

Abstract
Changes in DNA methylation status are not only important for regulating gene expression but are also suggested to induce chromosome instability. To reveal the correlation of DNA methylation status in heterochromatin regions with tumor histology and with chromosome alterations, DNA methylation status was examined by Southern blot analysis, and numerical and structural chromosome alterations, including the formation of der(16)t(1;16)/der(1;16), were examined by fluorescence in situ hybridization at the two loci in the pericentromeric satellite 2 regions of chromosomes 16 and 1 in 39 human breast carcinomas. DNA hypomethylation at the D16Z3 and the D1Z1 loci was detected in 31% (12 of 39) and 36% (12 of 33) of carcinomas, respectively, and mostly concurred. DNA hypomethylation was more frequent in the carcinoma group of more aggressive histological types or grade 3 than in the carcinoma of less aggressive histological types or grades 1 and 2, and tended to be more frequent in carcinomas with ≥4 copies of chromosomes 16 and/or 1 than in carcinomas with ≤3 copies of any of these chromosomes. The frequency of DNA hypomethylation at the D16Z3 and the D1Z1 loci was 45% (10 of 22) and 53% (9 of 17) in carcinomas without der(16)t(1;16)/der(1;16), formation, but only 12% (2 of 17) and 19% (3 of 16) in carcinoma with der(16)t(1;16)/der(1;16), respectively (P = 0.036 and 0.070). The 16q breakage was almost equally detected between carcinoma groups with and without the DNA hypomethylation. DNA hypomethylation in the satellite 2 regions was suggested to be associated with the accumulation of a large number of numerical chromosome alterations and involved in the development of breast carcinomas of aggressive histological features. On the contrary, chromosome instability induced by mechanisms other than DNA hypomethylation in heterochromatin regions might cause the formation of der(16)t(1;16)/der(1;16) and less aggressive breast carcinomas.
CpG islands are regions of the genome with a high density of CpG dinucleotides. These regions are frequently gene-associated and occur throughout the genome. In cancer, abnormal hypermethylation of the CpG islands in the promoter regions of some genes is linked to changes in gene silencing, especially in tumor-suppressor genes, eg, WT2 (H16), p16/CDKN2A/INK4A, E-cadherin (ECDH1), and hMLH1.1-10
Hypomethyaltion of other regions of the genome, including pericentromeric heterochromatin, constitutes another methylation abnormality associated with cancer. One can often find both changes in the same tumors, suggesting that hypermethylation events at specific genes and hypomethylation of pericentromeric heterochromatin can occur concurrently.
Pericentromeric heterochromatin regions are abundant in chromosomes 16 and 1 and contain satellite 2 sequences, noncoding repetitive DNA.11 In the analyses of Drosophila, Arabidopsis, and Homo sapiens, CpG dinucleotides and genes have been found to be relatively common in these heterochromatin regions.12-14 The CpG dinucleotides in the satellite 2 sequences are normally heavily methylated, but DNA hypomethylation in the heterochromatin regions has been reported to occur in cancers of the breast, ovary, and liver15-18 and has been linked in some cases to genomic and chromosomal instability.19-21 In lymphoblastoid cell lines or cancer cell lines, DNA uncoiling and rearrangements at the heterochromatin regions in chromosomes 16 and 1 are shown to be induced by DNA hypomethylation in these regions.21-24
In breast cancer, numerical and structural alterations of chromosomes 16 and 1 occur frequently, eg, aneusomies, 16q loss, and der(16)t(1;16)/der(1;16).25-27 These chromosome alterations are associated with histological features of breast carcinomas. Chromosome 16 aneusomies are frequently detected in high-grade (grade 3) carcinomas, whereas chromosome 16 disomy and der(16)t(1;16)/der(1;16) formation are usually detected in low-grade (grade 1) or intermediate-grade (grade 2) carcinomas and certain lower grade histological types, eg, papillary or tubular-type invasive ductal carcinoma (IDC) and invasive lobular carcinoma (ILC).27 These structural abnormalities involving chromosomes 16 and/or 1 are frequently generated by DNA rearrangements flanking the pericentromeric satellite 2 regions of 16q and 1q.25-27 We speculated that alterations in the DNA methylation status in these regions might play a role in the generation of numerical and structural alterations of chromosomes 16 and 1 in breast carcinomas. To validate this speculation, we studied the correlation between DNA methylation status in two satellite 2 regions, D16Z3 and D1Z1, detected by Southern blot analysis and numerical and structural alterations of chromosomes 16 and 1 detected by fluorescence in situ hybridization (FISH) in 43 breast tumors.
Materials and Methods
Tissue Samples
Primary breast tumors and nontumor breast tissues were obtained from 43 women who underwent biopsy or mastectomy at the National Cancer Center Hospital, Tokyo, for treatment and/or diagnosis of breast tumors. Resected tissue specimens were processed routinely for pathological diagnosis. Histological typing and grading were performed according to the criteria described by Sato and colleagues28 and by Elston.29 Thirty-nine tumors were diagnosed as carcinomas: 31 IDCs comprising 12 papillotubular-type (or papillary and tubular types), 9 solid tubular-type (or solid type), 10 scirrhous-type (or strand type), 5 intraductal carcinomas, 1 IDC with a predominantly intraductal component, and 2 ILCs. As for histological grade, 7 were grade 1, 15 were grade 2, and 17 were grade 3. Four were diagnosed to be benign tumors: three benign phyllodes tumors and one fibroadenoma.
