

Abstract
Understanding the electrostatic forces and features within highly heterogeneous, anisotropic, and chemically complex enzyme active sites and their connection to biological catalysis remains a longstanding challenge, in part due to the paucity of incisive experimental probes of electrostatic properties within proteins. To quantitatively assess the landscape of electrostatic fields at discrete locations and orientations within an enzyme active site, we have incorporated site-specific thiocyanate vibrational probes into multiple positions within bacterial ketosteroid isomerase. A battery of X-ray crystallographic, vibrational Stark spectroscopy, and NMR studies revealed electrostatic field heterogeneity of 8 MV/cm between active site probe locations and widely differing sensitivities of discrete probes to common electrostatic perturbations from mutation, ligand binding, and pH changes. Electrostatic calculations based on active site ionization states assigned by literature precedent and computational pKa prediction were unable to quantitatively account for the observed vibrational band shifts. However, electrostatic models of the D40N mutant gave qualitative agreement with the observed vibrational effects when an unusual ionization of an active site tyrosine with a pKa near 7 was included. UV-absorbance and 13C NMR experiments confirmed the presence of a tyrosinate in the active site, in agreement with electrostatic models. This work provides the most direct measure of the heterogeneous and anisotropic nature of the electrostatic environment within an enzyme active site, and these measurements provide incisive benchmarks for further developing accurate computational models and a foundation for future tests of electrostatics in enzymatic catalysis.
Extensive structural studies of enzymes have revealed that biological catalysis occurs within sequestered active site crevices that solvate reacting substrates with an anisotropic and chemically complex constellation of charged, polar, and hydrophobic groups. But beyond visualization of the chemical composition and architecture of active sites permitted by the rich library of available protein structures, our understanding of the electrostatic nature and properties of this highly heterogeneous environment and its role in molecular recognition and catalysis is largely based on simulations. Computations using three-dimensional structures routinely provide electrostatic potentials, as in Fig. 1A. These potentials have been used to identify and characterize protein–protein and protein–ligand binding sites, and interaction energies derived from these calculations have suggested key contributions from the electrostatic environment to enzymatic rate enhancement and specificity (1–5). However, even the most sophisticated computational methods have multiple approximations and limitations (6–8) and, most importantly, have not been rigorously evaluated through cycles of nontrivial computational predictions and experimental tests. Incisive and quantitative experimental measures of the electrostatic fields within proteins are needed to deepen our understanding of these environments and allow evaluation of computational predictions of electrostatic contributions to binding and catalysis.
Fig. 1.

Electrostatic potential map, catalytic scheme, and experimental X-ray structural models of KSI. (A) Electrostatic potential map of the D40N active site generated by solving the Poisson–Boltzmann equation using the program DelPhi (55, 56) and plotted on the solvent-accessible surface of the X-ray structural model for M116C-CN/D40N (apoprotein, PDB ID code 3OXA). Equilenin is modeled in the active site, shown in stick representation, with carbon atoms in green. The foreground residues have been removed to allow viewing into the active site. A drop in potential from 80 mV to −80 mV in the space of two Å equates to an 8 MV/cm field. (B) Reaction mechanism for pKSI catalysis of steroid isomerization, shown for the substrate 5-androstene-3,17-dione. (C) Superposition of the aligned X-ray crystal structures of M116C-CN/D40N, F86C-CN/D40N (equilenin bound, PDB ID code 3OWU) and M105C-CN/D40N (equilenin bound, PDB ID code 3OWY). The carbon atoms of Tyr16, Asp103, Asn40, and the bound ligand equilenin are shown in gray and derived from 3OWU. The carbon atoms of the Cys-CN residue for M116C-CN, M105C-CN, and F86C-CN are shown as green, magenta, and cyan, respectively. Atoms for nitrogen, oxygen, and sulfur appear as blue, red, and gold, respectively, for all structures. Inset: View of the thiocyanate groups of the three constructs translated in the same axis system so that sulfur atoms (yellow) coincide at the same location. The common Cartesian coordinate system is shown with gray sticks, demonstrating the relative orientation of each probe’s bond vectors to the coordinate system of the protein structure. (D) The local environment surrounding each of the three probes is shown in semitransparent surface representation. Note that cavities in the surface result in the nitrile probe being solvent-exposed for M116C-CN and F86C-CN, whereas M105C-CN lies beneath the surface.
Numerous biophysical studies have provided important qualitative insights into the differences between solvation environments sampled within proteins versus bulk water or organic solvents. These studies have inferred electrostatic potentials based on free energy measurements [e.g., pKa shifts of ionizable residues (9–11), reactivity differences among cysteines (12, 13), equilibrium binding constants of charged ligands (14), and redox potential shifts (15)] or spectroscopic variations in probes incorporated into or bound to proteins [e.g., fluorescent dyes (16), 19F and 13C chemical shifts (17–21), and EPR-measured g-factor perturbations of nitroxides (22)]. These studies have spurred the ongoing development of electrostatic calculation methodology (6, 7) and contributed greatly to our general recognition that the solvation environment within proteins is very different from water. However, each of these experimental observables is influenced by additional environmental factors beyond electric field effects, without independent experimental means to distinguish these contributions (8, 12, 23). For example, local electrostatic fields can perturb pKa values, but the observed pKa is also sensitive to the making or breaking of hydrogen bonds between the titrating group and nearby groups and to the access of the site to water. These pervasive limitations have precluded accurate quantitative measurement of electrostatic fields in proteins, how such fields vary as a function of position and orientation within enzyme active sites, and how local electrostatic perturbations influence local fields.
Vibrational probes incorporated into proteins at defined positions can provide sensitive and directional reporters of the local electrostatic environment. Nitrile (-CN) groups provide a particularly incisive and powerful probe. Their small size allows incorporation with minimal structural perturbation; they can be easily introduced at discrete and well-defined positions within proteins; their strong absorbance bands lie in an uncluttered region of the IR spectrum; and they are sensitive to both the magnitude and direction of the local electric field (24). Furthermore, in contrast to many of the preceding examples, the field sensitivity of the nitrile stretching frequency can be calibrated in situ, obviating the need for a computational model or calibration in terms of an equivalent solution dielectric value that may not be transferable from bulk solvent to the heterogeneous structure of proteins.
The probe’s electric field sensitivity is largely due to the anharmonicity of the nitrile bond that leads to an increase in dipole moment, 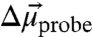 , between the ground and first excited vibrational state. This difference dipole moment results in a dependence of the transition energy on the size and direction of the local electrostatic field (Fig. S1 and SI Text). To quantify the electric field sensitivity of a specific vibrational probe, a known external electric field is applied and the effect of the field on the IR absorption spectrum is measured (25–27), a method known as vibrational Stark effect (VSE) spectroscopy. The electric field sensitivity of a vibrational probe directly calibrated in this fashion is called the linear Stark tuning rate,
, between the ground and first excited vibrational state. This difference dipole moment results in a dependence of the transition energy on the size and direction of the local electrostatic field (Fig. S1 and SI Text). To quantify the electric field sensitivity of a specific vibrational probe, a known external electric field is applied and the effect of the field on the IR absorption spectrum is measured (25–27), a method known as vibrational Stark effect (VSE) spectroscopy. The electric field sensitivity of a vibrational probe directly calibrated in this fashion is called the linear Stark tuning rate, 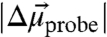 [in units of cm-1/(MV/cm)]. Eq. 1 can then be employed to translate observed IR frequency shifts,
[in units of cm-1/(MV/cm)]. Eq. 1 can then be employed to translate observed IR frequency shifts, 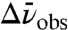 (in cm-1), into changes in the local protein electrostatic field,
(in cm-1), into changes in the local protein electrostatic field, 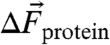 (in MV/cm), experienced by a probe at two different sites in a protein or as a result of a side-chain ionization, mutation or ligand binding:
(in MV/cm), experienced by a probe at two different sites in a protein or as a result of a side-chain ionization, mutation or ligand binding:
| [1] |
where h is Planck’s constant, c the speed of light, and θ is the angle between the vectors defined by 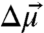 and
and 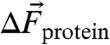 . The direction of the vibrational probe
. The direction of the vibrational probe 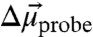 is typically along the oscillator bond axis for simple, linear nitriles (27). Thus, X-ray crystallography can provide the orientation of
is typically along the oscillator bond axis for simple, linear nitriles (27). Thus, X-ray crystallography can provide the orientation of 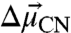 within a nitrile-modified protein and the VSE in an external field calibrates the magnitude of
within a nitrile-modified protein and the VSE in an external field calibrates the magnitude of 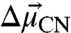 . Finally, although specific chemical interactions, such as hydrogen bonds, between nitrile probes and the surrounding environment can shift the nitrile IR frequency via a nonelectrostatic mechanism not captured by Eq. 1, we previously published an experimental method to detect and quantitatively correct for such effects (28). Thus, nitrile probes can provide quantitative measures of local electrostatic fields in the presence of specific interactions with surrounding groups.
. Finally, although specific chemical interactions, such as hydrogen bonds, between nitrile probes and the surrounding environment can shift the nitrile IR frequency via a nonelectrostatic mechanism not captured by Eq. 1, we previously published an experimental method to detect and quantitatively correct for such effects (28). Thus, nitrile probes can provide quantitative measures of local electrostatic fields in the presence of specific interactions with surrounding groups.
