyi Biotech, Auburn, CA). Resulting T cell purity was > 85%. 1 or 5 × 107 enriched T cells were transferred i.v. to B6 RAG−/− or splenectomized aly/aly recipients, respectively, 50 days after transplantation. Isolation of B-1 cells was accomplished by harvesting resident peritoneal cells of BALB/c wt mice by peritoneal lavage and subsequent high-speed sorting of CD5highIgMhighCD23low cells using a FACS ARIA (BD Biosciences, San Jose, CA). Resulting purity was >95%. 1×106 B-1 cells were transferred i.p. to BALB/c RAG−/− recipients. Serum IgM levels were measured at the indicated times by ELISA according to the manufacturer’s instructions (Bethyl Laboratories, Montgomery, TX).
3.6. Statistical analysis
All data are presented as mean ± SD and were analyzed by either Student’s t-test or one-way analysis of variance (ANOVA). Significance was set at p < 0.05.
4. Results
4.1. Absence of acute or chronic heart allograft rejection in RAG−/− recipients
To test whether the innate immune system alone is sufficient for chronic allograft rejection, we performed heterotopic, vascularized, heart transplantation from B10.BR (H-2k) donors to B6 (H-2b) RAG−/− recipients. All grafts continued to beat and contract for the duration of the experiment (Table 1). Grafts were harvested between 79 and 82 days after transplantation and analyzed for the presence of chronic rejection by light microscopy. Although minimal intragraft inflammation and fibrosis were observed, these changes were not significantly different from those detected in control syngeneic grafts (B6 RAG−/− donors to B6 RAG−/− recipients) (Fig. 1). As previously published by our group [17] and others [4], we confirmed that acute allograft rejection occurs in B6 RAG−/− recipients upon adoptive transfer of B6 T cells (Table 1).
Figure 1. Absence of chronic rejection in cardiac allografts transplanted to RAG−/− recipients.

Allogeneic B10.BR hearts or syngeneic B6 RAG−/− hearts were transplanted to B6 RAG−/− mice (n=7 and 6, respectively). Grafts were harvested 72 – 82 days later and stained with H&E, MT, and VVG. Syngeneic (left panel) and allogeneic (right panel) grafts appeared identical upon microscopic examination. There was no appreciable inflammatory infiltrate, myocyte dropout, or obliterative arteriopathy in either graft type. Representative whole heart sections stained with H&E are shown.
4.2. Absence of acute or chronic heart allograft rejection in RAG−/− recipients following innate stimulation or reconstitution with B-1 lymphocytes
Since laboratory mice are bred and housed in SPF facilities, it is conceivable that their innate responsiveness may be altered due to reduced exposure to pathogens. We therefore stimulated BALB/c RAG −/− recipients with a variety of innate ligands before or after transplanting heart grafts from B6 RAG−/− donors (Table 1). RAG−/− mice were used as heart donors to avoid the inadvertent transfer and activation of adaptive immune cells (T and B lymphocytes) to the host. Recipient stimulation with CFA (which contains a mixture of TLR and non-TLR ligands) [18], CpG (TLR-9 ligand), or polyI:C (TLR-3 ligand) failed to precipitate either acute or chronic heart allograft rejection as determined by histopathologic analysis (Table 1). Similarly, prior immunization of RAG−/− mice with allogeneic donor RAG−/− splenocytes, a procedure previously shown to stimulate monocyte-dependent innate alloimmunity [19], did not cause either acute or chronic heart allograft rejection (Table 1).
RAG−/− mice lack B-1 lymphocytes, which are the source of natural IgM antibodies. Natural IgM antibodies have been shown to bind otherwise cryptic tissue antigens that are exposed after ischemia-reperfusion injury leading to activation of the complement system [20]. We therefore asked whether reconstituting RAG−/− mice with B-1 cells would lead to chronic allograft rejection in the absence of adaptive immunity. CD5highIgMhighCD23low B-1 cells were sorted from resident peritoneal cells of BALB/c wt mice and transferred i.p. to BALB/c RAG−/− hosts. Six weeks later, the mice received B6 heart transplants. Serum IgM levels had reached 81 ± 24 ng/ml at the time of transplantation (n = 4), and had risen to 130 and 330 ng/ml 50 days later in the two mice that were tested. Despite the presence of significant circulating IgM in the recipients (corresponding to 25 – 65% of serum IgM level in wildtype animals), we did not observe histological hallmarks of chronic rejection at either 50 or 90 days after transplantation.
