

Abstract
A protein destined for export from the cell cytoplasm is synthesized as a preprotein with an amino-terminal signal peptide. In Escherichia coli, signal peptides that guide preproteins into the SecYEG protein conduction channel are typically subsequently removed by signal peptidase I. To understand the mechanism of this critical step, we have assessed the conformation of the signal peptide when bound to signal peptidase by solution NMR. We employed a soluble form of signal peptidase without its two transmembrane domains (SPase I Δ2-75) and the E. coli alkaline phosphatase signal peptide. Using a transferred NOE approach, we found clear evidence of weak peptide-enzyme complex formation. The peptide adopts a “U-turn” shape originating from the proline residues within the primary sequence that is stabilized by its interaction with the peptidase and leaves key residues of the cleavage region exposed for proteolysis. In dodecylphosphocholine (DPC) micelles the signal peptide also adopts a U-turn shape comparable to that observed in association with the enzyme. In both environments this conformation is stabilized by the signal peptide phenylalanine side chain-interaction with enzyme or lipid mimetic. Moreover, in the presence of DPC, the N-terminal core region residues of the peptide adopt a helical motif and, based on PRE (paramagnetic relaxation enhancement) experiments, are shown to be buried within the membrane. Taken together, this is consistent with proteolysis of the preprotein occurring while the signal peptide remains in the bilayer and the enzyme active site functioning at the membrane surface.
Keywords: NMR spectroscopy, transferred NOE, dodecylphosphocholine, SPase I Δ2-75, signal peptide
Introduction
Proteins destined for extracytoplasmic locations are synthesized with an amino-terminal signal peptide that serves as the signature of a preprotein to be exported. The signal peptide distinguishes an exported protein from a cytoplasmic one, facilitates entrance to the protein translocation pathway, and subsequently is cleaved from the preprotein so the mature region can complete folding and localization.1,2
In Escherichia coli, the signal peptidase I (SPase I) catalyzes the cleavage of the signal peptide to yield the mature form of the exported protein. It is membrane bound via its two amino-terminal transmembrane segments and contains a large carboxyl-terminal catalytic domain that protrudes into the periplasm.3 The SPase I catalytic mechanism is thought to function through a catalytic dyad involving Ser90 for nucleophilic attack and Lys145 as the general base for peptide bond hydrolysis.4 Several studies indicate that enzyme recognition of an appropriate signal peptide substrate requires residues with small side chains at the -1 and -3 position of the signal peptide.5,6 Indeed, von Heijne and colleagues found that the amino acids Ala, Ser, and Gly are most common at these positions.7 Consistent with this idea, crystal structures of the soluble catalytic domain (SPase I Δ2-75) reveal two shallow hydrophobic cavities neighboring the Ser-Lys dyad that could accommodate the signal peptide -1 and -3 residue side chains.8,9 In addition, NMR analysis indicates the SPase I Δ2-75 amide resonances shift upon peptide binding, a finding which has facilitated docking of the amino acids at these positions.10 Although there is considerable evidence regarding the importance of the signal peptide -1 and -3 residues for SPase I interaction, little is known about how the signal peptide interacts with the enzyme for cleavage. The structure of the Methanococcus jannaschii SecYEG channel suggests that the signal peptide region of the preprotein enters the SecYEG protein conducting channel by promoting the displacement of a domain of SecY that serves as the channel plug.11 Studies suggest the signal peptide intercalates with SecY and also contacts lipid.12 For the translocating polypeptide, both exit through the hour-glass pore and lateral movement out of the channel into the lipid milieu have been postulated.13 It is not clear, however, how the signal peptide disengages from the channel, nor the mechanism by which SPase I recognizes the signal peptide region for cleavage from the remainder of the translocating protein. Addressing these questions is important for understanding the mechanism of protein export and for developing antimicrobial agents against SPase I.
In this study, we have employed 2D proton NMR spectroscopy to examine the signal peptide conformation upon binding to SPase I Δ2-75 and in the presence of a membrane mimetic environment. Using the E. coli alkaline phosphatase signal peptide we demonstrate that its weak interaction with SPase I Δ2-75 can be studied and structurally characterized using transferred NOE methodology. We find that the peptide adopts an irregular “U-turn” shape originating from the proline residues within the primary sequence which is further stabilized by its interaction with the peptidase. We have also defined the signal peptide structure in DPC micelles and its potential location with respect to the cellular membrane. The strong parallels for the structure of the peptide in DPC and in association with the enzyme are consistent with its mechanism of action at the membrane-water interface.
Materials and Methods
Materials
All reagents were purchased from Thermo-Fisher Scientific Inc. (Pittsburg, PA) unless specified otherwise. The plasmid pET23b coding for the full–length SPase I was kindly provided by Ross Dalbey (The Ohio State University, Columbus, OH).
Expression and Purification of SPase I Δ2-75
The plasmid pET23b was modified to delete the DNA sequence coding for the two transmembranes domains of SPase I to give the soluble form of the enzyme. SPase I Δ2-75 was expressed and purified as reported elsewhere.14 Briefly, BL21(DE3) cells transformed with the IPTG-inducible plasmid coding SPase I Δ2-75 were grown to late log phase in Lauria-Bertani (LB) media containing ampicillin (0.125 mg/mL), centrifuged and suspended in 20 mM sodium phosphate (buffer A), pH 7.5, containing 0.5 mM phenylmethanesulfonylfluoride (Alexis Biochemicals, San Diego, CA) and 1 mM dithiothreitol. After French press lysis, the cells were spun down for 5 min at 12000 g and washed four times with 0.5 % Triton X-100 in buffer A. The pellet was dissolved in 6M guanidine chloride, then triton was removed by mixing the sample with SM-2 Biobeads (Bio-Rad, Hercules CA). Finally, buffer A was added to the solution to reduce the guanidine concentration to 4M, and then the solution was dialyzed 5 times against buffer A containing successively decreasing guanidine concentration (1M, 0.5 M, 0.1 M and twice with no guanidine).