Bacterial ketosteroid isomerase (KSI) has provided a powerful model system for probing fundamental aspects of enzymatic catalysis. The basic catalytic mechanism (Fig. 1B) is well established, many high resolution structures are available, good transition state analogs exist, and the importance of electrostatics to catalysis is the subject of extensive and ongoing study (29–32). In earlier work, we demonstrated that vibrational probes could be introduced into the active site in proximity to key catalytic residues (28, 33). We now report detailed studies that use vibrational probes to measure the differences in local electrostatic fields sampled at discrete positions within the active site and to test the sensitivity of local fields to nearby electrostatic perturbations from ligand binding, pH changes, and mutation. Using Eq. 1, we make direct comparisons between observed and computed changes in the electrostatic potential. Discrepancies, even at a qualitative level, between the predicted and observed effects led us to reconsider the ionization states of key residues. Our results provide quantitative insight into the fundamental electrostatic properties of a protein interior, identify unexpected features of the KSI active site, establish a benchmark for future computations, and provide a basis for designing future tests of electrostatic contributions to catalysis.
Results
Incorporation of Nitrile Electric Field Probes into Ketosteroid Isomerase.
Bacterial KSI from Pseudomonas putida catalyzes double-bond isomerization in steroids using a general base, D40, to deprotonate the substrate and form a dienolate reaction intermediate. This intermediate is stabilized by hydrogen bonds formed to Y16 and protonated D103 within an active site oxyanion hole that is linked via a hydrogen bond network to Y57 and Y32 (Fig. 1B) (34–37). We incorporated thiocyanate vibrational probes into KSI from P. putida at positions M116, M105, and F86 (Fig. 1C). These positions were selected on the basis of their proximity (within 3–11 Å) to the key catalytic residues listed above. Sites M116 and M105 were used in prior investigations of the effects of solvation on active site electrostatic properties (28, 33). In the analysis that follows, we also refer to data for a thiocyanate probe incorporated at residue L61 obtained in a previous study (28). This residue is located near the mouth of the active site and forms part of the solvent-accessible surface of the steroid binding pocket. Thiocyanate labeling of KSI was accomplished by engineering single cysteine mutations at each of the above positions in the cysteine-free variant C69S/C81S/C97S to ensure unique labeling. The Cys residues at positions 69, 81, and 97 are located on the enzyme’s surface, and their removal has a negligible effect on catalysis (38).
The single Cys-SH group introduced in each mutant was converted to Cys-S-CN using previously published methods (33, 39), and the individual probe-labeled enzymes are referred to as M116C-CN, M105C-CN, F86C-CN, and L61C-CN. Labeled KSI variants were prepared with the general base D40 intact or mutated to D40N to mimic the protonated D40 present in the dienolate intermediate complex (Fig. 1B). Functional assays with the thiocyanate-modified enzymes revealed activities (kcat/KM) within three- to sixfold of Cys-free KSI with D40 present or affinities within twofold for binding of the transition state analog equilenin to the D40N mutants, suggesting minimal perturbation to KSI structure and function.
X-Ray Crystal Structures of KSI-CN Variants.
We determined X-ray crystal structures for the D40N variants of M116C-CN, M105C-CN, and F86C-CN with or without the bound steroid transition state analog equilenin (Table 1, complete data collection and refinement statistics for each structure are listed in Table S1). The overall structures obtained for these variants were the same as those determined previously for P. putida KSI bound to equilenin (1OH0) or phenol (2PZV), with average rmsd values of 0.26 Å, 0.31 Å, and 0.31 Å, respectively, for main-chain atoms of apo M116C-CN, M105C-CN•equilenin, and F86C-CN•equilenin from the previously determined structures (which have a main-chain rmsd of 0.27 Å from each other). Thus, thiocyanate incorporation does not perturb the global structure of KSI.
Table 1.
X-ray crystallographic data for nitrile-modified KSI variants
| D40N Variant | Ligand | Resolution (Å) | Rwork/Rfree (%) | PDB ID code |
| M116C-CN | – | 1.9 | 24.4/29.0 | 3OXA |
| M105C-CN | equilenin | 2.3 | 28.0/34.6 | 3OWY |
| F86C-CN | – | 2.0 | 26.5/34.1 | 3OX9 |
| F86C-CN | equilenin | 1.7 | 21.6/27.0 | 3OWU |
Focusing on the nitrile probes, the electron-density maps determined for each structure (Fig. S2) supported refinement of the thiocyanate group to a single well-ordered conformation (Fig. 1C). Superposition of active site residues observed in the probe modified versus unmodified KSI structures revealed conformational differences on the same scale as the 0.1–0.2 Å estimated coordinate uncertainty in these structures, indicating minimal structural rearrangements within the active site in response to probe insertion. The thiocyanate group in both the M116C-CN and M105C-CN structures refined to a position very similar to that occupied by the parent methionine residue in the unmodified structures, indicating a sterically conservative substitution. For F86C-CN the thiocyanate group refined to a position distinct from that occupied by the phenyl ring with F86 present. Nevertheless, we did not observe structural rearrangement of nearby groups to fill this space, nor did this change perturb the position of bound equilenin. Finally, comparison of F86C-CN structures with and without bound equilenin revealed identical thiocyanate positions, within error. We conclude that thiocyanate incorporation at each of these three positions minimally perturbs the structure and function of the KSI active site and results in ordered and well-defined nitrile probe conformations.
Each of the three probes is positioned within a unique local environment. The nitrile of M105C-CN is buried well below the solvent-accessible surface of the KSI active site and is located 6 to 7 Å from the polar side-chain hydroxyl moieties of Tyr 16, 32 and 57, Asp 103, and the oxyanion of the bound steroid (Fig. 1C). Its immediate solvation environment is thus composed of a tightly packed shell of hydrophobic aliphatic and aromatic residues (Fig. 1D).
The cyano group in M116C-CN has the most central active site location of the probes, with the nitrile positioned less than 5 Å from the polar side-chain hydroxyl groups of Tyr 16, 32 and 57 and Asp 103, and less than 4 Å from the general base, Asp 40, and the expected position of the steroid oxyanion (Fig. 1C). It forms part of the solvent-accessible active site surface and is thus susceptible to hydrogen bond formation to adventitious water molecules within the active site.
The nitrile of F86C-CN refined to a position orthogonal to that of the original F86 phenyl ring, projecting away from the bound substrate and into a small recess contoured by the backbone groups of D103 and M84 (Fig. 1C). The 3-Å distance and triangular arrangement between the nitrile and amide groups of D103 and M84 suggested formation of a bifurcated hydrogen bond in which lone pairs on both the nitrile nitrogen and M84 carbonyl oxygen overlap with the amide proton of D103. IR and 13C NMR measurements described below support hydrogen bond formation to the probe. Positioned within this recess, the nitrile of F86C-CN is located 4 Å from a side-chain oxygen of D103, 5 Å from the oxyanion of the bound steroid, and 7–10 Å from the active site tyrosines.
These three probes represent distinct orientations relative to the molecular frame of the enzyme (Fig. 1C, Inset), span a range of distances to the polar groups of the active site, and experience varying degrees of solvent sequestration, hydrogen bond formation, and hydrophobic packing interactions. The unique and anisotropic chemical environment observed at each site therefore suggested that each probe samples a discrete electrostatic environment that, via Eq. 1, would lead to dispersion in the vibrational stretching frequency from site to site. In the sections that follow, we first present IR spectra for the three probes that demonstrate substantial -CN frequency dispersion. We then describe 13C NMR measurements for each nitrile that allowed us to rule out hydrogen bonding to the M105C-CN probe and identify and correct for hydrogen bond formation to the M116C-CN, and F86C-CN probes. Then we present in situ calibration of 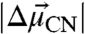 for each nitrile to determine the inherent electric field sensitivity of each probe. With these measurements in hand, we are able to quantitatively determine the differences in electric fields sampled by the three probes in their unique positions and orientations within the active site and the response to changes in the local electrostatic environment.
for each nitrile to determine the inherent electric field sensitivity of each probe. With these measurements in hand, we are able to quantitatively determine the differences in electric fields sampled by the three probes in their unique positions and orientations within the active site and the response to changes in the local electrostatic environment.
Tandem Measurement of IR Stretch Frequency and 13C NMR Chemical Shift.
FTIR spectra were acquired for each apo KSI-CN variant with either D40 or D40N present. These spectra, shown in Fig. 2A–C and summarized in Table 2, reveal symmetric peaks that span a 12 cm-1 range in stretching frequency and exhibit a broad range in linewidth. Although linewidths are determined by the complex distribution of inhomogeneous environments and the time scale for interconversion between them and are often difficult to interpret, our structural studies and NMR data provide a guide for understanding the observed linewidth differences. The narrow linewidth of the M105C-CN peak relative to the other probes, or compared to the spectrum for urea-unfolded protein (33), is consistent with the tight packing of hydrophobic groups around the probe within a region of the active site previously suggested to have limited dynamic mobility (40). The relatively broad F86C-CN spectrum may reflect that its substitution for bulkier phenylalanine results in looser packing around this probe than for the other probes. The observation of the same ordering of linewidths for the 13C NMR peaks for each probe (Fig. S3) supports the hypothesis that broader linewidth reflects greater conformational inhomogeneity (additional discussion in SI Text).