4.3. NK cells predispose healed allografts to T cell-mediated chronic rejection
We have previously shown that cardiac allografts parked for an extended period of time in RAG−/− mice undergo acute rejection upon T cell reconstitution of the host [17]. Moreover, cardiac allografts parked in either immunosuppressed, wildtype mice or splenectomized aly/aly mice (which lack all secondary lymphoid tissues) are not rejected by the host but undergo chronic rejection upon transfer of alloactivated T cells [21]. We therefore hypothesized that NK cells are responsible for persistent innate immune activation in the allograft and thus, set the stage for T cell-mediated chronic rejection. To test this hypothesis, we first quantitated and phenotyped NK cells that infiltrated syngeneic (B6) and allogeneic (BALB/c) heart grafts 50 days after transplantation to splenectomized aly/aly mice (n = 3 mice/group; grafts pooled prior to analysis). We found approximately two-fold more NK cells in allogeneic than syngeneic grafts (1.1 × 104 vs. 5.3 × 103 NK cells/graft, respectively). Moreover, NK cells that infiltrated allogeneic grafts had upregulated the activation marker CD69, albeit modestly (Fig. 2). To investigate whether NK cells contribute to chronic rejection in this model, we transplanted either BALB/c (H-2d) or (B6 × BALB/c)F1 (H-2b) hearts into splenectomized aly/aly (H-2b) mice and graft histology was studied 50 days after the transfer of alloactivated T cells from B6 mice immunized with BALB/c splenocytes. As shown in Fig. 3, F1 (H-2b/d) hearts displayed significantly less chronic allograft vasculopathy (% intimal thickening) than BALB/c (H-2d) hearts, suggesting that inhibition of NK cell activation by F1 grafts ameliorated chronic rejection. Alternatively, reduction of vasculopathy in these experiments could have been the result of reduced expression of Balb/c alloantigens in the hearts rendering them less immunogenic. Therefore, to provide further support for a role for NK cells in chronic allograft vasculopathy, we studied a second model in which BALB/c cardiac allografts were transplanted to splenectomized aly/aly mice and the recipients received either NK-depleting or isotype- control antibody for 7 weeks (n = 6/group). Alloactivated T cells were transferred one day after the last antibody injection (50 days after transplantation) and cardiac allografts were analyzed for the presence of chronic rejection 70 – 100 days after T cell transfer. All NK-depleted recipients exhibited mild acute and chronic rejection with low-grade inflammation, arteritis, and mild obliterative arteriopathy (Fig. 4). In contrast, 3/6 NK-replete recipients exhibited moderate to severe acute and chronic rejection, including significant graft fibrosis (Fig. 4B). Quantitation of vasculopathy (% intimal thickening) revealed significantly less chronic vasculopathy in the NK- depleted compared to the NK-replete group as shown in Fig. 4C. Commensurate with previously published data [22], control mice that did not receive activated T cells (n = 3) or mice that received naïve T cells 50 days after transplantation (n = 4) did not develop either acute or chronic rejection (Fig. 4C and D). These data suggest that NK cells contribute to persistent innate immune activation in the allograft that sets the stage for chronic rejection mediated by activated T cells.
Figure 2. Phenotype of graft-infiltrating NK cells.

Cells were isolated from allogeneic (BALB/c) and syngeneic (B6) heart grafts parked for 50 days in splenectomized B6 aly/aly mice. NK1.1 cells present within the grafts were identified by flow cytometry by gating on the lineage negative (B220−CD4−CD8−), NK1.1+ population (left panel). Expression of the activation marker CD69 on the gated cells was then determined (right panel). N=3/group, pooled prior to analysis.
Figure 3. Reduced chronic allograft vasculopathy in the absence of NK activity against the donor (F1 to parental strain transplantation model).