Signal Peptide
Wild-type signal peptides are marginally soluble at high concentration in aqueous media. To circumvent this issue, we previously reported that the addition of three lysine residues to each termini is a good strategy to design highly soluble signal peptides,10,15 which can be studied by NMR or other biophysical techniques. In this study, we used a comparable approach to examine the E. Coli alkaline phosphatase signal peptide (KAP25). In addition to the signal sequence, this derivative also contains four residues of the mature protein and it ends in a carboxyl-terminal amide (Figure 1A). This peptide was purchased by NeoBioSciences (Cambridge, MA). For binding analysis only, KAP21BpaC peptide derivative was obtained from Biomolecules Midwest Inc. (Waterloo, IL). This shorter derivative contains a benzoylphenylalanine (Bpa) moiety in place of the wild-type phenylalanine for photolabeling and was biotinylated via the terminal cysteine thiol group as reported elsewhere for detection on western blots.16
Figure 1.
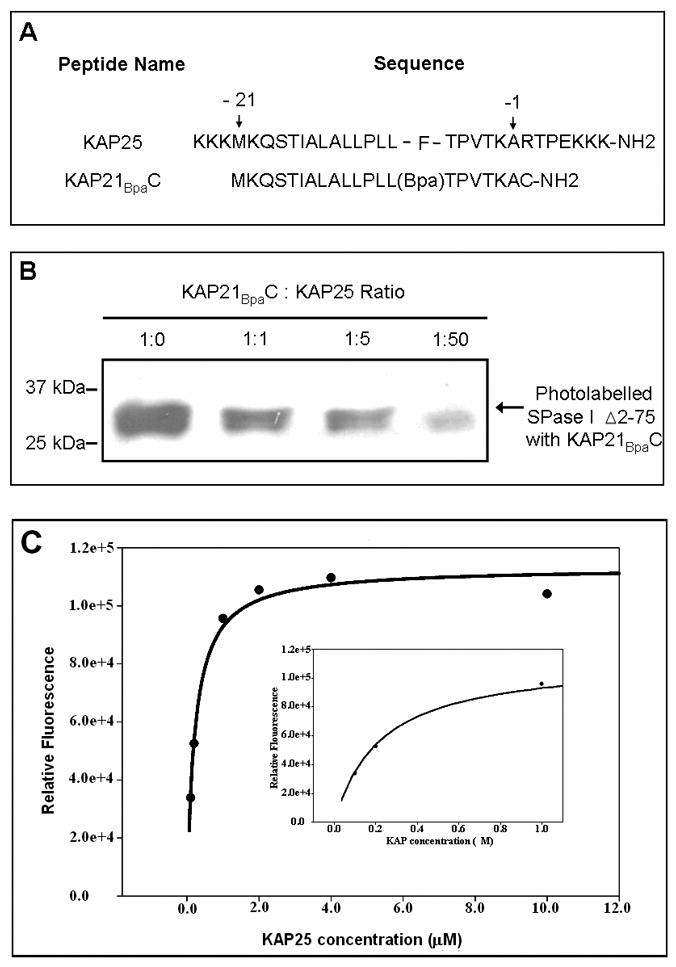
(A) The amino acid sequences of the KAP25 and KAP21BpaC peptides. Residues -21 and -1 mark the first and last residue of the alkaline phosphatase signal peptide, respectively (using the classical numbering scheme for the signal peptide region). For the KAP25 peptide, four residues of the mature protein (RTPE) are also included. (B) Western blot of the SPase I Δ2-75 photolabeled with the biotinylated KA21BpaC. SPase I Δ2-75 at 6.4 μM, was incubated with KAP21BpaC at 6.4μM in the presence and absence of increasing concentrations of KAP25. A decrease in the intensity of the photolabeled enzyme band is observed with an increase in KAP25 concentration. 25kDa and 37kDa molecular weight markers are shown for reference. (C) Intensity of 0.2 μM SPase I Δ2-75 tryptophan emission at 338 nm upon titration with KAP25 after subtraction of the control (0.2 μM SPase I Δ2-75 in the absence of peptide). Inset provides a higher scale resolution between 0 and 1 μM of KAP25.
Signal Peptide Binding Assay
The binding of KAP25 to the SPase I Δ2-75 was verified by two different techniques: a competition assay between KAP25 and the biotin-labeled KAP21BpaC peptide and by using fluorometry to monitor changes in tryptophan emission of SPase I Δ2-75 in samples having different KAP25 to SPase I Δ2-75 ratios.
For the competition assay, SPase I Δ2-75 and KAP21BpaC (both at 6.4 μM) in 20 mM phosphate buffer, pH 6.5 were incubated in the presence of different amounts of KAP25 peptide (1:1, 1:5 and 1:50 KAP21BpaC:KAP25 ratios) for 30 minutes in the dark at 25°C. The photolabeling reaction was achieved by irradiating the sample for 1 min with a Super-High-Intensity UV Lamp (Spectroline, Westbury, NY). Finally, SDS-PAGE loading buffer was added and the samples resolved by SDS-PAGE followed by transferring onto Immobilon PVDF membrane (Millipore) using a BioRad semi-dry transfer cell at 10 V for 60 min. After transfer, the membrane was incubated with 20 ml of Superblock T20 (Thermo-Fisher) for 60 min and washed 4 times with PBS containing 0.05% Tween. The membrane was incubated for 2 hr with streptavidin-HRP antibody (1:2500) in Superblock T20 and washed as reported above. Chemiluminescence was detected using the SuperSignal West Pico kit (Thermo). Competition by the KAP25 peptide is an indicator of enzyme binding.
The tryptophan fluorescence emission spectra of SPase I Δ2-75 (0.2 μM in 20 mM sodium phosphate buffer at pH 7) samples, in the presence of different concentrations of KAP25, ranging from 0.1 to 10 μM, were collected using a Fluoromax-3 spectrofluorometer (Horiba Jobin Yvon), after 3 hours of incubation at room temperature in the darkness. Spectra were recorded from 310 to 420 nm, in a 0.3 cm quartz cuvette, upon excitation at 290 nm at 25°C. Excitation and emission bandwidths were set to 3 and 4 nm respectively.