DNA Probes
Two satellite 2 sequence clones, pHuR195 and pUC1.77, localized at the D16Z3 locus on 16q11.230 and at the D1Z1 locus on 1q12,31 respectively, were used as DNA probes. These DNA probes were 32P-labeled using a random primer kit (Boehringer, Mannheim, Germany) for Southern blot analysis. The DNA clones used as probes for FISH were pHuR195, pUC1.77, a pSE16-2 clone composed of an α-satellite repetitive sequence localized at the D16Z2 locus on 16cen,32 and a cCJ52-105 clone localized at the D16S154 locus on 16q24.3.33 These four DNA probes were labeled with biotin or digoxigenin using a nick translation kit (Boehringer) and precipitated in ethanol with 20 μg each of salmon testis DNA (Sigma, St. Louis, MO) and Escherichia coli tRNA (Sigma).
CpG Methylation in Satellite 2 Regions of Chromosomes 16 and 1
A part of fresh tumor tissue was embedded in an OCT compound (Miles Inc., Elkhart, IN), and the predominant (>50%) distribution of tumor cells was confirmed microscopically on frozen sections. Carcinomas with marked proliferation of fibroblasts or inflammatory cells were not included in the analysis. In invasive carcinomas, invasive components were examined, whereas predominantly intraductal carcinoma and intraductal carcinoma, intraductal components were examined. From these tumor tissue samples and corresponding nontumor breast tissues, high-molecular weight DNA was extracted as described previously.34 The DNA methylation status was examined by digesting the tumor and nontumor DNA with MspI (Takara, Kyoto, Japan), which cuts the sequence CCGG regardless of the methylation of internal cytosine, or by digesting these DNAs with HpaII (Takara), which cuts the CCGG when the internal cytosine is not methylated but does not cut the CCGG when the internal cytosine is methylated. In parallel, the methylation status was also examined by digesting tumor and nontumor DNA with BstBI (New England Biolabs, Beverly, MA), which cuts the sequence TTCGAA when the internal cytosine is not methylated but does not cut that sequence when the internal cytosine is methylated.15,16 A 5-μg DNA was digested for 24 hours with 100 U of the restriction enzyme, electrophoresed, transferred to nitrocellulose filters, and hybridized to 32P-labeled DNA probes in a hybridization solution (50% formamide, 5× Denhardt’s solution; 0.1 mol/L piperazine-N,N′-bis[2-ethanesulfonic acid], 5 mmol/L ethylenediaminetetraacetic acid/0.1% sodium dodecyl sulfate, 0.65 mol/L sodium chloride) for 12 to 18 hours, and washed at 65°C for 30 minutes in 0.1× standard saline citrate (SSC) (1× SSC being 0.15 mol/L NaCl and 0.015 mol/L sodium citrate), and 0.1% sodium dodecyl sulfate.34 The signals of hybridization were detected by autoradiography.
The pattern of the tumor DNA digested by HpaII was defined as normal level methylation, or normomethylation, when the pattern was the same as that of HpaII-digested nontumor DNA, and as hypomethylation when the pattern was similar to that of MspI-digested nontumor DNA34 (Figure 1A)▶ . The ratio of the intensity of signal of a HpaII-digested 8.0-kb DNA fragment from tumor tissue was compared with the intensity of signal of the corresponding MspI-digested 8.0-kb DNA derived from the nontumor tissue.34 The equality of DNA amount between lanes was confirmed by referring to the intensity of the DNA smear that was electrophoresed in agarose gel and was stained with ethidium bromide (Sigma). The quantitative analysis was performed with a NIH Image software version 1.62 (National Institutes of Health, Bethesda, MD) to the digitized images acquired from the autoradiograms using a Scantouch 210 scanner (Nikon, Tokyo, Japan). When the ratio was ≥0.1, the tumor DNA was defined as hypomethylated. When the ratio was <0.1, the tumor was defined as normomethylated.
Figure 1.
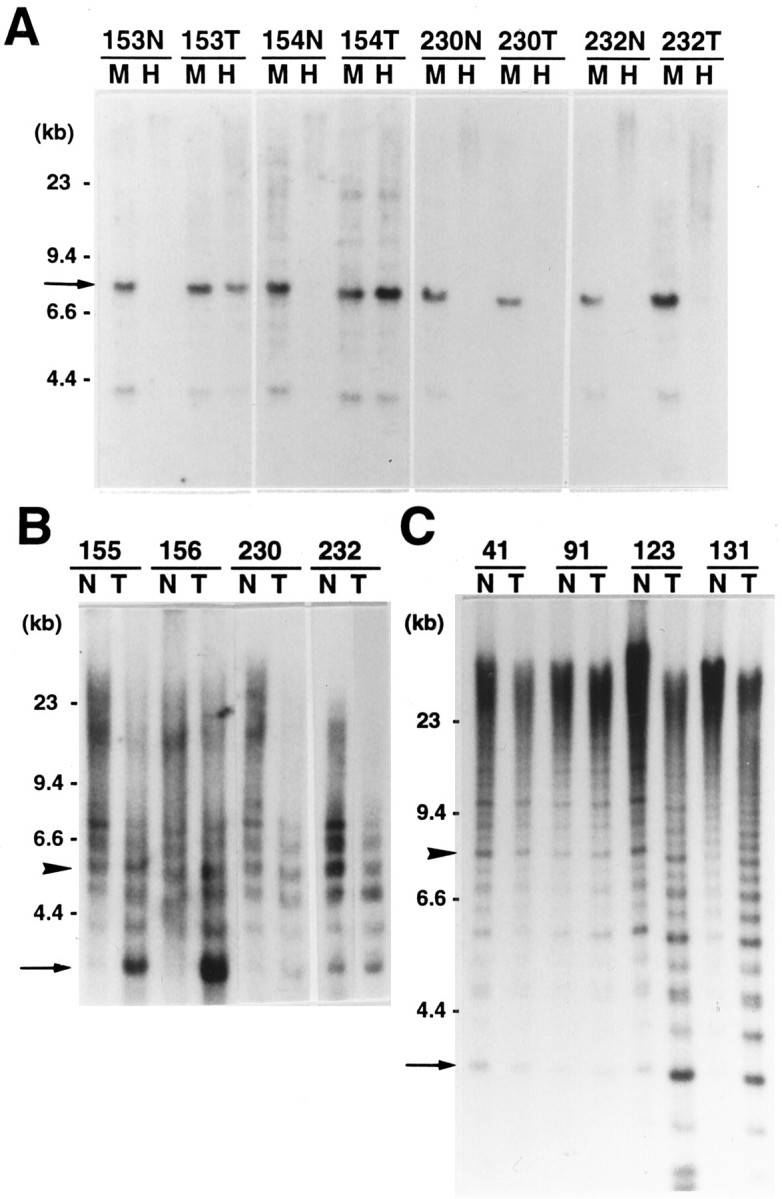