Fig. 2.

IR absorption spectra of KSI-CN variants in D40 versus D40N and at pH 6 versus 8. (A–C) X-ray structures of the ionizable groups in the region around each nitrile probe are shown with the Asn at position 40. In gray are the active site ionizable groups (Y32 omitted for clarity). The Cys-CN residue in each variant (carbon atoms colored as in Fig. 1) is aligned according to the axis system shown in the upper left inset of A. Dashed lines indicate the distance from the probe to the side chain of residue 40, which are 11, 9.2, and 5.7 Å, for positions 105, 86, and 116, respectively. (D–F) Absorption spectra of the nitrile stretching region for M105C-CN, F86C-CN, and M116C-CN (from left to right, respectively) in D40 (red) versus D40N (black) at pH 7.1. (G–I) A rotated view of the ionizable groups of D40N. D103 is circled in red in the cases where the observations are most at odds with the expectations for D103 ionization (positions 105 and 86). (J–L) Absorption spectra of the nitrile stretching region for KSI-CN variants at pH 5.6 (red) or pH 7.8 (black) in 100 mM MES buffer.
Table 2.
Vibrational frequencies and 13C NMR chemical shifts for nitrile-modified KSI variants at pH 7 in 40 mM potassium phosphate
| M116C-CN | F86C-CN | M105C-CN | L61C-CN | |||||||||
| νCN[cm-1] | νCN corrected [cm-1] | δ(13C)[ppm] | νCN[cm-1] | νCN corrected [cm-1] | δ(13C)[ppm] | νCN[cm-1] | νCN corrected [cm-1] | δ(13C)[ppm] | νCN[cm-1] | νCN corrected [cm-1] | δ(13C)[ppm] | |
| apoprotein | ||||||||||||
| D40N | 2,159.6* | 2,149.6 | 115.62 | 2,165.9 | 2,152.9 | 114.10 | 2,154.0 † | NA | 113.60 † | 2,158.6 † | 2,148.6 | 117.10 † |
| D40 | 2,161.3 | 2,151.6 | 114.43 | 2,165.3 | 2,152.3 | 113.98 | 2,153.2 | NA | 113.56 | __ | __ | __ |
| Bound to ligands | ||||||||||||
| 3-F,4-NO2-phe | 2,163.3 | 2,153.3 | 113.92 | 2,166.4 | 2,153.4 | __ | 2,153.3 | NA | 113.56 | __ | __ | __ |
| 4-NO2-phe | 2,163.8 | 2,153.8 | 113.94 | 2,166.6 | 2,153.6 | __ | 2,153.3 | NA | __ | __ | __ | __ |
| equilenin | 2,162.4* | 2,152.4 | 114.07 † | 2,167.7 | 2,154.7 | 112.78 | 2,153.9 † | NA | 113.61 † | __ | __ | __ |
Prior to any electrostatic analysis, it was necessary to first test and correct for effects from hydrogen bond formation to the nitrile probes. We recently published a method, based on tandem measurement of the nitrile IR frequency and 13C NMR chemical shift, that can detect the presence of specific probe interactions such as hydrogen bonds and quantitatively separate observed nitrile IR shifts into electrostatic and nonelectrostatic contributions (28). The common, linear electric field sensitivity of the IR and NMR observables leads to a correlation between them in the absence of specific interactions with the probe. Direct hydrogen bond formation to a nitrile leads to a departure from this linear correlation, ascribed to anomalous behavior in the IR but not the NMR dimension (41–43). By comparing tandem IR and NMR measurements for KSI-CN probes to a previously determined correlation of IR stretching frequency versus 13C NMR chemical shift of ethyl thiocyanate in aprotic solvents, such departures can be identified.
We previously applied this method to identify that the M116C-CN nitrile accepts a hydrogen bond and that the M105C-CN nitrile is free from hydrogen bonding (Table 2 and Fig. 3) (28). Hydrogen bond formation to the nitrile of M116C-CN but not M105C-CN is consistent with the solvent exposure of the former and hydrophobic burial of the latter observed in the X-ray structures (Fig. 1D). When we applied this method to F86C-CN, we observed a substantial offset in the IR frequency of F86C-CN from that expected based on its 13C NMR chemical shift and the trend line shown in green in Fig. 3, supporting the presence of the bifurcated hydrogen bond suggested from the X-ray structure.
Fig. 3.

13C-NMR Chemical shift versus nitrile stretching frequency for nitrile probes in apo or ligand-bound KSI-CN variants: F86C-CN (blue, -1.6 cm-1/ppm, R2 = 0.9), M116C-CN (black, -2.2 cm-1/ppm, R2 = 0.89), and M105C-CN (red), in solid circles (data from Table 2). These results are compared to data for the model compound ethylthiocyanate in different aprotic solvents, taken from ref. 28, in green diamonds (-1.7 cm-1/ppm, R2 = 0.68). Hydrogen bond corrections of −13 and -10 cm-1, applied to the nitrile stretching frequency of F86C-CN and M116C-CN, respectively, as described in the text, give the values shown with blue and black open circles, respectively. For a combined dataset including ethylthiocyanate, M105C-CN, and the corrected F86C-CN and M116C-CN values, the best fit line gives a slope of -1.8 cm-1/ppm, R2 = 0.83. Data points are numbered for ethylthiocyanate in dimethylsulfoxide (1), dimethylformamide (2), acetone (3), methylene chloride (4), tetrahydrofuran (5), chloroform (6), toluene (7), and cyclohexane (8). Data for KSI-CN variants are numbered 9–19: apo M116C-CN/D40N (9), apo M116C-CN (10), M116C-CN/D40N•equilenin (11), M116C-CN/D40N•3-F-4-NO2-phenol (12), M116C-CN/D40N•4-NO2-phenol (13), apo F86C-CN (14), apo F86C-CN/D40N (15), F86C-CN/D40N•equilenin (16), apo M105C-CN/D40N (17), apo M105C-CN (18), and M105C-CN/D40N•3-F-4-NO2-phenol (19).
Individual trend lines for the IR and 13C NMR data were determined by compiling multiple IR and 13C NMR measurements with mutants and bound ligands for each nitrile probe (Fig. 3). The slopes of all of these lines were the same, within error, strongly suggesting that in none of the examples of ligand binding or mutation studied by this method does the nitrile change from free to hydrogen-bonded or vice versa at any of the three sites. The larger 13 cm-1 offset observed for F86C-CN from the IR/NMR correlation line, relative to the 10 cm-1 offset observed for M116C-CN, may arise from the different hydrogen bond donors in the two cases: a backbone amide in the case of F86C-CN versus water for M116C-CN. Our prior conclusion (28), that IR shifts due to hydrogen bonding and local electrostatic effects are additive, is reinforced with the contribution here of additional data that strengthen the observed correlations. Based on this information, we can quantitatively correct for hydrogen bond formation to the F86C-CN and M116C-CN probes by subtracting their IR frequency offset from the IR/NMR correlation line (Fig. 3). Upon correction, the total dispersion in nitrile frequency between the three probes was reduced from 12 to 4.4 cm-1 (Table 2) with D40N present and 1.6 cm-1 with D40 present. This correction also reversed the relative peak positions of M105C-CN and F86C-CN, leaving the M105C-CN nitrile with the highest energy IR frequency.
In Situ Determination of the Stark Tuning Rate for Each Nitrile Probe.
While both NMR and IR are sensitive to electrostatic fields, the VSE experiment provides a quantitative experimental calibration of the sensitivity of the vibrational frequency with respect to a known, external electric field. Such a direct calibration is not currently possible for 13C NMR chemical shifts. Vibrational Stark tuning rates for individual probes are typically obtained using small molecules in frozen organic glasses (44–46). However, it remained untested whether the Stark tuning rate of a specific probe, such as a nitrile, varies substantially upon large perturbations to the local environment of the probe (47).
To address this question, we determined vibrational Stark spectra (field on minus field off) in situ for each KSI-CN variant embedded in a frozen glass at 80 K (Fig. 4). The Stark spectra were fit as a sum of derivatives of the absorption spectrum, and the contribution of the second derivative component was quantified to calculate 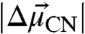 (see SI Materials and Methods). The values of
(see SI Materials and Methods). The values of 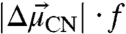 (where f is the local field correction factor, which for simplicity we assume to be equal to one*) determined for each probe were within 20% of each other and of the model compound ethyl thiocyanate (EtSCN) measured in frozen organic solvent (49) (Fig. 4). Thus, the electric field sensitivity of the three nitrile probes, quantified by the Stark tuning rate, appears to be an intrinsic property of the oscillator that varies minimally across the observed range of measured IR frequencies and unique structural environments, regardless of whether the probe is hydrogen-bonded or not. According to Eq. 1 and using the average calibrated value for
(where f is the local field correction factor, which for simplicity we assume to be equal to one*) determined for each probe were within 20% of each other and of the model compound ethyl thiocyanate (EtSCN) measured in frozen organic solvent (49) (Fig. 4). Thus, the electric field sensitivity of the three nitrile probes, quantified by the Stark tuning rate, appears to be an intrinsic property of the oscillator that varies minimally across the observed range of measured IR frequencies and unique structural environments, regardless of whether the probe is hydrogen-bonded or not. According to Eq. 1 and using the average calibrated value for 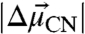 , a change in the projection of the field along the C-to-N bond vector of +1 MV/cm would shift the IR absorption,
, a change in the projection of the field along the C-to-N bond vector of +1 MV/cm would shift the IR absorption, 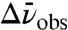 , by +0.65 cm-1.