NMR Spectroscopy
All the NMR experiments were performed in 20 mM sodium phosphate buffer, pH 6.1 on a Varian Inova 600MHz NMR equipped with inverse triple resonance cold probe. The transferred NOESY experiments were performed at 22 and 15°C at pH 6.1. Different ratios of the peptide to the binding partner along with different mixing times were investigated to find the optimal range for NOE transfer. All the spectra were processed with NMRPipe17 and analyzed by CCPN software suite.18 Resonance assignments were made using conventional 2D homonuclear TOCSY and NOESY spectra.19 All the NMR experiments involving membrane-mimetic conditions were performed at 40°C in the presence of perdeuterated dodecylphosphocholine (d38-DPC) micelles. The mole fraction partition coefficient, Kp, has been determined by titrating KAP25 with aliquots of 600 mM DPC, to a final peptide to micelle molar ratio of 1:1.4, and monitoring the changes in the chemical shifts in the TOCSY spectra, as reported by Grosssuer.20
To determine the location of KAP25 relative to the micelle surface, 16- and 5-doxyl stearic acids (16-DSA and 5-DSA) were employed for PRE (paramagnetic relaxation enhancement) experiments. These agents were dissolved in 50 mM DPC solution to make a 50mM stock solution, which was then added to the KAP25 in DPC samples to achieve the final DSA to DPC micelle ratio of approximately 1 to 2.5. The effects of the spin labels were observed by comparing the peak intensities of αH protons in the TOCSY spectra.
Structure Calculations
The structure calculations were performed using CYANA 2.1.21 Table I lists detailed structural statistics of the final 20 lowest energy conformers. None of these structures have NOE violation of more than 0.2 Å. The Protein Structure Software suite (PSVS; courtesy of CABM Structural Bioinformatics Laboratory, Rutgers State University of New Jersey) was used for structure quality assessment and validation. Molecular graphics images were produced using the MOLMOL program22 and UCSF Chimera.23
Table I.
Structural Statistics for KAP25 peptide: a) bound to SPase I Δ2-75 and b) in presence of DPC micelles
| Parameter | Ensemble | |
|---|---|---|
| Distance restraints | Bound a | DPCb |
| All | 207 | 246 |
| Short range (|i-j|<=1) | 185 | 162 |
| Medium-range (1<|i-j|<5) | 22 | 74 |
| Long-range (|i-j|>=5) | 0 | 10 |
| Average CYANA target function value | 0.16 | 0.15 |
| Violations | ||
| NOE (> 0.2 Å) | 0 | 0 |
| RMSDc | ||
| Average backbone RMSD to mean | 1.48 Å | 0.64 Å |
| Average heavy atom RMSD to mean | 1.96 Å | 0.96 Å |
| RMS Z score | ||
| Bond length | 1.065+/- 0.000 | 1.065+/- 0.000 |
| Bond angles | 0.175+/- 0.001 | 0.174+/- 0.001 |
| Omega angle restraints | 0.019+/- 0.005 | 0.017+/- 0.007 |
| Side chain planarity | 0.029+/- 0.007 | 0.028+/- 0.006 |
| Improper dihedral distribution | 0.304+/- 0.002 | 0.304+/- 0.001 |
| Ramchandran statisticsd | ||
| Residues in most favored regions | 64.0% | 62.9% |
| Residues in additional allowed regions | 35.4% | 37.1% |
| Residues in generously allowed regions | 0.6% | 0% |
| Residues in disallowed regions | 0% | 0% |
20 mM Sodium phosphate buffer, 22 °C
20 mM Sodium phosphate buffer, 40 °C, 75mM DPC (1:1.4 peptide:micelles molar ratio)
Residues -11 to -1
All residues
Results
To assess the bound conformation of the signal peptide, we employed a soluble form of signal peptidase that has its two transmembrane domains deleted (SPase I Δ2-75) and of the E. coli alkaline phosphatase derived signal peptide (KAP25). The sequence of the KAP25 signal peptide with four residues of the mature protein plus flanking lysine residues to enhance solubility is shown in Figure 1A. The binding of KAP25 was confirmed using a competition assay. This assay takes advantage of the signal peptide derivative, KAP21BpaC (Figure 1A), that is labeled with biotin for western blot detection and that has a Bpa photolabel incorporated in place of the naturally occurring phenylalanine. The binding of KAP21BpaC peptide was estimated by densitometry, after equilibration with SPase I Δ2-75 in the presence of different ratios of KAP25, following by uv-irradiation for photolabelling. Our data showed a reduction of the amount of photolabelled enzyme of 33, 48 and 80% for 1:1, 1:5 and 1:50, KAP21BpaC: KAP25 ratios, respectively. As shown in Figure 1B this indicates that the KAP25 efficiently competes with the KAP21BpaC peptide for interaction with SPase I Δ2-75. This finding also indicates that the positive charge added at the termini of the peptide to enhance solubility does not preclude its interaction with the enzyme. Furthermore, it is consistent with our previous work showing the interaction of this same peptide with SecA,15 another component of the Sec-dependent transport pathway, and a comparable peptide with SPase I Δ2-75.10 Moreover, we found that the enzyme slowly cleaves the KAP25 signal peptide with about 7, 19 and 27% cleavage observed at 24, 36 and 48 hours of incubation, respectively (data not shown). This slow extent of cleavage is consistent with previous studies that report a decrease in preprotein digestion by SPase I and the soluble form in the absence of Triton X-100 and for a small peptide relative to the preprotein.24,25
To further demonstrate binding of KAP25 to the signal peptidase, we recorded the tryptophan emission spectra of SPase I Δ2-75 in samples containing different concentrations of KAP25. Since the binding of a ligand to a protein involves conformational changes, tryptophan fluorescence is often used to evaluate this interaction, because its emission is very sensitive to chemical environment.26 In the case of SPase I Δ2-75, our data indicate that the binding of the signal peptide results in an increase of the emission intensity (Figure 1C). The binding is concentration dependent and saturable, indicative of a specific interaction between the enzyme and the peptide. A Kd of 0.25 μM was determined by regression analysis.
To examine the conformation of the KAP25 signal peptide we used 2D proton NMR and found that the presence of the three prolines (Pro-10, Pro-5 and Pro+3) in its primary amino acid sequence stabilized a somewhat rigid fold allowing complete backbone and side chain assignments of its “signaling” region. The corresponding TOCSY spectrum (with several assignments labeled) is presented in Figure 2A. Although it is not structured on its own in aqueous solution with most of the peaks in the corresponding 2D-1H NOESY spectrum representing intra-residual or sequential NOEs (Figure 2B, shown in black), its interaction with SPase I Δ2-75 induces the bound conformation which has a higher structural order as judged from emerging transferred NOEs (Figure 2B, shown in red).