Detection of DNA hypomethylation at satellite 2 regions in chromosomes 16 and 1 in breast carcinomas using Southern blot analysis. A: MspI/HpaII digestion analysis at the D16Z3 locus. Nontumor DNA (N) and tumor DNA (T) were digested completely with MspI (M) or HpaII (H). Nontumor DNA was fragmented by MspI that is resistant to DNA methylation but was not cut well with HpaII that is sensitive to DNA methylation. In cases 153 and 154, the signal intensities of an 8.0-kb fragment (arrow) in HpaII-digested tumor DNA were almost the same as those of the 8.0-kb fragment in MspI-digested nontumor DNA, and the tumor DNA was assessed as hypomethylated. In cases 230 and 232, the 8.0-kb fragment in HpaII-digested tumor DNA is not visible and shows the same signal pattern as HpaII-digested nontumor DNA; it was assessed as normomethylated. B: BstBI digestion analysis at the D16Z3 locus. In cases 155 and 156, the signal intensity of a shorter (≤4 kb) DNA fragment (arrow) in tumor DNA (T) was much higher (>10-fold) than that of the DNA fragment in nontumor DNA (N). As internal control, the intensity of a longer (>4 kb) DNA fragment (arrowhead) was compared between tumor and nontumor DNA. BstBI could cut the tumor DNA extensively because of hypomethylation in tumor DNA. In cases 230 and 232, the signal intensity of the shorter DNA fragments was similar between tumor DNA and nontumor DNA, assessed as normomethylated. C: BstBI digestion analysis at the D1Z1 locus. In cases 123 and 131, the signal intensity of a shorter (≤6 kb) DNA fragment (arrow) in tumor DNA (T) was much higher (>10-fold) than that in nontumor DNA (N), and the tumor DNA was assessed as hypomethylated. As internal control, the intensity of a longer (>6 kb) DNA fragment (arrowhead) was compared between tumor and nontumor DNA. In cases 41 and 91, the signal intensity of the shorter DNA fragments was similar between tumor DNA and nontumor DNA, assessed as normomethylated.
BstBI-digested tumor DNA was categorized as normomethylated when the pattern was the same as that of BstBI-digested nontumor DNA. For the D16Z3 locus, by modifying the criteria by Qu and colleagues,15 the tumor was categorized as hypomethylated when the intensity of signals that were 4 kb or shorter in tumor DNA was stronger than that of signals in nontumor DNA (Figure 1B)▶ . The intensity of signal of a 3.2-kb hybridizing fragment (a), representing ≤4-kb DNA fragments, in tumor DNA was compared with that of the 3.2-kb hybridizing fragment (b) in nontumor DNA. As internal control, the intensity of signal of a 5.5-kb hybridizing fragment, representing >4-kb DNA fragments, was compared between tumor DNA (c) and nontumor tissue DNA (d) using the NIH Image software. The ratio was calculated by the formula: (a/b)/(c/d) = ad/bc. When the ratio was >10.0, the tumor DNA was defined as hypomethylated.
For the D1Z1 locus, the tumor was categorized as hypomethylated when the ratio of the intensity of a 3.5-kb hybridizing fragment (a), representing ≤6-kb fragments, in tumor DNA to the intensity of the 3.5-kb hybridizing fragment (b) in nontumor DNA was >10.0 according to the calculation above. As internal control, the intensity of signal of an 8.0-kb hybridizing fragment, representing >6-kb DNA fragments, was compared between tumor DNA (c) and nontumor tissue DNA (d) (Figure 1C)▶ .
FISH
Fresh tumor tissue was imprinted on glass slides for FISH analysis. Two-color FISH analyses were performed using combinations of D16Z2 (biotin) and D1Z1 (digoxigenin), D16Z3 (digoxigenin) and D1Z1 (biotin), and D16Z3 (digoxigenin) and D16S154 (biotin).35 Briefly, the cell samples on glass slides were digested with 0.5 μg/ml of proteinase K (Merck, Darmstadt, Germany), fixed with paraformaldehyde, and denatured in 70% formamide/2× SSC at 75°C for 2 minutes. A total of 1 μg of two DNA probes at an appropriate ratio was mixed with 2.5 μg of Cot-1 DNA (Life Technologies, Inc., Grand Island, NY), denatured at 70°C, mixed with an equal volume of 2× SSC/20% dextran sulfate, incubated at 37°C for 15 minutes, and hybridized with the samples on glass slides at 37°C for 12 to 16 hours. After a serial wash in 2×, 1×, and 4× SSC, the slides were reacted with avidin-fluorescein isothiocyanate and anti-digoxigenin rhodamine (Boehringer), washed, stained with 4,6-diamidino-2-phenylindole, and mounted with an antifade solution. Using a BX50-34-FLA-1 fluorescence microscope (Nikon), the fluorescent signals of 100 to 400 nuclei were counted separately by two observers (HT and TT).