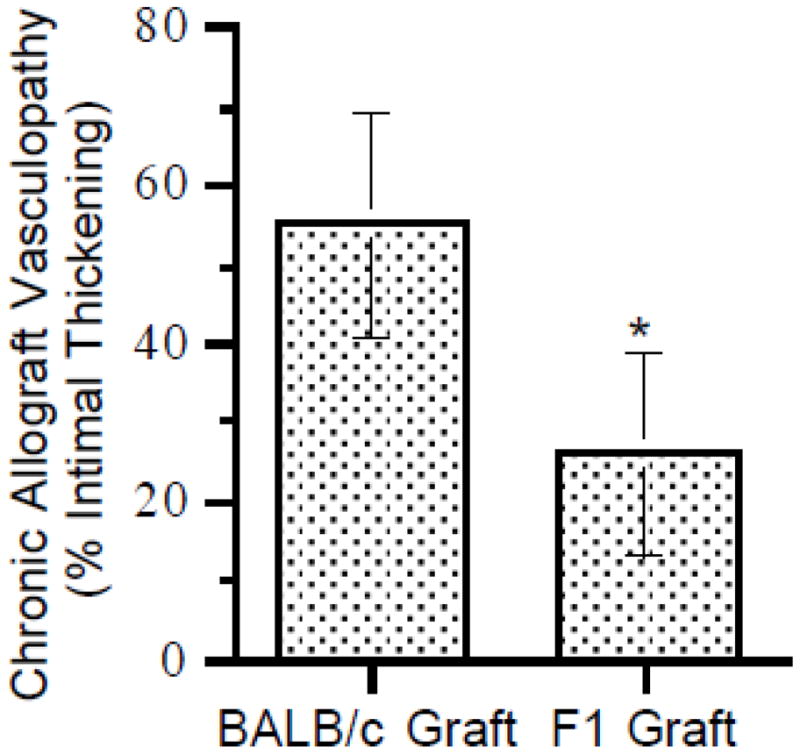
Alloactivated T cells were transferred to B6 aly/aly-spleen recipients of either (BALB/c × B6)F1 or BALB/c cardiac allografts 50 days after transplantation. Grafts were harvested 90 – 100 days after T cell transfer and stained with VVG. Vasculopathy (ratio of intima to vessel wall) in intra-parenchymal vessels was quantitated by histomorphometry. Data shown are mean ± SD (n = 5/group, p<0.05).
Figure 4. NK depletion reduces chronic rejection in a fully allogeneic transplantation model.

BALB/c cardiac allografts were parked for 50 days in splenectomized B6 aly/aly mice prior to the transfer of alloactivated T cells. Recipients were either NK-depleted during the parking period or remained NK-replete (isotype control). Grafts were harvested 70 – 100 days after T cell transfer and stained with H&E, MT, and VVG. (A) Representative low power micrographs of H&E and MT stained whole allograft sections of NK-depleted (left panel, n=6) and NK-replete mice (right panel, n=6) are shown. Note large areas of myocyte dropout (*) and early fibrosis (blue on MT stain) present in the NK-replete but not in the NK-depleted mouse. (B) Representative higher magnification (15x) of H&E, MT and VVG stained sections from NK-replete mice showing intense and widespread inflammation (arrowhead), evolving fibrosis (*) and obliterative with superimposed inflammatory arteritis (arrow). (C) Obliterative arteriopathy (ratio of intima to vessel wall) in intra-parenchymal vessels was quantitated by histomorphometry in NK-replete and NK-depleted mice (n = 6/group). Pooled results from control mice that either received no T cells (n=3) or mice that received naïve T cells 70 days following transplantation (n=4) are also shown. Data are mean ± SD. ** p<0.001, *** p<0.0001. (D) Representative low power micrographs of H&E and VVG stained allograft sections from control mice that received no T cells.
5. Discussion
We addressed in this report the fundamental question whether chronic allograft pathology can occur in the absence of an adaptive immune response. We found that heart allografts transplanted to mice that lack T and B cells do not develop histopathologic features of chronic rejection, even when the recipient’s innate immune system was stimulated with microbial ligands that activate pattern recognition receptors or if mice were reconstituted with natural IgM antibodies. We also found that NK cells, although not sufficient to cause graft pathology, predispose healed allografts to T cell-mediated chronic rejection long after transplantation.
Our finding that NK cells are not sufficient for either acute or chronic rejection of a solid organ allograft is consistent with the biology of NK cells. NK cell activation is dependent on both the absence of inhibitory signals delivered by self-MHC (missing self) and the presence of stimulatory ligands on the target tissue [23]. Although NK stimulatory ligands such as the MHC I-related proteins MICA and MICB have been detected in solid grafts [24], they may not be sufficient for full-scale activation of NK cells typically observed after viral infection [25]. In addition, the limited proliferative capacity of NK cells could restrict the extent of tissue damage they may cause even if they were to be activated. Exceptions could arise however if additional factors that induce the expansion and activation of NK cells are present in a transplant recipient. For example, Graham et al demonstrated that NK-mediated chronic coronary allograft vasculopathy develops in RAG−/− mice infected with LCMV but not in control uninfected recipients [10]. In addition, Kroemer et al. observed that RAG−/− mice acutely reject skin allografts if NK cells are massively expanded by excess, exogenous IL-15 [14]. We conclude that the most likely role of NK cells in solid organ transplantation is the maintenance of an inflammatory state that facilitates the migration of primed T cells to the graft. One weakness of our data is that we did not conduct extensive quantitation and phenotypic analysis of NK cells from individual allografts instead of pooled heart transplants.