Figure 2.
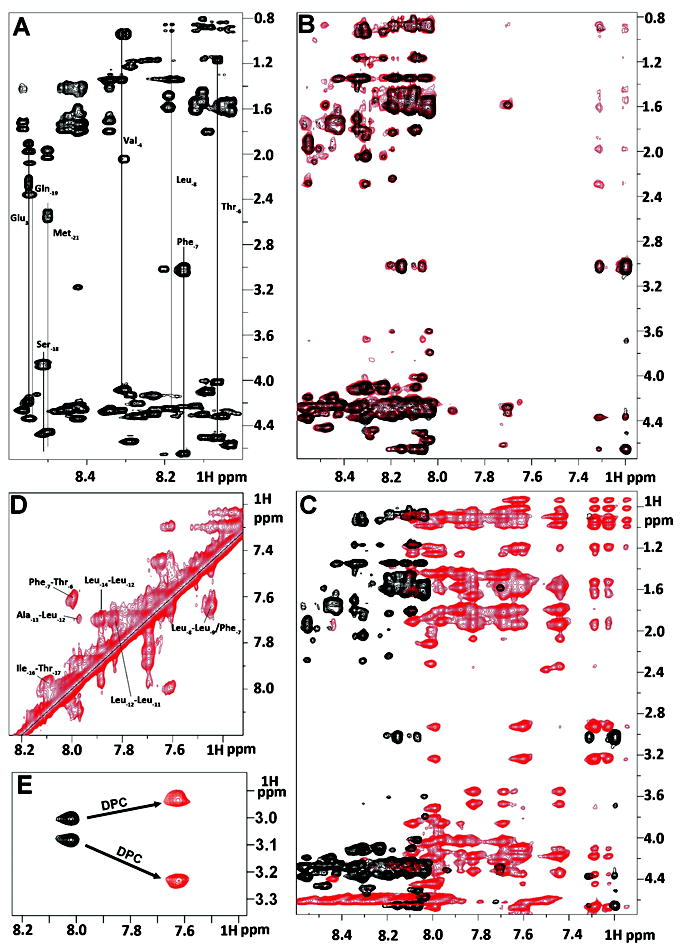
Summary of NMR spectral perturbation for KAP25 peptide upon binding to SPase I Δ2-75 or to DPC micelles. (A) 1H-1H TOCSY spectrum of KAP25 peptide. (B) transferred NOE evidence of the interaction between the KAP25 peptide and SPase I Δ2-75. Superimposition of 1H-1H NOESY spectra for KAP25 in absence (black) and presence (red) of SPase I Δ2-75 at 150:1 ratio, 22°C, 400 ms mixing time. (C) NOESY spectral perturbation upon KAP25 binding to DPC. 1H-1H NOESY spectra for KAP25 in absence (black) and presence (red) of DPC micelles at 1:1.4 molar ratio, 40°C, 200 ms mixing time. (D) The expanded amide region of the NOESY spectrum for KAP25 peptide in DPC showing few HN (i, i+1) and (i, i+2) connections. (E) NH-Hβ phenylalanine cross-peaks in 1H-1H TOCSY spectra in the absence (black) or in the presence of DPC.
The basis of transferred NOE spectroscopy27 is that during weak peptide-protein interactions characterized by a fast dissociation rate, the cross relaxation between protons of the peptide in the bound state, which is governed by the large correlation time of the complex, is transferred to the peptide in the free state through chemical exchange. This phenomenon is manifested by the appearance of additional peaks in the NOESY spectrum of the peptide mixed with a small molar portion of the target protein. Indeed, the NOESY spectrum of KAP25 mixed with SPase I Δ2-75 at the molar ratio of 150:1 shows additional peaks (see Figure 2B, red), providing a clear notification of weak binding.
Interestingly, most of the additional peaks are found within the aromatic part of the spectrum and are related to Phe-7 indicating that side chain motions of this amino acid are limited due to its interaction with the enzyme. The distance restraints that originated from the NOEs of this complex NOESY spectrum have been used for structural characterization of the SPase I Δ2-75 induced conformation of the KAP25 peptide. Figure 3A and 3B, illustrates an ensemble of the 20 lowest energy structures with statistics presented in Table I. The overall fold does not carry any secondary structural elements and is instead represented by an irregular U-shape composed of two turns which are primarily defined by Pro-10 and Pro-5 (both in the cis-conformation). Although there were no long-range NOEs found, medium range NOEs have provided a sufficient number of distance restraints to characterize a well-defined backbone of the U-turn in the converged structural ensemble.
Figure 3.
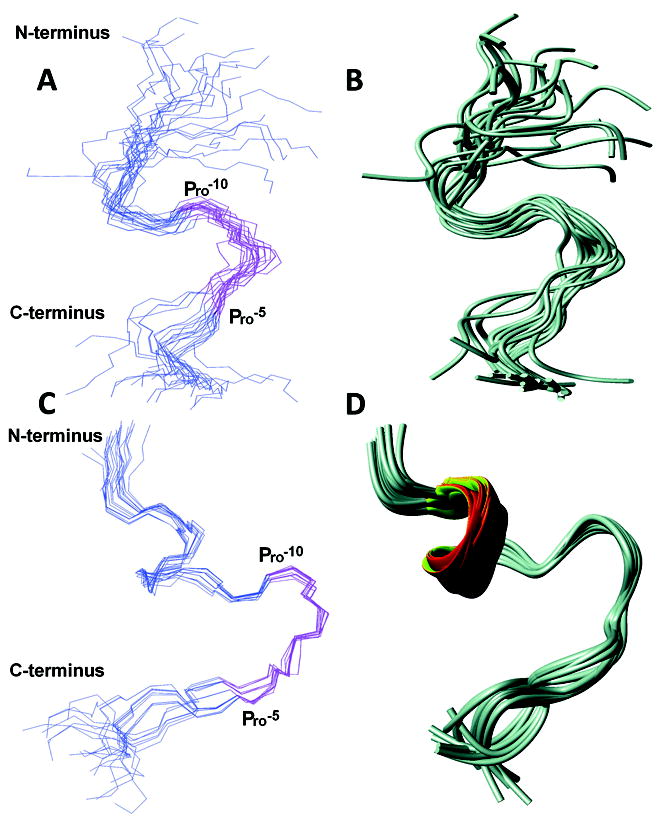
NMR structures of KAP25 in presence of SPase I Δ2-75 (A and B) and DPC (C and D). Disordered regions (residues up to -18 and above +4) are excluded for clarity. The backbone residues are represented in blue and the -10PLLFTP-5 region is highlighted in pink. Residues -15 to +3 are used for superimposition. (A) Backbone superimposition of 20 lowest energy conformers of KAP25 in presence of SPase I Δ2-75. (B) Ribbon representation of the ensemble. (C) Backbone superimposition of 20 lowest energy conformers of KAP25 in the presence of DPC micelles at a molar ratio of 1:1.4. (D) Ribbon representation of the ensemble. Molecular graphics images were produced by MOLMOL.