The numerical alterations of chromosomes 16 and 1 and the breakage of 16q were assessed as follows. The total numbers of each of the D1Z1, D16Z2, D16Z3, and D16S154 signals in 100 to 200 interphase tumor cell nuclei were counted independently by the two observers. Although the number of signals at the D1Z1 and D16Z2 only shows disomy or aneusomies of 1q12 and 16cen, respectively, we regarded that these numbers represented primarily the numbers of chromosomes 1 and 16. The mean signal counts of chromosomes 16 and 1 in individual tumors were calculated by dividing the total counts of D16Z2 signals and those of D1Z1 signals, respectively, by the total number of counted nuclei. The mean copy score, which was regarded to represent the mean number of chromosome 16 or chromosome 1 per tumor cell, was defined as 1, 2, 3, 4, 5, and ≥6 when the mean D1Z1 or D16Z2 signal count was ≤1.4, 1.5 to 2.4, 2.5 to 3.4, 3.5 to 4.4, 4.5 to 5.4, and ≥5.5, respectively. The mean copy score of 2 was assessed to be disomy, and other mean copy scores were assessed as aneusomy. The proximal breakage of chromosome 16q was defined as a difference of one or more per cell in the mean copy scores between the D16Z2 and the D16Z3 loci.27 The distal 16q breakage was defined as a difference of one or more per cell in the mean copy scores between the D16Z3 and the D16S154 loci.27
In a previous study, 10 adjacent normal tissue specimens were used as a control, and mean and SD were 16.2 and 3.33 for D16Z2-D1Z1 co-localization, and 18.5 and 3.89 for D16Z3-D1Z1 co-localization per 100 nuclei. In 10 lymphocyte specimens from peripheral blood, mean and SD were 13.3 and 2.98 for D16Z2-D1Z1 co-localization, and 11.2 and 2.49 for D16Z3-D1Z1 co-localization per 100 nuclei. Therefore, tumors in which the percentage of co-localized signals was greater than the mean of control specimens plus 3 SD (24% for the former co-localization and 30% for the latter co-localization) were judged as having a cell clone with der(16)t(1;16)/der(1;16).35
We included a lymphocyte specimen as a control in each assay and confirmed the efficiency of hybridization of probes and that the percentage of D16Z2-D1Z1 or D16Z3-D1Z1 co-localization was under the cutoff value. On the other hand, the co-localization was detected in ≥50% of tumor cells in all tumors that were judged to have der(16)t(1;16)/der(1;16) formation clonally.
Statistical Analysis
The correlation of DNA hypomethylation in the satellite 2 regions with other parameters was analyzed by a chi-square test or Fisher’s exact test.
Results
HpaII/MspI and BstBI digestion analyses could reveal the status of DNA methylation at the D16Z3 locus in 43 and 42 tumors, but only BstBI digestion analysis could reveal the status at the D1Z1 locus in 37 tumors. The concordance between the DNA methylation status acquired by MspI/HpaII digestion and that by BstBI digestion was 95% (40 of 42). In four benign tumors, the DNA methylation status at the D16Z3 or D1Z1 locus was estimated as a normal level by both of these analyses. DNA hypomethylation at the D16Z3 locus was detected in 12 (31%) of 39 carcinomas by MspI/HpaII digestion and/or by BstBI digestion. In two carcinomas, DNA was assessed as hypomethylated by BstBI but as normomethylated by MspI/HpaII, and these tumors were regarded to have DNA hypomethylation.
DNA hypomethylation at the D1Z1 locus was detected in 12 (36%) of 33 carcinomas. The statuses of DNA hypomethylation were concordant between the D16Z3 and the D1Z1 loci in 30 (91%) of 33 carcinomas (P < 0.0001) (Table 1)▶ .
Table 1.
Correlation of DNA Hypomethylation between the D1Z1 Locus and the D16Z3 Locus in Individual Breast Carcinomas
| Hypomethylation at D1Z1 | Number of tumors (%) | P value | |
|---|---|---|---|
| Hypomethylation at D16Z3 | |||
| Present | Absent | ||
| Present | 10 | 2 | <0.0001 |
| Absent | 1 | 20 | |
A strong correlation was seen between DNA hypomethylation at each of the two satellite 2 loci and the histological type of breast carcinoma. DNA hypomethylation at the D16Z3 locus was not detected in the carcinoma group comprising intraductal/predominantly intraductal carcinomas and papillotubular-type IDC (0 of 18). On the other hand, DNA hypomethylation at that locus was detected in 57% (12 of 21) of the carcinomas in the group comprising solid tubular-type IDC, scirrhous-type IDC, and ILC (P < 0.0001) (Table 2)▶ . Similarly, DNA hypomethylation at the D1Z1 locus was detected only in 6% (1 of 17) of carcinomas in the former group, but it was detected in 69% (11 of 16) of carcinomas in the latter group (P = 0.0002) (Table 2A)▶ .
Table 2.