, by +0.65 cm-1.
Fig. 4.

Absorption (A) and Stark (B) spectra for KSI-CN variants. (A) Absorption spectra of the apo enzymes at 77 K in 50% glycerol/water at 10–15 mM, scaled to a common concentration and path-length. From left to right, M105C-CN, M106C-CN, and F86C-CN in red, black, and blue, respectively. (B) Vibrational Stark spectra of the same samples, scaled to a common value of applied field equal to 1 MV/cm. M116C-CN and F86C-CN are further scaled 4X and 8X, respectively, for ease of visualization (broader absorption features necessarily result in smaller features in a Stark spectrum as the Stark spectrum is proportional to the second derivative of the absorption). Analytical frequency-weighted second derivatives of the absorption spectra of A are plotted with dashed lines next to the corresponding Stark spectrum.
Quantitative Measurement of Electrostatic Field Heterogeneity Within the KSI Active Site.
We first consider the data with the D40N mutant, because our results (X-ray, 13C NMR, and Stark) are most extensive in this background. After correction for hydrogen bonding, we observed a 4.4 cm-1 range in IR frequencies for the nitrile probes at positions 116, 105, and 86 in the D40N background (Table 2). According to Eq. 1, this dispersion in IR frequency corresponds to a 7 ± 3 MV/cm variation in the magnitude of the local electrostatic field projected onto each probe (error analysis described in SI Materials and Methods). When the corrected IR frequency of L61C-CN was included in the analysis (28), the observed IR range expanded to 5.4 cm-1 (L61C-CN/D40N vs. M105C-CN/D40N; Table 2), which translates to an 8 ± 4 MV/cm difference in the projection of the local electric field on each nitrile. Comparing only the hydrogen-bonded probes, a difference of 7 ± 4 MV/cm (L61C-CN/D40N vs. F86C-CN/D40N; Table 2) is observed. This dispersion in local electric field sampled at discrete probe positions and orientations provides direct quantitative evidence for a heterogeneous and anisotropic electrostatic environment within KSI. To understand how this electric field range depends on local charge groups within the KSI active site, in the following sections we measure the electrostatic effects on each probe of common charge perturbations due to mutation, pH titration, and ligand binding.
Electrostatic Fields Within the KSI Active Site Are Altered by Restoring Asp at Position 40.
Upon changing residue 40 from Asn back to Asp, IR shifts of different magnitude and direction were registered by each probe (Fig. 2
D–F). The peak widths in either background were similar, suggesting that mutations at residue 40 do not affect the dynamics or flexibility at any of the probe sites. Relatively small shifts, defined as 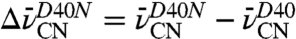 , of +0.6 and +0.8 cm-1 were observed for the nitriles of F86C-CN and M105C-CN, respectively, and a larger -1.7 cm-1 shift was observed for the M116C-CN nitrile. These shifts by each probe, none of which involves a change in hydrogen bonding state (Fig. 3 and Table 2), indicate that each nitrile experiences the electrostatic effects of this mutation differently. On the basis of these shifts, we determined that the 7 MV/cm field dispersion sampled in D40N by probes at positions 86, 105, and 116 is reduced to 3 MV/cm with the negatively charged Asp a position 40.
, of +0.6 and +0.8 cm-1 were observed for the nitriles of F86C-CN and M105C-CN, respectively, and a larger -1.7 cm-1 shift was observed for the M116C-CN nitrile. These shifts by each probe, none of which involves a change in hydrogen bonding state (Fig. 3 and Table 2), indicate that each nitrile experiences the electrostatic effects of this mutation differently. On the basis of these shifts, we determined that the 7 MV/cm field dispersion sampled in D40N by probes at positions 86, 105, and 116 is reduced to 3 MV/cm with the negatively charged Asp a position 40.
To understand the electrostatic effects of the D40N mutation, we first had to establish the protonation states of the ionizable residues in the active site before and after the mutation. Prior functional studies of catalysis by wild-type KSI from P. putida revealed a pKa of approximately 4 that was assigned to the general base, D40 (50). Upon mutation of D40 to Asn, phenolate and equilenin binding studies suggested an enzymatic pKa of approximately 6 that was provisionally assigned to the oxyanion hole residue, D103 (Fig. 1B) (32). This assignment was based on work with the homologous KSI from Comamonas testosteroni that suggested that D103 has an elevated pKa of approximately 9 in WT, due to electrostatic repulsion from negative charge on D40 and burial in a hydrophobic pocket, but a lower pKa in D40N in the absence of charge repulsion from D40 (51). We tested these assignments using state-of-the-art algorithms to predict pKa values for active site residues [H++ (52), PROPKA (53), and Karlsberg+ (54); see SI Materials and Methods and Table S2] in wild type and D40N KSI from P. putida. These computational methods predicted that the D40N mutation, which ablates the negative charge at position 40, lowers the pKa of D103 from > 9 in wild type to 7 ± 1 in D40N (Table S2). The D40N mutation is thus predicted by these computational methods to result in charge migration from position 40 to position 103 in apo P. putida KSI at neutral pH. All other residues in and around the active site, including Y16, Y32, and Y57 (Fig. 1), were predicted by computation to titrate with pKa values greater than 11 (Table S2).
Electrostatic Fields Within the KSI Active Site Are Altered by pH Titiration.
The functional results described above provide evidence that the D40N mutation is accompanied by ionization of a nearby residue whose pKa has been lowered to a value near 7 due to this mutation. To study the effects of this ionization alone and thus to understand its contribution to the net electrostatic effect of the D40N mutation, we compared the changes sensed by the probes in response to raising the buffer pH from 5.6 to 7.8, values at which we expected this nearby residue to be neutral or ionized, respectively. Shifts, defined as 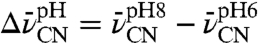 , of +1, 0, and -2 cm-1 were observed for F86C-CN, M105C-CN, and M116C-CN, respectively (Fig. 2
J–L). These values recapitulate the approximate size and direction of the IR shifts observed above (Fig. 2
D–F) due to mutation of residue 40 from Asp to Asn. This suggests that the ionization of this nearby residue, affected either by pH or as an adventitious consequence of the D40N mutation, makes a larger contribution to the observed IR shifts of our probes than the ablation of charge at position 40 due to the D40N mutation itself.
, of +1, 0, and -2 cm-1 were observed for F86C-CN, M105C-CN, and M116C-CN, respectively (Fig. 2
J–L). These values recapitulate the approximate size and direction of the IR shifts observed above (Fig. 2
D–F) due to mutation of residue 40 from Asp to Asn. This suggests that the ionization of this nearby residue, affected either by pH or as an adventitious consequence of the D40N mutation, makes a larger contribution to the observed IR shifts of our probes than the ablation of charge at position 40 due to the D40N mutation itself.
Electrostatic Modeling Suggests Ionization of an Active Site Tyr in D40N.
Literature precedent (32), pKa prediction calculations, and simple chemical reasoning all suggested that the D40N mutation resulted in charge transfer from D40 to D103, with a resulting pKa for D103 near 7 in D40N. However, the following analysis motivated us to question this assumption. We compared the observed frequency shifts to those predicted from continuum electrostatic modeling with DelPhi (55, 56), to test whether these predicted charge configurations were consistent with the electrostatic field changes reported by the IR probes above. As summarized in Fig. 5, the predicted shifts assuming that D103 was the ionizing residue in D40N disagreed with the experimental results for each probe position. Even though we would not expect quantitative agreement between calculation and experiment, we were surprised by the enormity of the discrepancies at each probe position.
Fig. 5.

Comparison of experimental electrostatic field changes (in MV/cm) observed at each probe site and predicted field changes calculated for each probe site based on ionization of different active site residues using DelPhi and the experimental Stark tuning rate. Comparison of experimental versus calculated field changes are made for D40 versus D40N (A–B) and for raising the pH from 6 to 8 (C–D). A and C show the sign and magnitude of the observed (black) and predicted shifts side by side, with predictions for the scenarios in which D103, Y16, Y32, or Y57 (red, green, blue, and magenta, respectively) lose a proton due to mutation or raising the pH. B and D show the magnitude of the error in the predicted shift depending on the choice for the group undergoing ionization at each of the probe sites, 105, 86, and 116 (orange, light blue, and gray, respectively).