Since the native signal peptidase I is an integral membrane protein which likely binds its ligands within the context of the lipid bilayer, or at the cytoplasmic interface, we investigated how a membrane mimetic, in the form of DPC micelles, might affect the conformation of our representative signal peptide KAP25.
The NOESY spectrum of KAP25 recorded in the presence of DPC micelles at a molar ratio of 1:1.4, assuming 54 molecules of the detergent per micelle,28 (see Figure 2C, in red) shows large chemical shift perturbations indicative of its interaction with the membrane. The HN-HN region of this NOESY spectrum (with several labeled amide-amide connections) is presented in Figure 2D. The shifts are predominantly up-field in the amide region, which is typically associated with the increase in α-helical content. Indeed, according to our structural calculations, the N-terminus of the peptide signaling region forms one turn of a helix, which is followed by the U-shape conformation very similar to the one found for the peptide bound to SPase I Δ2-75. The DPC-induced structure of KAP25 peptide is illustrated in Figure 3, panels C and D, as a back-bone superposition of the 20 lowest energy structures with statistics for this ensemble shown in Table I. The number of distance restraints derived from the available NOE peaks was limited, probably due to the dynamic nature of the peptide C-terminus even within this membrane-mimicking environment. Two equivalently populated conformations of the peptide, characterized by approximately a 200° difference (-130° or 70°) in the φ dihedral angle of Thr-6, satisfy all the distance restraints during structural calculations. Furthermore, the DPC addition caused not only the up-field shifts in the amide region, particularly Phe-7 NH resonance shifts from 8.02 to 7.63 ppm, but the micelle environment also increased the anisotropy of the Phe-7 Hβ. Indeed, the difference in chemical shift of the two phenylalanine β-protons is 0.07 and 0.3 ppm for the membrane-free and membrane-bound peptide, respectively (see Figure 2E). The increase of the anisotropy of the Phe-7 Hβ indicates that the aromatic ring strongly interacts with the micelles and its local motion is restricted by this interaction. Finally, the KAP25 affinity for the DPC micelles was quantified in terms of mole fraction partition coefficient obtained by least square fitting of the observed chemical shift of the Phe-7 NH and Hβ as a function of DPC micelle concentration. We determined a Kp value of 3 × 106 for this residue, which is similar to the reported Kp for other membrane-bound peptides.20
To probe more precisely how KAP25 interacts with the membrane, we performed additional experiments where the paramagnetic relaxation agents, 5-DSA and 16-DSA,29 have been introduced into the micelles. The consequent drop in the intensities of the peaks corresponding to the residues in close proximity to these reagents has been followed to gain insight on amino acids most profoundly interacting with the membrane. The intensity ratios were mapped on the surface of the peptide structure determined in DPC (Figure 4), representing its direct contacts with the hydrophobic aliphatic tails of this membrane mimetic. The differences in data sets acquired with two paramagnetic labels allow us to estimate, although only qualitatively due to the relatively high dynamics of the detergent system, the depth of peptide insertion and to distinguish between simple surface contacts and more profound membrane imbedding. While in 5-DSA the paramagnetic tag is associated with the part of the tail which is close to the head group and the membrane-water interface, in 16-DSA the paramagnetic tag is attached to the very end of the tail and positioned within the most hydrophobic part of the membrane. According to our combined PRE data KAP25 peptide is mostly associated with the membrane surface. The majority of its 1H alpha resonances are affected more by the interaction with 5-DSA rather than 16-DSA, with the exception of Leu-11, which penetrates deep within the micelle core. The Phe-7 is located at a more intermediate position, as judged by the similar reduction in its resonance intensities caused by the both agents. This is the same Phe-7 side chain that has been involved in many of the emerging tr-NOE peaks for the peptide in complex with SPase I Δ2-75. The N-terminal part of the peptide, partially inserted into the micelles, is restricted by interaction with DPC and characterized by a single conformation. In contrast, the C-terminus, that includes the cleavage site, has additional degrees of freedom manifested by two distinct conformations found in our structure and shows a smaller overall effect of the paramagnetic agents with residues Ala-1 and Val-4 affected the most.
Figure 4.
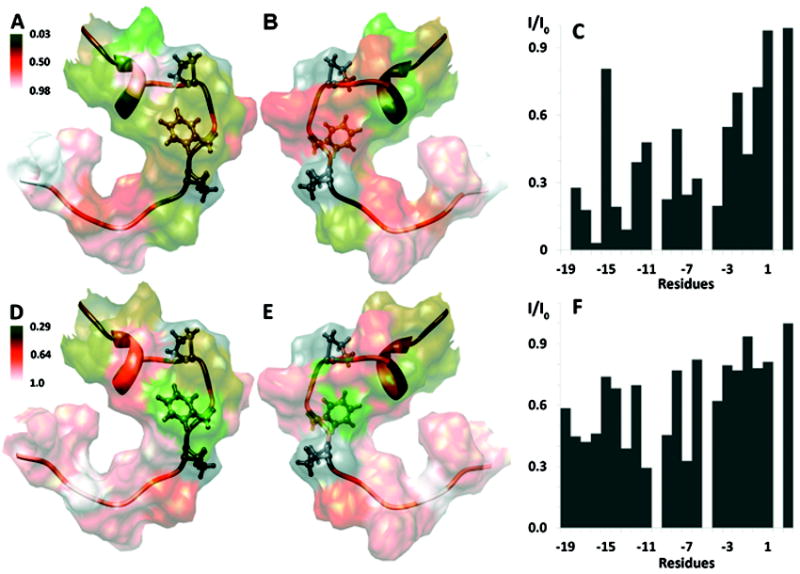
Summary of KAP25 (in ribbon representation) interactions with DPC micelles. The disordered regions (N-terminal residues to -18 and C-terminal to +4) are excluded for clarity. Pro-10, Phe-7, Pro-5 are highlighted in ball and stick representation. The intensity ratios from the PRE experiments measured in the presence of 5- and 16-DSA are mapped onto the surface of Kap25 (A/B and D/E respectively, with 180° rotation between corresponding panels). A green-orange-white color gradient is used (respective color keys are shown adjacent to the figure). The normalized experimental intensity ratios for the KAP25 backbone Hα protons in the presence (I) and in the absence of (I0) of paramagnetic agents are plotted against residue numbers in panels C and F for 5- and 16-DSA, respectively. An intensity ratio of one indicates no effect of the spin label. Molecular graphics images were produced by UCSF Chimera.