Correlation of DNA Hypomethylation at the D16Z3 Locus and at the D1Z1 Locus with Histological Features of and Changes in Chromosomes 16 and 1 in Breast Carcinoma
| Parameters | No. of cases (%) | |||
|---|---|---|---|---|
| DNA hypomethylation | ||||
| D16Z3 | P | D1Z1 | P | |
| A. Histological type | ||||
| Intraductal/predominantly intraductal carcinoma | 0/6 (0) | <0.0001 | 0/6 (0) | 0.0002 |
| IDC, papillotubular type | 0/12 (0) | 1/11 (9) | ||
| IDC, solid tubular type | 5/9 (56) | 5/7 (71) | ||
| IDC, scirrhous type | 6/10 (60) | 5/7 (71) | ||
| Invasive lobular carcinoma | 1/2 (50) | 1/2 (50) | ||
| B. Histological grade | ||||
| 1 | 0/7 (0) | 0.019 | 0/6 (0) | 0.044 |
| 2 | 3/15 (20) | 4/13 (31) | ||
| 3 | 9/17 (53) | 8/14 (57) | ||
| C. Clinical stage | ||||
| I | 2/11 (18) | NS | 4/9 (44) | NS |
| II | 7/21 (33) | 6/18 (33) | ||
| III,IV | 3/7 (43) | 2/6 (33) | ||
| D. Mean copy score at D16Z2 | ||||
| ≤ 3 | 4/23 (17) | 0.034 | 5/18 (28) | NS |
| ≥ 4 | 8/16 (50) | 7/15 (47) | ||
| E. Mean copy score at D1Z1 | ||||
| ≤ 3 | 2/15 (13) | 0.065 | 3/12 (25) | NS |
| ≥ 4 | 10/24 (42) | 9/21 (43) | ||
| F. der(16)t(1;16)/der(1;16) formation | ||||
| Present | 2/17 (12) | 0.036 | 3/16 (19) | 0.070 |
| Absent | 10/22 (45) | 9/17 (53) | ||
| G. 16q breakages | ||||
| Detected | 9/30 (30) | 9/26 (35) | ||
| der(16)t(1;16)-positive | 2/17 (12) | 0.019 | 3/16 (19) | 0.046 |
| der(16)t(1;16)-negative | 7/13 (54) | 6/10 (60) | ||
| Not detected | 3/9 (33) | 3/7 (43) | ||
IDC, invasive ductal carcinoma; NS, not significant.
DNA hypomethylation in the satellite 2 regions was also more frequent in carcinomas of higher histological grade. DNA hypomethylation at the D16Z3 and the D1Z1 loci was detected in none of the grade 1 carcinomas but in 20% and 23% of the grade 2 carcinomas and in 53% and 57% of the grade 3 carcinomas, respectively (P = 0.019 and P = 0.044) (Table 2B)▶ . Clinical stages were not correlated with DNA methylation status at these loci (Table 2C)▶ .
The assessments of the FISH results by two observers were always agreed. FISH analysis for four benign tumors always revealed 16cen disomy without 16q breakage or der(16)t(1;16)/der(1;16) formation. Of 39 carcinomas, disomy and aneusomy at the D16Z2 locus were detected in 14 (36%) and 25 (64%) and, at the D1Z1 locus, in 7 (18%) and 32 (82%), respectively (Figure 2)▶ . The mean copy scores were ≤3 in 23 (59%) and ≥4 in 16 (41%) at the D16Z2 locus and ≤3 in 15 (38%) and ≥4 in 24 (62%) at the D1Z1 locus, respectively. The frequency of DNA hypomethylation at the D16Z3 locus was higher in the group with mean copy scores of ≥4 at the D16Z2 locus (50%) than in the group with mean copy scores of ≤3 (17%) (P = 0.034) (Table 2D)▶ . DNA hypomethylation at the D16Z3 locus also tended to be higher in the group with mean copy scores of ≥4 at the D1Z1 (42%) than in the group with mean copy scores of ≤3 (13%) (P = 0.065). (Table 2E)▶ .
Figure 2.
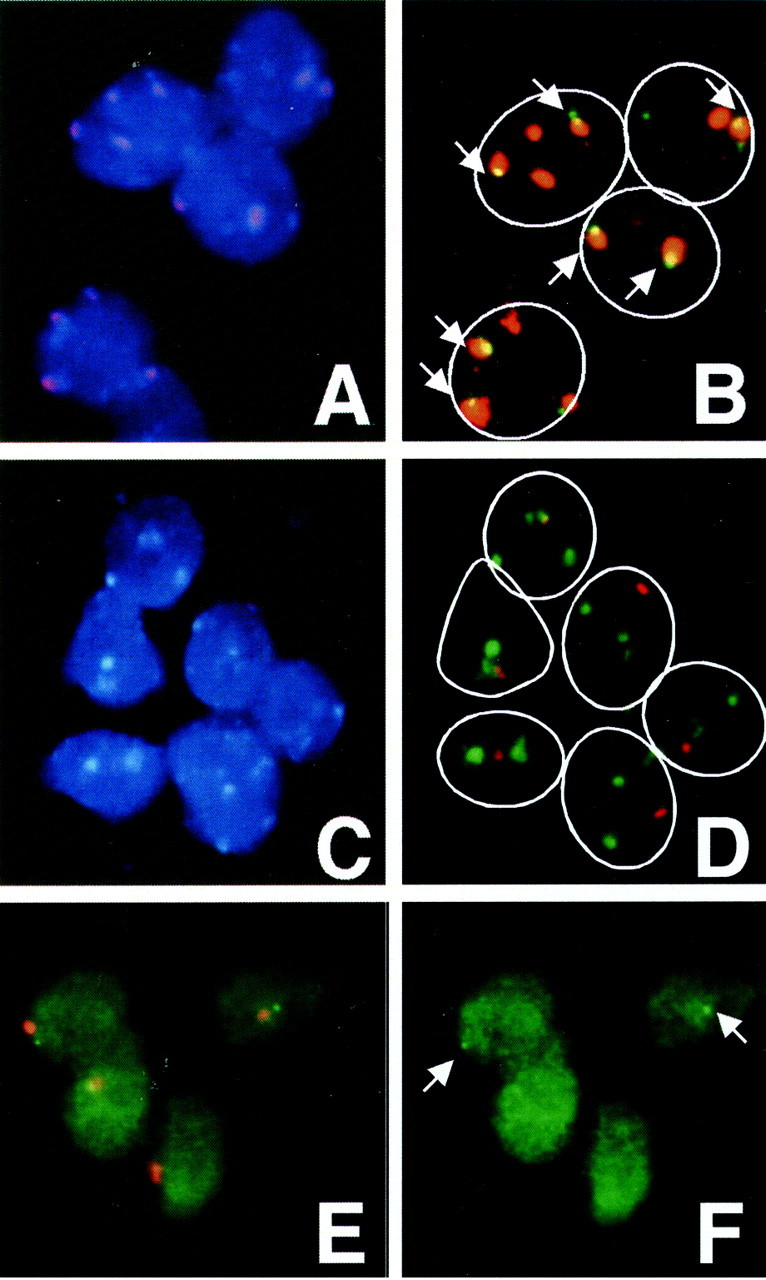
Numerical and structural alterations of chromosomes 16 and 1 in breast carcinomas detected by FISH. A and B: The two-color FISH visualizes two or three green signals of the D16Z2 locus and three or four red signals of the D1Z1 locus per nucleus. One or two signals are co-localized per nucleus (arrows). C and D: The two-color FISH visualizes one red signal of the D16Z3 locus and three or four green signals of the D1Z1 locus per nucleus. E and F: The two-color FISH visualizes one red signal of the D16Z3 locus and one green signal of the D16S154 locus (arrows) per nucleus. This carcinoma was assessed to have the mean copy score of 3 of chromosomes 16 and the score of 4 of chromosome 1 and to carry proximal 16q breakage and der(16)t(1;16)/der(1;16). The contour of tumor cell nuclei is surrounded by white lines in B and D. A and C: DAPI-FITC-Texas Red triple-band-pass filter; B, D, and E: FITC-Texas Red double-band-pass filter; F: FITC filter.