The disagreement between measured field changes and the predicted effects due to D103 ionization can be readily appreciated using Coulomb’s law, Eq. 1, and the distances and angles between each probe and D103 shown in Fig. 2 G and H (for a list of angles, distances and Coulomb’s law calculations, see Table S3). Based on the 5.7- and 3.5-Å proximity of D103 (circled in red) to M105C-CN and F86C-CN, respectively, and the 107° and 68° angles formed by the nitrile bond vector and the vector between the probe and the carboxylate group, the ionization of D103 would be expected to result in large −15 and +22 MV/cm fields at these probe positions. In contrast to this expectation, only small shifts are observed for these probes (Fig. 2 J and K). The failure of electrostatic modeling based on ionization of D103 to account for our observed IR shifts motivated us to consider whether a residue other than D103 might be ionizing in the D40N mutant.
The only other titratable residues within the active site are Y16, Y32, and Y57, which form an extended hydrogen bond network in the oxyanion hole (Fig. 1B). Although computational pKa predictions for these Tyr residues all gave values greater than 11 (see above), we nonetheless used Delphi to predict IR shifts based on ionization of each of these residues, as done above for D103, and compared these predictions to the observed shifts for mutation of D40 to Asn (Fig. 5). Surprisingly, ionization at any of these residues did a better job of accounting for the observed frequency shifts than ionization of D103, with residue Y57 giving the closest congruence. We therefore turned to experiments that could directly assess whether an ionized tyrosine was present in the enzyme at neutral pH.
UV and Tyr 13C NMR Spectra Identify an Ionized Tyr in the D40N Active Site.
UV absorbance provides a sensitive probe of possible tyrosine ionization within KSI, as the absorbance peak centered at 280 nm for neutral tyrosine shifts to 295 nm upon ionization (57). As shown in Fig. 6 there is an increase in absorbance at 295 nm in the UV spectrum of the D40N mutant that titrated with an apparent pKa of 6.3 ± 0.1 as the solution pH was raised from 4.7 to 10.5, suggesting ionization of a Tyr residue within unliganded KSI D40N with this pKa.†
Fig. 6.

pH titration of the UV absorbance at 300 nm for pKSI D40N. The molar absorptivity change at 300 nm is plotted relative to the value observed with an external buffer pH of 4.7. A two-stage sigmoidal curve was fit to the data, yielding a pKa of 6.3 for the lower pKa. The dashed line shows the molar absorptivity change at 300 nm observed for the tyrosine deprotonation in aqueous buffer. Inset: UV-vis absorption difference spectra for KSI D40N (solid) and aqueous tyrosine (dashed), calculated from the pH 9.1 minus pH 5.5, and pH 14 minus pH 4.7 spectra, respectively.
To directly test for the presence of an ionized Tyr in pKSI at neutral pH, we turned to 13C NMR studies of KSI containing Tyr residues labeled with 13C at position Cζ, the carbon adjacent to the hydroxyl group, as the chemical shift of this carbon is exquisitely sensitive to the ionization state of Tyr. For tyrosine-containing peptides in neutral water, Cζ has a 13C chemical shift of 155.5 ppm for a protonated Tyr that shifts downfield nearly 11 ppm to 166.3 ppm upon ionization (58). Wild-type and the D40N mutant of pKSI were expressed in cells grown in a minimal M9 medium containing 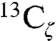 -labeled tyrosine, and 13C-NMR spectra were obtained. KSI from P. putida contains four tyrosines: Y16, Y32, and Y57 form an active site hydrogen bond network with each other, and Y119 is located on the protein surface near the dimer interface and far from the active site. For WT KSI, D40 is expected to be the only ionized active site residue. Consistent with this expectation, we observed a cluster of peaks in the 13C NMR spectrum of WT KSI with chemical shifts around 158 ppm, very similar to the expected position for a protonated Tyr (Fig. 7A). The peak at 157.5 ppm is approximately twice as intense as the flanking peaks at 157.0 and 158.5 ppm, suggesting that two of the Tyr residues in WT KSI have identical chemical shifts.
-labeled tyrosine, and 13C-NMR spectra were obtained. KSI from P. putida contains four tyrosines: Y16, Y32, and Y57 form an active site hydrogen bond network with each other, and Y119 is located on the protein surface near the dimer interface and far from the active site. For WT KSI, D40 is expected to be the only ionized active site residue. Consistent with this expectation, we observed a cluster of peaks in the 13C NMR spectrum of WT KSI with chemical shifts around 158 ppm, very similar to the expected position for a protonated Tyr (Fig. 7A). The peak at 157.5 ppm is approximately twice as intense as the flanking peaks at 157.0 and 158.5 ppm, suggesting that two of the Tyr residues in WT KSI have identical chemical shifts.
Fig. 7.

13C-NMR spectra for 13Cζ-Tyr-labled KSI. (A–C) apo-WT, apo-D40N and D40N in complex with 4-NO2-phenolate, respectively. Spectra were taken in 40 mM potassium phosphate, pH 7.1, 23 °C. Chemical shifts for the neutral and ionized tyrosine in small unstructured peptides (58) are shown with dashed lines.
In contrast, the 13C NMR spectrum of D40N at pH 7 displays four well-resolved peaks (Fig. 7B). The most upfield peak at 157.5 ppm is very similar to the expected chemical shift for a neutral tyrosine in water. This peak can be assigned to the surface residue Y119 based on its selective disappearance in the 13C spectrum of the Y119F/D40N mutant (Fig. S4). The three remaining peaks are observed at 159.7 ppm, 161.6 ppm, and 165.4 ppm, all downfield of the chemical shift expected for neutral Tyr. The peak at 165.4 ppm is deshielded by nearly 8 ppm relative to the peak at 157.5 ppm for neutral Y119 and is within 1 ppm of the chemical shift expected for a fully ionized Tyr in aqueous solution.‡ On the basis of our UV and 13C NMR results, we conclude that an active site Tyr is indeed ionized in unliganded D40N at neutral pH, consistent with our prediction above based on quantitative modeling of the IR peak shifts for our nitrile probes.
Electrostatic Fields Within the KSI Active Site Are Altered by Binding a Charged Ligand.
As a final test of how fields in the KSI active site are affected by a local charge perturbation, we measured the electric field changes associated with binding of a charged ligand. The ligands 3-fluoro-4-nitrophenol (3F,4-NO2-phe), and 4-nitrophenol (4NO2-phe) were previously shown to bind KSI D40N as anions and have been studied as transition state analogs (32, 59). As shown in Fig. 7C, binding of 4NO2-phe to D40N shifts the observed 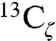 resonances for KSI Tyr residues into the chemical shift range expected for neutral tyrosine. Thus, binding of 4NO2-phe to D40N results in migration of negative charge from an active site Tyr to the phenolate oxygen. Using the IR frequency difference upon ligand binding, defined as
resonances for KSI Tyr residues into the chemical shift range expected for neutral tyrosine. Thus, binding of 4NO2-phe to D40N results in migration of negative charge from an active site Tyr to the phenolate oxygen. Using the IR frequency difference upon ligand binding, defined as 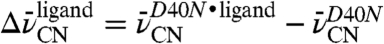 , the average Stark tuning rate of 0.65 cm-1/(MV/cm), and Eq. 1, we determined that binding of 4-nitrophenol results in field shifts of +6.5, −1.1, and +1.1 MV/cm for probes at position 116, 105, and 86, respectively. Similar values are observed for 3F,4-NO2-phe (Table 2). The large +6.5 MV/cm shift observed for M116C-CN upon 4-NO2-phe binding is consistent with the close proximity (< 4 Å) of this probe to the phenolate oxygen and is nearly as large as the 8 MV/cm site-to-site field variation observed in apo D40N.
, the average Stark tuning rate of 0.65 cm-1/(MV/cm), and Eq. 1, we determined that binding of 4-nitrophenol results in field shifts of +6.5, −1.1, and +1.1 MV/cm for probes at position 116, 105, and 86, respectively. Similar values are observed for 3F,4-NO2-phe (Table 2). The large +6.5 MV/cm shift observed for M116C-CN upon 4-NO2-phe binding is consistent with the close proximity (< 4 Å) of this probe to the phenolate oxygen and is nearly as large as the 8 MV/cm site-to-site field variation observed in apo D40N.
Discussion and Conclusions
Electrostatic Heterogeneity Within an Enzyme Active Site.
X-ray crystallography has contributed enormously to our understanding of protein structure. Nevertheless, translating the pictures of the chemically heterogeneous milieu of charged, polar, and hydrophobic groups yielded by X-ray crystallography into a quantitative understanding of the highly heterogeneous and anisotropic electrostatic environment has remained one of the most challenging and elusive goals of protein biophysics. Using thiocyanate vibrational probes incorporated at multiple positions and orientations within the active site of bacterial ketosteroid isomerase, we have exploited the directional nature of the nitrile stretching vibration and its exquisite electric field sensitivity to determine that electric fields within the KSI active site vary up to 8 MV/cm, and future studies of additional probe positions may give a larger range.