Discussion
The signal peptide of an exported protein has a tripartite structure: an N-terminal region with positively charged residues, a central hydrophobic core region, and a more polar C-terminal region preceding the site of cleavage from the remainder of the protein. Positions -1 and -3 relative to the cleavage site are important for signal peptidase recognition and are often occupied by alanine residues or other amino acids with small side chains. The E. coli alkaline phosphatase signal peptide consists of Ala-1 and Thr-3 (Figure 1A). At the -5 position a residue with a small side chain is also frequently found such as Ala (25%), Gly (19%), Ser (26%), and Pro (20%),30 the latter of which is found at this position in the alkaline phosphatase signal peptide.
Using transferred NOE, we find that the peptide adopts an irregular “U-turn” shape originating from the proline residues within the primary sequence. This leaves a signal peptide cleavage region that is not highly structured and exposed for interaction with the enzyme. This is appropriate for access of the -1 and -3 residues permitting cleavage C-terminal to the -1 residue. It is also consistent with the proposed orientation of the translocating preprotein with the signal peptide extending across membrane translocation sites and at least transiently forming a loop about the cleavage region with the mature part of the preprotein being progressively translocated through the translocon from the cytoplasm. We do not know when the signal peptide disengages the SecYEG translocon but evidence indicates it is in direct contact with lipid12 and this may expose the cleavage region for enzymatic cleavage. Given the occurrence of other residues at position -5 of other signal peptides, it is likely that it is its small nature that is critical, rather than the Pro per se, for forming the U-turn. That the same shape is observed upon interaction with SPase I Δ2-75 and in DPC micelles yet stabilized in the latter underscores the possibility that the lipid environment may promote this conformation and place it ideally for signal peptidase recognition. This hypothesis is supported by recent studies indicating the SPase I Δ2-75 catalytic site is located in a hydrophobic β-sheet domain thought capable of inserting into the membrane for cleavage of the signal peptide from the mature protein.8,31 Moreover, site-directed mutagenesis studies have shown Trp300 and Trp310, which are predicted to associate with the membrane surface, to be important for proteolytic activity.32 These data, in addition to the well-known necessity of detergents for optimal proteolytic activity and crystallization of SPase I Δ2-75, support a model where the enzyme cleaves the signal peptide in the membrane or on its surface.
The main difference between the micelle-bound and the SPase-bound KAP25 conformations is that the N-terminus of the core region adopts a helical turn in the presence of lipids. This helix is formed from residue Ile-16 to Ala-13 and may play a role in the interaction with the membrane since it is located far from the cleavage site and this region is not affected by the enzyme. As suggested by our PRE experiments, the Leu-11 is the most membrane penetrating residue and it may play a role in anchoring the peptide to the membrane. The middle portion of the peptide folds in a similar way, adopting an irregular U-turn. The interaction with micelles seems to induce a more rigid conformation for Phe-7, which was similar to the one found in the presence of the enzyme. Moreover, our PRE data indicate that the Phe-7 does not penetrate as deeply into the micelle as Leu-11, thereby it may be available to stabilize the complex with SPase by forming a weak interaction with the hydrophobic residues of the enzyme. The peptide C-terminus, that includes the cleavage site, is not structured under both conditions investigated.
In Figure 5, the superimposition of the two sets of KAP25 structures (representatives of the SPase I Δ2-75 bound ensemble are shown in black, and representatives of the ensemble determined in DPC micelles shown in red) are zoomed over the U-turn (residues -10PLLFTP-5) region. The similarities between the two models are highlighted by the spontaneous alignment of the Phe-7 side-chains in a number of the representative conformers shown from both ensembles even though the superimposition was performed specifically for the backbones. One of the two predominant Phe-7 rotomers, from the loosely defined SPase I Δ2-75-KAP25 complex, overlays very well with the Phe-7 rotomer found in KAP25 under membrane-mimetic conditions. The NMR spectra also reveal that the Phe-7 gives rise to many of the additional peaks observed in the presence of the enzyme while in the DPC micelles containing paramagnetic agents, the signal reduction for the Phe-7 αH proton is particularly strong. Taken together, this suggests that this phenylalanine residue, at the C-terminal end of the signal peptide core region, may play a key role in tethering the peptide for presentation of the cleavage site to the enzyme. A highly hydrophobic residue is often found at this position (Leu 64%) in signal peptides in general.30 While not structured in the enzyme complex, one helical turn of the preceding signal peptide core region is also observed in micelles in good agreement with numerous studies indicating an α-helical conformation of the core region is important during signal peptide membrane insertion.33,34
Figure 5.