Natural IgM antibodies have recently been proposed to mediate complement activation and subsequent tissue pathology in rodent models of ischemia-reperfusion injury [26]. Likewise, the transfer of either complement-fixing or non-complement-fixing donor-specific IgG antibodies was shown to induce chronic arterial lesions in heart allografts transplanted to RAG−/− recipients [27]. Our finding that natural IgM antibodies are not sufficient to cause graft pathology may again indicate the inability of innate immune mediators to effect chronic rejection on their own. Alternatively, it is possible that higher titers of natural IgM antibodies observed in mice with normal B-1 lymphocyte numbers could contribute to chronic rejection either directly or through collaboration with adaptive immune mechanisms.
Our data also shed light on the question whether chronic allograft rejection in humans is principally due to an ongoing host anti-donor immune response, or whether antigen-independent factors have an equally important role [8]. Although the mouse heterotopic heart transplantation model used in our studies differs in some respects from human orthotopic heart transplantation, our findings demonstrate that chronic allograft rejection is an immunological process that depends on adaptive immune mechanisms. It is likely that components of the innate immune system, for example macrophages [28], serve as downstream mediators of the pathological lesions of chronic rejection but, alone, are not sufficient to initiate or cause chronic rejection.
Highlights.
Innate immunity alone does not cause chronic rejection of cardiac allografts in mice
Neither stimulation with adjuvants nor natural antibodies induce chronic rejection
NK cell alloreactivity predisposes allografts to T cell-mediated injury
Footnotes
D.Z. designed and performed experiments and contributed to the writing of the manuscript; Q.L. performed microsurgical procedures; G.C. performed experiments, provided conceptual and experimental advice, and reviewed the manuscript; F.K.B. and A.J.D. performed histological analyses; D.M.R. and W.D.S. provided conceptual and experimental advice and reviewed the manuscript; F.G.L. supervised the project and wrote the manuscript. This work was funded by National Institutes of Health grant AI064343 (to D.M.R., W.D.S., A.J.D., and F.G.L.); GC is funded by National Institutes of Health grant AI079177; D.Z. was funded by a grant from the Deutsche Forschungsgemeinschaft. The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hall BM. Cells mediating allograft rejection. Transplantation. 1991;51:1141–51. doi: 10.1097/00007890-199106000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Miller JFAP. Effect of neonatal thymectomy on the immunological responsiveness of the mouse. Proc R Soc Lond B. 1962;156:415–28. [Google Scholar]
- 3.Hall BM, Dorsch S, Roser B. The cellular basis of allograft rejection in vivo. I. The cellular requirements for first-set rejection of heart grafts. J Exp Med. 1978;148:878–89. doi: 10.1084/jem.148.4.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bingaman AW, Ha J, Waitze S-Y, Durham MM, Cho HR, Tucker-Burden C, et al. Vigorous allograft rejection in the absence of danger. J Immunol. 2000;164:3065–71. doi: 10.4049/jimmunol.164.6.3065. [DOI] [PubMed] [Google Scholar]
- 5.Russell PS, Chase CM, Sykes M, Ito H, Shaffer J, Colvin RB. Tolerance, mixed chimerism, and chronic transplant arteriopathy. J Immunol. 2001;167:5731–40. doi: 10.4049/jimmunol.167.10.5731. [DOI] [PubMed] [Google Scholar]
- 6.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–8. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu K, Schonbeck U, Mach F, Libby P, Mitchell RN. Host CD40 ligand deficiency induces long-term allograft survival and donor-specific tolerance in mouse cardiac transplantation but does not prevent graft arteriosclerosis. J Immunol. 2000;165:3506–18. doi: 10.4049/jimmunol.165.6.3506. [DOI] [PubMed] [Google Scholar]