To put this magnitude of site-to-site field heterogeneity in context, consider that a positive and a negative elementary unit of charge separated from each other by 1 Å represents a dipole of 5 Debye. The polar covalent bonds present within amino acids or the KSI steroid substrate typically have bond dipoles in the range of 1–4 Debye. The difference in electrostatic stabilization of a 5 Debye dipole situated with the position and orientation of the nitrile of L61C-CN versus that of M105C-CN can be calculated from 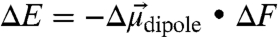 and equals 2 kcal/mol. Thus, measured differences in the local electrostatic fields within the KSI active site due to differences in location or orientation are of sufficient magnitude to result in significant energetic effects on individual bond dipoles, if properly aligned and if the surrounding dipoles are sufficiently restricted from rearrangement. Similar arguments apply to differential stabilization of dipolar transition states.
and equals 2 kcal/mol. Thus, measured differences in the local electrostatic fields within the KSI active site due to differences in location or orientation are of sufficient magnitude to result in significant energetic effects on individual bond dipoles, if properly aligned and if the surrounding dipoles are sufficiently restricted from rearrangement. Similar arguments apply to differential stabilization of dipolar transition states.
Prior reports of electrostatic variation sampled within proteins generally explored either changes in response to charge perturbation, or site-to-site differences, whereas this study encompassed both features. In the first case, the stretching vibration of CO bound to myoglobin (17, 60) and a nitrile-containing inhibitor bound at the active site of human aldose reductase (46) exhibited a range of frequencies in response to mutations in the vicinity of the probe. The vibrational Stark model presented for these band shifts translated into electrostatic changes of 15 and 13 MV/cm, respectively. However, the largest shifts coincided with cases that likely involved the making or breaking of hydrogen bonds to the probe, as hypothesized on structural grounds (46, 61), which would convolute the observed effects (28). In the second case, the chemical shift dispersion for 19F-labeled amino acids incorporated at multiple locations within galactose binding protein were calibrated using a computational model to suggest that site-to-site variations in the projection of the electrostatic field on the C─F bond vector as large as 40 MV/cm occur (20). However, conflicting computational arguments have been made that short-range chemical interactions (i.e., van der Waals interactions) between fluorine atoms and surrounding groups can dominate the chemical shift dispersion rather than local electrostatics (19, 62, 63).
While it may not be possible to completely isolate long-range electrostatic effects from these examples of specific local interactions, our work suggests that, in select cases, such effects can be controlled and corrected for by determining Stark tuning rates (Fig. 4), vibrational frequencies, and 13C NMR chemical shifts (Fig. 3 and Table 2) for the nitrile probes. In particular, the relationship between the nitrile’s NMR chemical shift and vibrational frequency allows for a separate accounting for hydrogen bond contributions, in contrast to electric field observables such as those provided by modeling pKa shifts and for which no such straightforward corrections are currently possible. Furthermore, use of a spectator probe that experiences field changes remotely, rather than participating in a chemical change as part of the observation, greatly simplifies the interpretation. Thus, our measurements of both charge perturbations and site-to-site variations, based entirely on experimental data, represent the cleanest isolation of electrostatic field effects in proteins obtained to date. Nevertheless, future experimental tests and computational comparisons will be needed to further define the capabilities and limitations of this approach.§
Local Versus Global Determinants of the Electrostatic Environment.
The large size of enzymes relative to simple chemical catalysts has prompted many questions regarding the role of the global protein structure in shaping the catalytic properties of active sites. Unlike the significant, collective contribution of distal or nonactive site residues to the positioning of binding and catalytic groups within active sites, a role for these distal residues in shaping electrostatic properties of active sites has remained uncertain. Electrostatic effects are additive and therefore always a collective property of all residues. However, the relative contributions of discrete charges and dipoles to the collective sum is unknown. For KSI, the 7 MV/cm field variation registered by probes at positions 86, 105, and 116 in apo D40N is reduced to 2 MV/cm upon reversion to D40, and reduced to zero within experimental error upon binding the ligand 4-nitrophenolate (Table 2). Thus, rearrangement of a single charge can result in local field changes that approach the magnitude of static field variation within the active site. This observation suggests that the site-to-site variability in local electrostatic fields in KSI is dominated by the specific location of nearby charges rather than the collective electrostatic effects exerted by many residues throughout the protein.
Limited Electrostatic Rearrangement Within Active Sites.
Two related observations in this report support the long-held view that structural organization of proteins during folding restricts electrostatic rearrangement and place quantitative limits on the extent of any such rearrangements. Our observation that charge perturbations separated by several angstroms from the probes can lead to substantial electrostatic changes (Fig. 2) provides evidence for limited electrostatic attenuation due to restricted motion of protein groups and limited water penetration into the protein interior. The observation of qualitative agreement between measured field changes and those calculated with continuum electrostatics, assuming a value of 2 for the internal dielectric, also argues for low electrostatic screening. However, we stress that computational models based on a uniform “protein dielectric” are an inadequate description of the heterogeneous protein environment, as noted by others (6, 11).
While the charge transfers explored in this manuscript represent changes that have equilibrated with the surrounding protein matrix, understanding nonequilibrium effects due to transient charge perturbations remains an important challenge. In a separate study, we have carried out time-resolved measurements of the changes in vibrational frequency of these same nitrile-modified KSI constructs following photoexcitation of a bound chromophore. These experiments provide a measure of the electrostatic changes within the protein matrix in response to the near-instantaneous creation of an excited state dipole in the KSI active site (65). The results of this study, especially the observation that the magnitude of the initial electric field-induced shift and the orientation of the nitrile remain constant over the picosecond time-scale of the experiment suggest an effectively rigid structural and electrostatic environment on this time scale. This observation complements the finding in the current study of limited steady-state electrostatic rearrangement in response to charge perturbation.
Observation of an Ionized Tyr in the KSI D40N Active Site.
An apparent pKa of 6.3, tentatively assigned to Y57 based on electrostatic modeling, represents a 3.7 unit perturbation from the solution pKa value of 10 for the tyrosine side chain in aqueous peptides (58) and occurs in the absence of a nearby stabilizing counter-ion. In contrast, ionized tyrosine residues in calmodulin, dTDP-glucose-4,6-dehydratase, UDP-galactose-4-epimerase and alanine racemase, the only prior literature reports to our knowledge for tyrosine residues with pKa values near 6, appear to be stabilized by proximal positively charged side-chains or metal atoms (66–69). The role played by the unique and extended hydrogen bond network, that includes Y16, Y57, Y32 and multiple water molecules, in shaping the unusual acidity of the tyrosine residue in D40N is the subject of an upcoming study.
Computational Prediction Versus Experimental Measurement of Protein Properties.
We have leveraged the strengths of computational modeling to extend and deepen our understanding of electrostatic fields and features within KSI, while also highlighting critical limitations in current computational approaches, as have been raised on theoretical grounds (7, 8). The algorithms we tested universally erred in predicting the relative pKa values for D103 and the experimentally identified titratable active site tyrosine in the D40N mutant of P. putida KSI. This failure is not a subtle effect due to ionization of the first of two residues of nearly equal pKa creating a charge-repulsion interaction that raises the pKa of the second residue; even for the D40N/D103N double mutant, the active site tyrosines are predicted to have pKa values above 11 (additional discussion in SI Text). In light of the complex hydrogen bond network that appears to underpin the unusually low tyrosine pKa observed, we speculate that simple continuum electrostatic models for the protein and solvent are inadequate to have predicted this observation.
Without experimental knowledge of the ionization states for all residues in the active site in all scenarios of mutation, ligand binding and pH (a problem common to all but the most exhaustively characterized proteins), we found irreconcilable discrepancies between the electrostatic field differences measured by our nitrile probes and continuum electrostatic calculations or even simple arguments based on Coulomb’s law. However, computational models for the change in electrostatic field felt by our nitrile probes gave qualitative agreement with the experimental results provided that an ionization at neutral pH was assumed for an active site tyrosine residue instead of D103, with the models giving the best agreement for ionization of Y57. These computational results inspired UV and 13C NMR experiments in this work that tested and confirmed ionization of a Tyr in apo D40N with a pKa of 6.3. Combined molecular mechanics sampling and continuum methods, the so-called MM-GBSA approach (70), the inclusion of explicit water molecules in the active site, molecular dynamics with polarizable force fields or a complete quantum mechanical treatment may improve the accuracy of calculated properties.
This synergism between directional IR field measurements, computational modeling, and spectroscopic probes of side-chain ionization states highlights the productive interplay between experiment and theory and the power of utilizing directional probes at multiple positions to triangulate local electrostatic effects. The directional sensitivity of our electrostatic probes was indispensable in modeling and distinguishing the effects of ionization at Y57 versus other positions, and contrasts with common electrostatic characterizations of protein interiors in terms of nondirectional or uniform “effective dielectric” descriptions. Finally, our study directly measuring field differences from multiple positions and orientations provides important benchmarks for improving computation modeling of proteins and can serve as a guide for designing future incisive tests of the functional roles and contributions of protein electrostatics.
Materials and Methods.
See SI Materials and Methods for complete experimental methods including the procedure for thiocyanate and 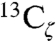 -tyrosine labeling of KSI, the conditions of crystal growth for KSI-CN variants, the details of X-ray acquisition and structure solving, the parameters for continuum electrostatic calculations with the program DelPhi and the description of instrumentation and sample preparation for UV-vis, IR, Stark-effect and NMR spectroscopies.