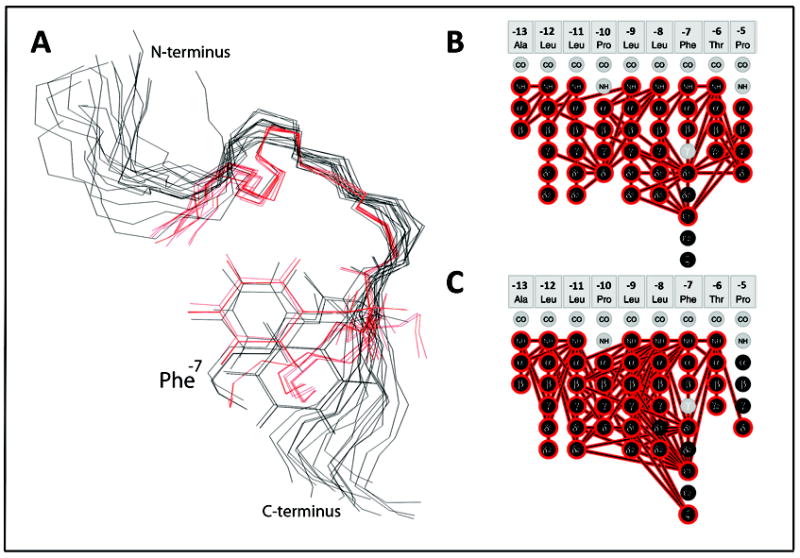
Structural comparison of the two KAP25 folds. (A) The overlay of the KAP25 bound to SPase I Δ2-75, shown in black, and KAP25 in DPC micelles, shown in red. Only the “U-turn” region, structurally restricted by two prolines, Pro-10 and Pro-5, was used for the backbone superposition. The Phe-7 side chains of several representative conformers from both ensembles are shown for comparison. (B) and (C) panels illustrate distance restraints, derived from the observed NOE peaks, which have been used in structural calculations for KAP25 bound to SPase I Δ2-75 or DPC, respectively.
To date several crystal structures of the signal peptidase, including some in complex with an inhibitor, have been reported and the enzyme residues 81-85, 142-149 were proposed to form the binding site including the catalytic dyad Ser90 and Lys145.9 In addition, the membrane association of SPase I was proposed through Trp300, an important residue for peptidase activity.31 We have attempted in silico docking to test whether the KAP25 U-shape conformation, identified by transferred NOE, allows fitting of its C-terminus into the binding pocket of SPase I Δ2-75. We used HADDOCK webserver for these calculations (http://haddock.chem.uu.nl/enmr/eNMR-portal.html). The residues from the binding site of the enzyme and the residues Leu-9 through Glu4 of KAP25 were set as active during the docking. Within the most populated HADDOCK cluster, characterized by the best score and no violations of the distance restraints acquired from transferred NOE data, the C-terminus of the peptide is located inside the catalytic dyad pocket of the enzyme (Figure 6). The stretch of the peptide hydrophobic residues Leu-9, Leu-8 and Phe-7 is close to the corresponding hydrophobic patch on the SPase I Δ2-75 surface which is proposed to associate with a membrane. The N-terminus residues found inside of the DPC micelle are also oriented toward a membrane.
Figure 6.
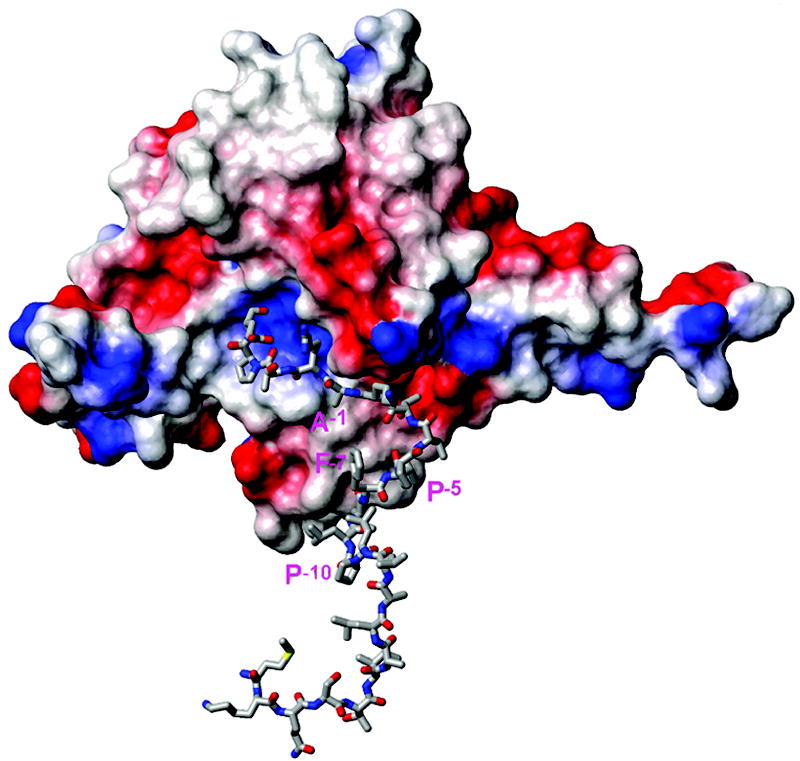
Representative structure of the complex of SPase I Δ2-75 (PDB ID: 1KN9, shown in surface presentation colored by charge) with the KAP25 structure (represented by sticks) as obtained from HADDOCK docking. Residues Ala-1, Pro-5, Phe-7 and Pro-10 of KAP25 are marked in pink (using one letter code). Note that residues from the N-terminus to Pro-10 of the peptide are shown but do not interact with the enzyme and are likely to reside within the membrane.
The SPase I is essential for viability of bacterial cells. Moreover, since its catalytic mechanism employs a Ser-Lys dyad not common to higher organisms, the enzyme is a valuable target for antimicrobial agents. To pursue this possibility, we must delineate the mechanism of recognition of the signal peptide region of a translocating preprotein. The structure of the signal peptide revealed here provides a key step toward the long-term development of inhibitors of signal peptide-signal peptidase I interactions.
Acknowledgments
This work was supported in part by National Institutes of Health grant GM037639 (to D.A.K.) and an American Heart Association grant-in-aid (to O.V.). The eNMR project (European FP7 e-Infrastructure grant, contract no. 213010, www.enmr.eu), supported by the national GRID Initiatives of Italy, Germany and the Dutch BiG Grid project (Netherlands Organization for Scientific Research), is acknowledged for the use of web portals, computing and storage facilities.