- 8.Salama A, Remuzzi G, Harmon W, Sayegh M. Challenges to achieving clinical transplantation tolerance. J Clin Invest. 2001;108:943–8. doi: 10.1172/JCI14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–97. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 10.Graham JA, Wilkinson RA, Hirohashi T, Chase CM, Colvin RB, Madsen JC, et al. Viral infection induces de novo lesions of coronary allograft vasculopathy through a natural killer cell-dependent pathway. Am J Transplant. 2009;9:2479–84. doi: 10.1111/j.1600-6143.2009.02801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uehara S, Chase CM, Kitchens WH, Rose HS, Colvin RB, Russell PS, et al. NK cells can trigger allograft vasculopathy: the role of hybrid resistance in solid organ allografts. J Immunol. 2005;175:3424–30. doi: 10.4049/jimmunol.175.5.3424. [DOI] [PubMed] [Google Scholar]
- 12.Maier S, Tertilt C, Chambron N, Gerauer K, Huser N, Heidecke CD, et al. Inhibition of natural killer cells results in acceptance of cardiac allografts in CD28−/− mice. Nat Med. 2001;7:557–62. doi: 10.1038/87880. [DOI] [PubMed] [Google Scholar]
- 13.McNerney ME, Lee KM, Zhou P, Molinero L, Mashayekhi M, Guzior D, et al. Role of natural killer cell subsets in cardiac allograft rejection. Am J Transplant. 2006;6:505–13. doi: 10.1111/j.1600-6143.2005.01226.x. [DOI] [PubMed] [Google Scholar]
- 14.Kroemer A, Xiao X, Degauque N, Edtinger K, Wei H, Demirci G, et al. The innate NK cells, allograft rejection, and a key role for IL-15. J Immunol. 2008;180:7818–26. doi: 10.4049/jimmunol.180.12.7818. [DOI] [PubMed] [Google Scholar]
- 15.Corry RJ, Winn HJ, Russel PS. Primarily vascularized allografts of hearts in mice: The role of H-2D, H-2K, and non H-2 antigens. Transplantation. 1973;16:343–50. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Chalasani G, Dai Z, Konieczny BT, Baddoura FK, Lakkis FG. Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proc Natl Acad Sci USA. 2002;99:6175–80. doi: 10.1073/pnas.092596999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zecher D, Li Q, Oberbarnscheidt MH, Demetris AJ, Shlomchik WD, Rothstein DM, et al. NK cells delay allograft rejection in lymphopenic hosts by downregulating the homeostatic proliferation of CD8+ T cells. J Immunol. 2010;184:6649–57. doi: 10.4049/jimmunol.0903729. [DOI] [PubMed] [Google Scholar]
- 18.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, et al. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–8. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zecher D, van Rooijen N, Rothstein DM, Shlomchik WD, Lakkis FG. An innate response to allogeneic nonself mediated by monocytes. J Immunol. 2009;183:7810–6. doi: 10.4049/jimmunol.0902194. [DOI] [PubMed] [Google Scholar]
- 20.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–52. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chalasani G, Li Q, Konieczny BT, Smith-Diggs L, Wrobel B, Dai Z, et al. The allograft defines the type of rejection (acute versus chronic) in the face of an established effector immune response. J Immunol. 2004;172:7813–20. doi: 10.4049/jimmunol.172.12.7813. [DOI] [PubMed] [Google Scholar]
- 22.Lakkis F. Reply to “Confounding Variables Complicate Conclusions in aly Model”. Nature Med. 2001;7:1165–6. doi: 10.1038/nm1101-1165a. [DOI] [PubMed] [Google Scholar]
- 23.Lanier L. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 24.Hankey KG, Drachenberg CB, Papadimitriou JC, Klassen DK, Philosophe B, Bartlett ST, et al. MIC expression in renal and pancreatic allografts. Transplantation. 2002;73:304–6. doi: 10.1097/00007890-200201270-00029. [DOI] [PubMed] [Google Scholar]
- 25.Kitchens WH, Uehara S, Chase CM, Colvin RB, Russell PS, Madsen JC. The changing role of natural killer cells in solid organ rejection and tolerance. Transplantation. 2006;81:811–7. doi: 10.1097/01.tp.0000202844.33794.0e. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Austen WJ, Chiu I, Alicot E, Hung R, Ma M, et al. identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci (USA) 2004;101:3886–91. doi: 10.1073/pnas.0400347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, et al. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant. 2010;10:510–7. doi: 10.1111/j.1600-6143.2009.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitchens WH, Chase CM, Uehara S, Cornell LD, Colvin RB, Russell PS, et al. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am J Transplant. 2007;7:2675–82. doi: 10.1111/j.1600-6143.2007.01997.x. [DOI] [PubMed] [Google Scholar]