-tyrosine labeling of KSI, the conditions of crystal growth for KSI-CN variants, the details of X-ray acquisition and structure solving, the parameters for continuum electrostatic calculations with the program DelPhi and the description of instrumentation and sample preparation for UV-vis, IR, Stark-effect and NMR spectroscopies.
Supplementary Material
Acknowledgments.
We thank Corey Liu and Steve Lynch for assistance with NMR experiments and Axel Brunger for helpful discussions. P.A.S. was supported in part by HHMI and G. Lieberman predoctoral fellowships. Funding was provided by grants to S.G.B. (National Institutes of Health Grant GM27738) and D.H. (National Science Foundation Grant MCB-1121778). Portions of this research were conducted at the Advanced Light Source, a national user facility operated by the Lawrence Berkeley National Laboratory, and at the Stanford Magnetic Resonance Laboratory, which is supported in part by the Stanford University Medical School.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 1824.
Data deposition: Atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3OXA, 3OWY, 3OX9, 3OWU).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111566109/-/DCSupplemental.
*The local field correction factor describes the enhancement of the local field in the vicinity of the probe, with respect to the applied field in a Stark experiment, due to differences in the solvent and solute dielectric properties. In general, it is a tensor property but can be approximated with a scalar and has been calculated (48) to fall within a range of 1 < f < 1.3, with the larger value for larger dielectric differences.
‡We attempted to titrate the observed downfield peak by recording NMR spectra from pH 8 to pH 6. However, as the pH was lowered below 7 the downfield shifted peaks were observed to broaden, becoming difficult to distinguish from noise below pH 6.4, suggesting a complex exchange mechanism. A more detailed discussion can be found in SI Text.
§While it is theoretically possible to also extract the absolute value of the electric field projection with this model by comparing the measured frequency with a gas-phase (i.e., zero external field) frequency measurement, there are multiple features of the gas-phase to condensed-phase frequency shift that are not captured by electrostatics alone. For an introduction to this topic see ref. 64.
References
- 1.Warshel A. Energetics of enzyme catalysis. Proc Natl Acad Sci USA. 1978;75:5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selzer T, Schreiber G. Predicting the rate enhancement of protein complex formation from the electrostatic energy of interaction. J Mol Biol. 1999;287:409–419. doi: 10.1006/jmbi.1999.2615. [DOI] [PubMed] [Google Scholar]
- 3.Lee L-P, Tidor B. Barstar is electrostatically optimized for tight binding to barnase. Nat Struct Mol Biol. 2001;8:73–76. doi: 10.1038/83082. [DOI] [PubMed] [Google Scholar]
- 4.Getzoff ED, et al. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 5.Dong F, Zhou H. Electrostatic contribution to the binding stability of protein-protein complexes. Proteins. 2006;65:87–102. doi: 10.1002/prot.21070. [DOI] [PubMed] [Google Scholar]
- 6.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 7.Schnieders MJ, Baker NA, Ren P, Ponder JW. Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum. J Chem Phys. 2007;126:124114. doi: 10.1063/1.2714528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antosiewicz J, McCammon JA, Gilson MK. The determinants of pKas in proteins. Biochemistry. 1996;35:7819–7833. doi: 10.1021/bi9601565. [DOI] [PubMed] [Google Scholar]
- 9.Sharp K, Honig B. Electrostatic interactions in macromolecules: Theory and applications. Annu Rev Biophys Biophys Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- 10.Fersht AR, Sternberg MJE. Can a simple function for the dielectric response model electrostatic effects in globular proteins? Protein Eng. 1989;2:527–530. doi: 10.1093/protein/2.7.527. [DOI] [PubMed] [Google Scholar]
- 11.Chimenti MS, Castañeda CA, Majumdar A, García-Moreno B. Structural origins of high apparent dielectric constants experienced by ionizable groups in the hydrophobic core of a protein. J Mol Biol. 2011;405:361–377. doi: 10.1016/j.jmb.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Britto PJ, Knipling L, Wolff J. The local electrostatic environment determines cysteine reactivity of tubulin. J Biol Chem. 2002;277:29018–29027. doi: 10.1074/jbc.M204263200. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, Kobertz WR, Deutsch C. Mapping the electrostatic potential within the ribosomal exit tunnel. J Mol Biol. 2007;371:1378–1391. doi: 10.1016/j.jmb.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 14.Kangas E, Tidor B. Charge optimization leads to favorable electrostatic binding free energy. Phys Rev E Stat Nonlin Soft Matter Phys. 1999;59:5958–5961. doi: 10.1103/physreve.59.5958. [DOI] [PubMed] [Google Scholar]
- 15.Mao JJ, Hauser K, Gunner MR. How cytochromes with different folds control heme redox potentials. Biochemistry. 2003;42:9829–9840. doi: 10.1021/bi027288k. [DOI] [PubMed] [Google Scholar]
- 16.Abbyad P, et al. Measurement of solvation responses at multiple sites in a globular protein. J Phys Chem B. 2007;111:8269–8276. doi: 10.1021/jp0709104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Augspurger JD, Dykstra CE, Oldfield E. Correlation of C-13 and O-17 chemical-shifts and the vibrational frequency of electrically perturbed carbon-monoxide—a possible model for distal ligand effects in carbonmonoxyheme proteins. J Am Chem Soc. 1991;113:2447–2451. [Google Scholar]
- 18.Park KD, et al. Distal and proximal ligand interactions in heme-proteins—correlations between C-O And Fe-C vibrational frequencies, O-17 and C-13 nuclear-magnetic-resonance chemical-shifts. Biochemistry. 1991;30:2333–2347. doi: 10.1021/bi00223a007. [DOI] [PubMed] [Google Scholar]
- 19.Feeney J, et al. F-19 nuclear magnetic resonance chemical shifts of fluorine containing aliphatic amino acids in proteins: Studies on Lactobacillus casei dihydrofolate reductase containing (2S,4S)-5-fluoroleucine. J Am Chem Soc. 1996;118:8700–8706. [Google Scholar]
- 20.Pearson JG, Oldfield E, Lee FS, Warshel A. Chemical-shifts in proteins—a shielding trajectory analysis of the fluorine nuclear-magnetic-resonance spectrum of the Escherichia-coli galactose binding-protein using a multipole shielding polarizability local reaction field molecular-dynamics approach. J Am Chem Soc. 1993;115:6851–6862. [Google Scholar]
- 21.Augspurger JD, Dykstra CE. Correlation of F-19 chemical shielding and chemical-shift nonequivalence. J Am Chem Soc. 1993;115:12016–12019. [Google Scholar]
- 22.Voinov MA, Ruuge A, Reznikov VA, Grigor’ev IA, Smirnov AI. Mapping local protein electrostatics by EPR of pH-sensitive thiol-specific nitroxide. Biochemistry. 2008;47:5626–5637. doi: 10.1021/bi800272f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czerwinski RM, et al. Effects of mutations of the active site arginine residues in 4-oxalocrotonate tautomerase on the pKa values of active site residues and on the ph dependence of catalysis. Biochemistry. 1999;38:12358–12366. doi: 10.1021/bi9911177. [DOI] [PubMed] [Google Scholar]
- 24.Suydam IT, Boxer SG. Vibrational Stark effects calibrate the sensitivity of vibrational probes for electric fields in proteins. Biochemistry. 2003;42:12050–12055. doi: 10.1021/bi0352926. [DOI] [PubMed] [Google Scholar]
- 25.Chattopadhyay A, Boxer SG. Vibrational Stark effect spectroscopy. J Am Chem Soc. 1995;117:1449–1450. [Google Scholar]
- 26.Andrews SS, Boxer SG. Vibrational stark effects of nitriles I. Methods and experimental results. J Phys Chem A. 2000;104:11853–11863. [Google Scholar]
- 27.Andrews SS, Boxer SG. Vibrational Stark effects of nitriles II. Physical origins of stark effects from experiment and perturbation models. J Phys Chem A. 2002;106:469–477. [Google Scholar]
- 28.Fafarman AT, Sigala PA, Herschlag D, Boxer SG. Decomposition of vibrational shifts of nitriles into electrostatic and hydrogen-bonding effects. J Am Chem Soc. 2010;132:12811–12813. doi: 10.1021/ja104573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feierberg I, Aqvist J. The catalytic power of ketosteroid isomerase investigated by computer simulation. Biochemistry. 2002;41:15728–15735. doi: 10.1021/bi026873i. [DOI] [PubMed] [Google Scholar]
- 30.Warshel A, Sharma P, Chu Z, Aqvist J. Electrostatic contributions to binding of transition state analogues can be very different from the corresponding contributions to catalysis: Phenolates binding to the oxyanion hole of ketosteroid isomerase. Biochemistry. 2007;46:1466–1476. doi: 10.1021/bi061752u. [DOI] [PubMed] [Google Scholar]