References
- 1.Paetzel M, Karla A, Strynadka NC, Dalbey RE. Signal peptidases. Chem Rev. 2002;102:4549–4580. doi: 10.1021/cr010166y. [DOI] [PubMed] [Google Scholar]
- 2.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 3.Bilgin N, Lee JI, Zhu HY, Dalbey R, von Heijne G. Mapping of catalytically important domains in Escherichia coli leader peptidase. EMBO J. 1990;9:2717–2722. doi: 10.1002/j.1460-2075.1990.tb07458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paetzel M, Strynadka NC, Tschantz WR, Casareno R, Bullinger PR, Dalbey RE. Use of site-directed chemical modification to study an essential lysine in Escherichia coli leader peptidase. J Biol Chem. 1997;272:9994–10003. doi: 10.1074/jbc.272.15.9994. [DOI] [PubMed] [Google Scholar]
- 5.von Heijne G. Patterns of amino acids near signal-sequence cleavage sites. Eur J Biochem. 1983;133:17–21. doi: 10.1111/j.1432-1033.1983.tb07424.x. [DOI] [PubMed] [Google Scholar]
- 6.Karamyshev AL, Karamysheva ZN, Kajava AV, Ksenzenko VN, Nesmeyanova MA. Processing of Escherichia coli alkaline phosphatase: role of the primary structure of the signal peptide cleavage region. J Mol Biol. 1998;277:859–870. doi: 10.1006/jmbi.1997.1617. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1998;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Paetzel M, Dalbey RE, Strynadka NC. Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature. 1998;396:186–190. doi: 10.1038/24196. [DOI] [PubMed] [Google Scholar]
- 9.Paetzel M, Dalbey RE, Strynadka NC. Crystal structure of a bacterial signal peptidase apoenzyme: implications for signal peptide binding and the Ser-Lys dyad mechanism. J Biol Chem. 2002;277:9512–9519. doi: 10.1074/jbc.M110983200. [DOI] [PubMed] [Google Scholar]
- 10.Musial-Siwek M, Yeagle PL, Kendall DA. A Small Subset of Signal Peptidase Residues are Perturbed by Signal Peptide Binding. Chem Biol Drug Des. 2008;72:140–146. doi: 10.1111/j.1747-0285.2008.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 12.Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell. 1998;94:795–807. doi: 10.1016/s0092-8674(00)81738-9. [DOI] [PubMed] [Google Scholar]
- 13.Rapoport TA, Goder V, Heinrich SU, Matlack KE. Membrane-protein integration and the role of the translocation channel. Trends Cell Biol. 2004;14:568–575. doi: 10.1016/j.tcb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Paetzel M, Chernaia M, Strynadka N, Tschantz W, Cao G, Dalbey RE, James MN. Crystallization of a soluble, catalytically active form of Escherichia coli leader peptidase. Proteins. 1995;23:122–125. doi: 10.1002/prot.340230115. [DOI] [PubMed] [Google Scholar]
- 15.Wowor AJ, Yu D, Kendall DA, Cole JL. Energetics of SecA Dimerization. J Mol Biol. 2011;408:87–98. doi: 10.1016/j.jmb.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Miller A, Rusch SL, Kendall DA. Demonstration of a specific Escherichia coli SecY-signal peptide interaction. Biochemistry. 2004;43:13185–13192. doi: 10.1021/bi049485k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 18.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR Spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 19.Wuthrich K. NMR of proteins and nucleic acids. New York: John Wiley & Sons; 1998. p. 320. [Google Scholar]
- 20.Grossauer J, Kosol S, Schrank E, Zangger K. The peptide hormone ghrelin binds to membrane-mimetics via its octanoyl chain and an adjacent phenylalanine. Bioorg Med Chem. 2010;18:5083–5088. doi: 10.1016/j.bmc.2010.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 22.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 23.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 24.Tschantz WR, Paetzel M, Cao G, Suciu D, Inouye M, Dalbey RE. Characterization of a soluble, catalytically active form of Escherichia coli leader peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry. 1995;34:3935–3941. doi: 10.1021/bi00012a010. [DOI] [PubMed] [Google Scholar]
- 25.Dev IK, Ray PH, Novak P. Minimum substrate sequence for signal peptidase I of Escherichia coli. J Biol Chem. 1990;265:20069–20072. [PubMed] [Google Scholar]
- 26.Ward LD. Measurement of ligand binding to proteins by fluorescence spectroscopy. Methods Enzymol. 1985;117:400–414. doi: 10.1016/s0076-6879(85)17024-2. [DOI] [PubMed] [Google Scholar]
- 27.Clore GM, Gronenborn AM. Theory and applications of the transferred nuclear Overhauser effect to the study of the conformations of small ligands bound to proteins. J Magn Reson. 1982;48:402–417. [Google Scholar]
- 28.Tieleman DP, van der Spoel D, Berendsen HJC. Molecular dynamics simulations of dodecyl phosphocholine micelles at three different aggregate sizes: micellar structure and lipid chain relaxation. J Phys Chem B. 2000;104:6380–6388. [Google Scholar]
- 29.Hilty C, Wider G, Fernández C, Wüthrich K. Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. Chembiochem. 2004;5:467–473. doi: 10.1002/cbic.200300815. [DOI] [PubMed] [Google Scholar]
- 30.von Heijne G. A new method for predicting signal peptide sequence cleavage sites. Nucl Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Klompenburg W, Paetzel M, de Jong JM, Dalbey RE, Demel RA, von Heijne G, de Kruijff B. Phosphatidylethanolamine mediated insertion of the catalytic domain of leader peptidase in membranes. FEBS Lett. 1998;431:75–79. doi: 10.1016/s0014-5793(98)00733-9. [DOI] [PubMed] [Google Scholar]
- 32.Kim YT, Muramatsu T, Takahashi K. Identification of Trp 300 as an important residue for Escherichia coli leader peptidase activity. Eur J Biochem. 1995;234:358–362. doi: 10.1111/j.1432-1033.1995.358_c.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Miller A, Kendall DA. Signal peptide determinants of SecA binding and stimulation of ATPase activity. J Biol Chem. 2000;275:10154–10159. doi: 10.1074/jbc.275.14.10154. [DOI] [PubMed] [Google Scholar]
- 34.Miller A, Wang L, Kendall DA. Synthetic signal peptides specifically recognize SecA and stimulated ATPase activity in the absence of preprotein. J Biol Chem. 1998;273:11409–11412. doi: 10.1074/jbc.273.19.11409. [DOI] [PubMed] [Google Scholar]