Of 39 carcinomas, 17 (44%) showed clonal der(16)t(1;16)/der(1;16) formation. The incidences of DNA hypomethylation at each of the D1Z1 and D16Z3 loci were lower in the tumor group with der(16)t(1;16)/der(1;16) formation than in the group without (P = 0.036 and 0.070) (Table 2F)▶ . Proximal and distal 16q breakages were detected by FISH in 17 (44%) and 21 (54%) carcinomas, respectively, and these 16q breakages were detected in 30 carcinomas (77%). There was no correlation between extensive DNA hypomethylation at the satellite 2 loci and the presence of proximal or distal 16q breakage (Table 2G)▶ .
Among the 30 cases with 16q breakages, 17 were accompanied by der(16)t(1;16)/der(1;16), but the other 13 were not. DNA hypomethylation at the D16Z3 locus was detected in 7 (54%) of 13 carcinomas with 16q breakages without der(16)t(1;16)/der(1;16) but in only 2 (12%) of 17 carcinomas with both 16q breakages and der(16)t(1;16)/der(1;16) (P = 0.019). DNA hypomethylation at the D1Z1 locus was detected in 6 (60%) of 10 carcinomas with 16q breakages without der(16)t(1;16)/der(1;16) but in only 3 (19%) of 16 carcinomas with both 16q breakages and der(16)t(1;16)/der(1;16) (P = 0. 046).
Discussion
In the present study, DNA hypomethylation at the D16Z3 and D1Z1 satellite 2 loci in heterochromatin regions was detected in 28% and 33% of breast carcinomas, respectively, and usually concurred but was not detected in four benign tumors. DNA hypomethylation at these loci was correlated with aggressive histological features, ie, solid tubular-type IDC, scirrhous-type IDC, and grade 3 carcinomas. In the carcinomas of less aggressive histological features, ie, intraductal carcinomas, papillotubular-type IDC, and grade 1 to 2 carcinomas, the DNA at these satellite 2 regions was usually normomethylated. DNA hypomethylation at these loci, therefore, seemed to be involved only in the genesis of breast carcinomas of aggressive histological features.
Numerical alterations of chromosome 16 are more frequent in breast carcinomas of grade 3 and of aggressive histological types than in those of grade 1 to 2 and of other histological types.27 DNA hypomethylation at the D16Z3 locus and mean copy scores of ≥4 of chromosome 16 tended to show correlation. Because the incidence of chromosome 16 and 1 aneusomies was much higher than that of DNA hypomethylation at the D16Z3 or the D1Z1 locus, the latter did not seem to always cause numerical chromosome alterations.
The 16q breakages, which indicate the loss of 16q arms, were not correlated with DNA hypomethylation at the D16Z3 or the D1Z1 locus. However, among the 16q breakages, those with fusion to 1q only showed an inverse correlation with DNA hypomethylation at these loci. The 16q breakages without der(16)t(1;16)/der(1;16) formation appeared to be frequently accompanied by DNA hypomethylation. Therefore, the manner of chromosome breakage and fusion, or rearrangements, seems to be influenced by, or to have an influence on, the DNA methylation status in these satellite 2 regions. In Wilms’ tumor, it is reported that there was a correlation between DNA hypomethylation at the satellite 2 regions on chromosomes 1 and 16 and loss of heterozygosity on 1q and 16q.36 Recently, Wong and colleagues37 also reported the correlation between the hypomethylation of chromosome 1 heterochromatin DNA and 1q copy gain in hepatocellular carcinoma. It is unclear whether these losses were accompanied by certain chromosome translocations, eg, der(16)t(1;16)/der(1;16).