- 31.Chakravorty DK, Hammes-Schiffer S. Impact of mutation on proton transfer reactions in ketosteroid isomerase: Insights from molecular dynamics simulations. J Am Chem Soc. 2010;132:7549–7555. doi: 10.1021/ja102714u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraut D, et al. Testing electrostatic complementarity in enzyme catalysis: Hydrogen bonding in the ketosteroid isomerase oxyanion hole. PLoS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sigala PA, Fafarman AT, Bogard PE, Boxer SG, Herschlag D. Do ligand binding and solvent exclusion alter the electrostatic character within the oxyanion hole of an enzymatic active site? J Am Chem Soc. 2007;129:12104–12105. doi: 10.1021/ja075605a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue L, Talalay P, Mildvan AS. Studies of the mechanism of the delta-5-3-ketosteroid isomerase reaction by substrate, solvent, and combined kinetic deuterium-isotope effects on wild-type and mutant enzymes. Biochemistry. 1990;29:7491–7500. doi: 10.1021/bi00484a019. [DOI] [PubMed] [Google Scholar]
- 35.Choi G, et al. Asp-99 donates a hydrogen bond not to tyr-14 but to the steroid directly in the catalytic mechanism of Delta(5)-3-ketosteroid isomerase from Pseudomonas putida biotype B. Biochemistry. 2000;39:903–909. doi: 10.1021/bi991579k. [DOI] [PubMed] [Google Scholar]
- 36.Kim SW, et al. High-resolution crystal structures of Delta(5)-3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry. 1997;36:14030–14036. doi: 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- 37.Pollack RM, Thornburg LD, Wu ZR, Summers MF. Mechanistic insights from the three-dimensional structure of 3-Oxo-[Delta]5-steroid isomerase. Arch Biochem Biophys. 1999;370:9–15. doi: 10.1006/abbi.1999.1384. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, et al. Mutational analysis of the three cysteines and active-site aspartic acid 103 of ketosteroid isomerase from Pseudomonas putida biotype B. J Bacteriol. 1997;179:7742–7747. doi: 10.1128/jb.179.24.7742-7747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fafarman AT, Webb LJ, Chuang JI, Boxer SG. Site-specific conversion of cysteine thiols into thiocyanate creates an IR probe for electric fields in proteins. J Am Chem Soc. 2006;128:13356–13357. doi: 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sigala P, et al. Testing geometrical discrimination within an enzyme active site: Constrained hydrogen bonding in the ketosterold isomerase oxyanion hole. J Am Chem Soc. 2008;130:13696–13708. doi: 10.1021/ja803928m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindquist BA, Corcelli SA. Nitrile groups as vibrational probes: Calculations of the C N infrared absorption line shape of acetonitrile in water and tetrahydrofuran. J Phys Chem B. 2008;112:6301–6303. doi: 10.1021/jp802039e. [DOI] [PubMed] [Google Scholar]
- 42.Choi J-H, Oh K-I, Lee H, Lee C, Cho M. Nitrile and thiocyanate IR probes: Quantum chemistry calculation studies and multivariate least-square fitting analysis. J Chem Phys. 2008;128:134506. doi: 10.1063/1.2844787. [DOI] [PubMed] [Google Scholar]
- 43.Reimers JR, Hall LE. The solvation of acetonitrile. J Am Chem Soc. 1999;121:3730–3744. [Google Scholar]
- 44.Suydam I, Snow C, Pande V, Boxer S. Electric fields at the active site of an enzyme: Direct comparison of experiment with theory. Science. 2006;313:200–204. doi: 10.1126/science.1127159. [DOI] [PubMed] [Google Scholar]
- 45.Stafford AJ, Ensign DL, Webb LJ. Vibrational Stark effect spectroscopy at the interface of Ras and Rap1A bound to the Ras binding domain of RalGDS reveals an electrostatic mechanism for protein-protein interaction. J Phys Chem B. 2010;114:15331–15344. doi: 10.1021/jp106974e. [DOI] [PubMed] [Google Scholar]
- 46.Webb LJ, Boxer SG. Electrostatic fields near the active site of human aldose reductase: 1. New inhibitors and vibrational stark effect measurements. Biochemistry. 2008;47:1588–1598. doi: 10.1021/bi701708u. [DOI] [PubMed] [Google Scholar]
- 47.Dalosto SD, Vanderkooi JM, Sharp KA. Vibrational stark effects on carbonyl, nitrile, and nitrosyl compounds including heme ligands, CO, CN, and NO, studied with density functional theory. J Phys Chem B. 2004;108:6450–6457. doi: 10.1021/jp0310697. [DOI] [PubMed] [Google Scholar]
- 48.Bublitz G, Boxer S. Stark spectroscopy: Applications in chemistry, biology, and materials science. Annu Rev Phys Chem. 1997;48:213–242. doi: 10.1146/annurev.physchem.48.1.213. [DOI] [PubMed] [Google Scholar]
- 49.Fafarman AT, Boxer SG. Nitrile bonds as infrared probes of electrostatics in ribonuclease S. J Phys Chem B. 2010;114:13536–13544. doi: 10.1021/jp106406p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yun YS, et al. Origin of the different pH activity profile in two homologous ketosteroid isomerases. J Biol Chem. 2003;278:28229–28236. doi: 10.1074/jbc.M302166200. [DOI] [PubMed] [Google Scholar]
- 51.Thornburg L, et al. Electrophilic assistance by Asp-99 of 3-oxo-Delta 5-steroid isomerase. Biochemistry. 1998;37:10499–10506. doi: 10.1021/bi980099a. [DOI] [PubMed] [Google Scholar]
- 52.Gordon JC, et al. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bas DC, Rogers DM, Jensen J. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 54.Kieseritzky G, Knapp E. Optimizing pKa computation in proteins with pH adapted conformations. Proteins. 2008;71:1335–1348. doi: 10.1002/prot.21820. [DOI] [PubMed] [Google Scholar]
- 55.Sitkoff D, Sharp K, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 56.Gilson MK, Sharp K, Honig B. Calculating the electrostatic potential of molecules in solution—method and error assessment. J Comput Chem. 1988;9:327–335. [Google Scholar]
- 57.Li YK, Kuliopulos A, Mildvan AS, Talalay P. Environments and mechanistic roles of the tyrosine residues of delta-5-3-ketosteroid isomerase. Biochemistry. 1993;32:1816–1824. doi: 10.1021/bi00058a016. [DOI] [PubMed] [Google Scholar]
- 58.Richarz R, Wuthrich K. C-13 NNMR chemical-shifts of common amino-acid residues measured in aqueous-solutions of linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers. 1978;17:2133–2141. [Google Scholar]
- 59.Petrounia I, Pollack R. Substituent effects on the binding of phenols to the D38N mutant of 3-oxo-Delta(5)-steroid isomerase. A probe for the nature of hydrogen bonding to the intermediate. Biochemistry. 1998;37:700–705. doi: 10.1021/bi972262s. [DOI] [PubMed] [Google Scholar]
- 60.Park ES, Andrews SS, Hu RB, Boxer SG. Vibrational stark spectroscopy in proteins: A probe and calibration for electrostatic fields. J Phys Chem B. 1999;103:9813–9817. [Google Scholar]
- 61.Decatur SM, Boxer SG. A test of the role of electrostatic interactions in determining the CO stretch frequency in carbonmonoxymyoglobin. Biochem Biophys Res Commun. 1995;212:159–164. doi: 10.1006/bbrc.1995.1950. [DOI] [PubMed] [Google Scholar]
- 62.Gregory DH, Gerig JT. Prediction of fluorine chemical-shifts in proteins. Biopolymers. 1991;31:845–858. doi: 10.1002/bip.360310705. [DOI] [PubMed] [Google Scholar]
- 63.Gregory DH, Gerig JT. Structural effects of fluorine substitution in proteins. J Comput Chem. 1991;12:180–185. [Google Scholar]
- 64.Schweizer KS, Chandler D. Vibrational dephasing and frequency-shifts of polyatomic-molecules in solution. J Chem Phys. 1982;76:2296–2314. [Google Scholar]
- 65.Jha SK, Ji M, Gaffney KJ, Boxer SG. Direct measurement of the protein response to an electrostatic perturbation that mimics the catalytic cycle in ketosteroid isomerase. Proc Natl Acad Sci USA. 2011;108:16612–16617. doi: 10.1073/pnas.1113874108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerratana B, Cleland WW, Frey PA. Mechanistic roles of Thr134, Tyr160, and Lys 164 in the reaction catalyzed by dTDP-glucose 4,6-dehydratase. Biochemistry. 2001;40:9187–9195. doi: 10.1021/bi0108249. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, et al. Mechanistic roles of tyrosine 149 and serine 124 in UDP-galactose 4-epimerase from Escherichia coli. Biochemistry. 1997;36:10675–10684. doi: 10.1021/bi970430a. [DOI] [PubMed] [Google Scholar]
- 68.Pundak S, Roche RS. Tyrosine and tyrosinate fluorescence of bovine testes calmodulin: Calcium and pH dependence. Biochemistry. 1984;23:1549–1555. doi: 10.1021/bi00302a032. [DOI] [PubMed] [Google Scholar]
- 69.Sun S, Toney MD. Evidence for a two-base mechanism involving tyrosine-265 from arginine-219 mutants of alanine racemase. Biochemistry. 1999;38:4058–4065. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- 70.Kuhn B, Kollman PA, Stahl M. Prediction of pKa shifts in proteins using a combination of molecular mechanical and continuum solvent calculations. J Comput Chem. 2004;25:1865–1872. doi: 10.1002/jcc.20111. [DOI] [PubMed] [Google Scholar]