In breast carcinomas that have a positive hormone receptor status and show the presence of hyperproliferative mammary glands in the background of the breast, der(16)t(1;16)/der(1;16) occurs frequently. Therefore, high sensitivity to estrogens of mammary glandular epithelial cells might induce der(16)t(1;16)/der(1;16)-positive breast carcinomas. Histologically, these carcinomas with der(16)t(1;16)/der(1;16) are mostly papillotubular-type IDC, ILC, intraductal carcinoma, and grade 1 to 2 carcinomas.27
Chromosome instability of different categories might be involved in the establishment of histological features of breast carcinomas. One category could be chromosome alterations that occur in association with DNA hypomethylation in heterochromatin and could make mostly solid tubular-type IDC or scirrhous-type IDC, and grade 3 carcinomas. DNA hypomethylation in the heterochromatin satellite 2 regions might induce, or occur in conjunction with, a larger number of numerical and structural chromosome alterations, eg, high-level polysomies of chromosome 16 and 1, and 16q loss without der(16)t(1;16)/der(1;16). Amplification of the 17q region including the ERBB2 (HER-2/neu) locus and loss of heterozygosity on chromosome arms 17p, 17q, 7q, and 11p might also be present in this pathway because these alterations are mostly detected in high-grade carcinomas.28,38-40 Another category of chromosome instability might be chromosome alterations that occur in association with DNA damage caused by estrogens.41,42 This category of chromosome instability might give rise to der(16)t(1;16)/der(1;16) and a smaller number of numerical and other structural alterations, and appeared to cause the genesis of breast carcinomas of less aggressive histological types and grades.
In conclusion, we could suggest that DNA hypomethylation in the heterochromatin regions of chromosomes 16 and 1 induced, or occurred in association with, numerous chromosome abnormalities and was involved in the genesis of breast carcinomas of aggressive histological features. However, another carcinogenetic pathway mediated by a different type of chromosome instability was suggested to exist in breast carcinomas of less aggressive histological features. In the latter pathway, DNA hypomethylation in the heterochromatin regions does not appear to be involved, and DNA damage induced by other mechanisms might cause the formation of der(16)t(1;16)/der(1;16) and breast carcinomas of less aggressive histological features.
Acknowledgments
We thank the staff of the Department of Surgery, National Cancer Center Hospital, for surgically resected specimens; Dr. N. I. McGill (MRC Human Genetic Unit, Western General Hospital, Edinburgh, UK), Dr. Y. Nakamura (Institute of Medical Science, Tokyo University, Tokyo), and Dr. H. F. Willard (Case Western Reserve University, Cleveland, OH) for providing DNA probes; and the American Type Culture Collection (Rockville, MD) for providing clone pHuR195.
Footnotes
Address reprint requests to Hitoshi Tsuda, M.D., Department of Pathology II, National Defense Medical College, 3-2 Namiki, Tokorozawa, Saitama 359-8513, Japan. E-mail: htsuda@cc.ndmc.ac.jp.
Supported in part by a Grant-in-Aid from the Ministry of Health, Labor, and Welfare of Japan (grant 10-3).
References
- 1.Feinberg AP, Vogelstein B: Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301:89-92 [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB: The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002, 3:415-428 [DOI] [PubMed] [Google Scholar]
- 3.Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP: Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nat Genet 1994, 7:433-439ibid 1994, 8:203 [DOI] [PubMed] [Google Scholar]
- 4.Moulton T, Crenshaw T, Hao Y, Moosikasuwan J, Lin N, Dembitzer F, Hensle T, Weiss L, McMorrow L, Loew T, et al: Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nat Genet 1994, 7:440-447 [DOI] [PubMed] [Google Scholar]
- 5.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D: 5′CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995, 1:686-692 [DOI] [PubMed] [Google Scholar]
- 6.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB: Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995, 55:4525-4530 [PubMed] [Google Scholar]
- 7.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA: Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995, 55:4531-4535 [PubMed] [Google Scholar]
- 8.Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S: Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA 1995, 92:7416-7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanai Y, Ushijima S, Hui A-M, Ochiai A, Tsuda H, Sakamoto M, Hirohashi S: The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer 1990, 71:355-359 [DOI] [PubMed] [Google Scholar]
- 10.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB: Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998, 95:6870-6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karpen GH, Allshire RC: The case of epigenetic effects on centromere identity and function. Trends Genet 1997, 13:489-496 [DOI] [PubMed] [Google Scholar]
- 12.Sun X, Wahlstrom J, Karpen G: Molecular structure of a functional Drosophila centromere. Cell 1997, 91:1007-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Copenhaver CP, Nickel K, Kuromori T, Benito N-I, Kaul S, Lin X, Bevan M, Murphy G, Harris B, Parnell LD, McCombie WR, Martienssen RA, Marra M, Preuss D: Genetic definition and sequence analysis of Arabidopsis centromeres. Science 1999, 286:2468-2474 [DOI] [PubMed] [Google Scholar]
- 14.Barry AE, Howman EV, Cancilla MR, Saffery R, Choo KHA: Sequence analysis of an 80-kb human neocentromere. Hum Mol Genet 1999, 8:217-227 [DOI] [PubMed] [Google Scholar]
- 15.Qu G, Dubeau L, Narayan A, Yu M-C, Ehrlich M: Satellite DNA hypomethylation vs. overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential. Mutation Res 1999, 423:91-101 [DOI] [PubMed] [Google Scholar]
- 16.Bernardino J, Roux C, Almeida A, Vogt N, Gibaud A, Gerbault-Seureau M, Magdelenat H, Bourgeois CA, Malfoy B, Dutrillaux B: DNA hypomethylation in breast cancer: an independent parameter of tumor progression? Cancer Genet Cytogenet 1997, 97:83-89 [DOI] [PubMed] [Google Scholar]
- 17.Narayan A, Ji W, Zhang X-Y, Marrogi A, Graff JR, Baylin SB, Ehrlich M: Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int J Cancer 1998, 77:833-838 [DOI] [PubMed] [Google Scholar]
- 18.Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S: Expression of mRNA for DNA methyltransferases and methyl-CpG-binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology 2001, 33:561-568 [DOI] [PubMed] [Google Scholar]
- 19.Lengauer C, Kinzler KW, Vogelstein B: DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci USA 1997, 94:2545-2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jeanisch R: DNA hypomethylation leads to elevated mutation rates. Nature 1998, 395:89-93 [DOI] [PubMed] [Google Scholar]
- 21.Almeida A, Kokalj-Vokac N, Lefrancois D, Viegas-Pequignot E, Jeanpierre M, Dutrillaux B, Malfoy B: Hypomethylation of classical satellite DNA and chromosome instability in lymphoblastoid cell lines. Hum Genet 1993, 91:538-546 [DOI] [PubMed] [Google Scholar]
- 22.Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Péquignot E: Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402:187-191 [DOI] [PubMed] [Google Scholar]
- 23.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM: The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA 1999, 96:14412-14417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilain A, Vogt N, Dutrillaux B, Malfoy B: DNA methylation and chromosome instability in breast cancer cell lines. FEBS Lett 1999, 460:231-234 [DOI] [PubMed] [Google Scholar]
- 25.Kokalj-Vokac N, Alemeida A, Gerbault-Seureau M, Malfoy B, Dutrillaux B: Two-color FISH characterization of i(1q) and der(1;16) in human breast cancer cells. Genes Chromosom Cancer 1993, 7:8-14 [DOI] [PubMed] [Google Scholar]
- 26.Tsarouha H, Pandis N, Bardi G, Teixeira MR, Andersen JA, Heim S: Karyotypic evolution in breast carcinomas with i(1)(q10) and der(1;16)(q10;p10) as the primary chromosome abnormality. Cancer Genet Cytogenet 1999, 113:156-161 [DOI] [PubMed] [Google Scholar]
- 27.Tsuda H, Takarabe T, Hirohashi S: Correlation of the numerical and structural status of chromosome 16 with the histological type and grade of non-invasive and invasive breast carcinomas. Int J Cancer 1999, 84:381-387 [DOI] [PubMed] [Google Scholar]
- 28.Sato T, Tanigami A, Yamakawa K, Akiyama F, Kasumi F, Sakamoto G, Nakamura Y: Allelotype of breast cancer: cumulative allele losses promote tumor progression in primary breast cancer. Cancer Res 1990, 50:7184-7189 [PubMed] [Google Scholar]
- 29.Elston C: Grading of invasive carcinoma of the breast. Page DL Anderson TJ eds. Diagnostic Histopathology of the Breast. 1987:pp 300-311 Churchill Livingstone, Edinburgh
- 30.Moyzis RK, Albright KL, Barthordi MF, Cram LS, Deaven LL, Hildebrand CE, Joste NE, Longmire JL, Meyne J, Schwarzacher-Robinson T: Human chromosome-specific repetitive DNA sequences: novel markers for genetic analysis. Chromosoma 1987, 95:375-386 [DOI] [PubMed] [Google Scholar]
- 31.Cooke HJ, Hindley J: Cloning of human satellite III DNA: different components are on different chromosomes. Nucleic Acids Res 1979, 6:3177-3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greig GM, England SB, Bedford M, Willard HF: Chromosome-specific alpha satellite DNA from the centromere of human chromosome 16. Am J Hum Genet 1989, 45:862-872 [PMC free article] [PubMed] [Google Scholar]
- 33.Julier C, Nakamura Y, Lathrop M, O’Connell P, Leppert M, Mohandas T, Lalouel J-M, White RA: Primary map of 24 loci on human chromosome 16. Genomics 1990, 6:419-427 [DOI] [PubMed] [Google Scholar]
- 34.Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Sugimura T, Hirohashi S: Aberrant DNA methylation on chromosome 16 is an early event in hepatocarcinogenesis. Jpn J Cancer Res 1996, 87:1210-1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuda H, Takarabe T, Susumu N, Inazawa J, Okada S, Hirohashi S: Detection of numerical and structural alterations and fusion of chromosomes 16 and 1 in low-grade papillary breast carcinoma by fluorescence in situ hybridization. Am J Pathol 1997, 151:1027-1034 [PMC free article] [PubMed] [Google Scholar]
- 36.Qu GZ, Grundy PE, Narayan A, Ehrlich M: Frequent hypomethylationin Wilms’ tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet Cytogenet 1999, 109:34-39 [DOI] [PubMed] [Google Scholar]
- 37.Wong N, Lam W-C, Lai PB-S, Pang E, Lau W-Y, Johnson PL: Hypomethylation of chromosome 1 heterochromatin DNA correlates with q-arm copy gain in human hepatocellular carcinoma. Am J Pathol 2001, 159:465-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buerger H, Otterbach F, Simon R, Schäfer K-L, Poremba C, Diallo R, Brinkschmidt C, Dockhorn-Dworniczak B, Boecker W: Different genetic pathways in the evolution of invasive breast cancer are associated with distinct morphological subtypes. J Pathol 1999, 189:521-526 [DOI] [PubMed] [Google Scholar]
- 39.Bièche I, Champème MH, Matifas F, Hacène K, Callahan R, Lidereau R: Loss of heterozygosity on chromosome 7q and aggressive primary breast cancer. Lancet 1992, 339:139-143 [DOI] [PubMed] [Google Scholar]
- 40.Ali IU, Lidereau R, Theillet C, Callahan R: Reduction to homozygosity of genes on chromosome 11 in human breast neoplasia. Science 1987, 238:185-188 [DOI] [PubMed] [Google Scholar]
- 41.Telang NT, Suto A, Wong GY, Osborne MP, Bradlow L: Induction by estrogen metabolite 16β-hydroxyestrone of genotoxic damage and aberrant proliferation in mouse mammary epithelial cells. J Natl Cancer Inst 1992, 84:634-638 [DOI] [PubMed] [Google Scholar]
- 42.Yager JD: Endogenous estrogens as carcinogenesis through metabolic activation. J Natl Cancer Inst Monogr 2000, 27:67-73 [DOI] [PubMed] [Google Scholar]