

Abstract
The generation of a B cell repertoire involves producing and subsequently purging autoreactive B cells. Receptor editing, clonal deletion and anergy are key mechanisms of central B cell tolerance. Somatic mutation of antigen-activated B cells within the germinal center produces a second wave of autoreactivity; but the regulatory mechanisms that operate at this phase of B cell activation are poorly understood. We recently identified a post germinal center tolerance checkpoint, where receptor editing is re-induced to extinguish autoreactivity that is generated by somatic hypermutation. Re-induction of the recombinase genes RAG1 and RAG2 in antigen-activated B cells requires antigen to engage the B cell receptor and IL-7 to signal through the IL-7 receptor. We demonstrate that this process requires IL-6 to upregulate IL-7 receptor expression on post germinal center B cells. Diminishing IL-6 by blocking antibody or haplo-insufficiency leads to reduced expression of the IL-7 receptor and RAG and increased titers of anti-DNA antibodies following immunization with a peptide mimetope of DNA. The dependence on IL-6 to initiate receptor editing is B cell intrinsic. Interestingly, estradiol decreases IL-6 expression thereby increasing the anti-DNA response. Our data reveal a novel regulatory cascade to control post germinal center B cell autoreactivity.
1. Introduction
It is well established that the process of V (D) J rearrangement for the generation of the B cell receptor (BCR) leads to a high frequency of autoreactive B cells [1]. These B cells must be purged from the repertoire prior to the acquisition of immunocompetence in order to avoid autoimmune pathology. Numerous elegant studies have revealed mechanisms for the removal or neutralization of such B cells during early B cell development. These mechanisms all rely on engagement of the BCR and include apoptosis, anergy and receptor editing. They occur in the bone marrow and during the transitional B cell stage when B cells exit the bone marrow and migrate to the spleen to complete the process of selection and maturation. Apoptosis rather than proliferation occurs in immature B cells following BCR cross-linking because the events downstream of BCR ligation differ in the immature B cell from those events downstream of BCR ligation in the mature, immunocompetent B cell [2]. The nature of the anergic B cell, now termed transitional 3 (T3), remains controversial. There appear to be a number of phenotypes of anergic B cells, yet all share the features of failure to respond to BCR ligation and a shortened life span unless rescued from the state of anergy [3]. Receptor editing refers to the re-expression of RAG genes once a complete BCR has been formed to generate most commonly a new light chain, but occasionally a new heavy chain [4, 5]. The new heavy and light chain combination has a new antigenic specificity; if no longer autoreactive, the B cell will continue a maturation program to immunocompetence. Receptor editing is initiated in the bone marrow and represents the most frequently occurring mechanism of negative selection or tolerance induction [6].
Studies in mice have shown that both immature and transitional B cells are subject to tolerance induction and studies of human bone marrow and peripheral blood B point to a tolerance checkpoint at the junction of immature and transitional B cells and again at the junction of transitional and mature B cells [7].
Numerous studies have also shown that autoreactivity is generated de novo by somatic hypermutation during the germinal center response following antigen activation [8]. In our own studies of the B cell response to the hapten phosphorylcholine (PC) coupled to a protein carrier, we observed that 40% of the PC-reactive B cells displayed cross-reactivity with double-stranded (ds) DNA and can be rescued from apoptosis through increased expression of the anti-apoptotic molecule Bc1-2 [9]. The mechanisms that operate at this moment to mediate negative selection are of particular interest because many studies of the autoantibodies derived from peripheral blood B cells of patients with autoimmune disease demonstrate the presence of extensive somatic mutation and back mutation to the germline variable region gene sequence often abrogates the autoreactivity [10–13]. We have recently identified a post germinal center tolerance checkpoint, where receptor editing is re-induced to diminish the frequency of autoreactive B cells generated by somatic hypermutation [14]. This process reduces the autoreactivity in a primary response and even more markedly diminishes autoreactivity in the memory compartment [15].
In order to study germinal center B cell selection, we generated a model for antigen-induced anti-DNA antibody production. Immunization of BALB/c mice with a peptide mimetope of DNA, DWEYS, multimerized on a polylysine backbone, MAP-peptide, leads to the productions of anti-DNA antibodies [12]. These lupus-like anti-DNA antibodies have pathogenic potential as they bind glomeruli and neurons [16]. The production of autoantibodies is T cell dependent, Ed restricted and genetically determined by MHC haplotype as DBA/2 mice, which express H-2d like BALB/c mice, mount a robust T cell response to peptide but do not produce autoantibodies [17]. Using fluorochrome-tagged tetramers composed of biotinylated peptide and fluorochrome-tagged streptavidin, we have been able to visualize and isolate antigen-specific B cells. Approximately 75% of tetramer-reactive B cells produce an antibody that cross-reacts with DNA [12]. Some produce an antibody specific for peptide with no DNA reactivity. The tetramer-reactive population can be subdivided into an early and late stage of differentiation based on expression of B220. B220hi cells express GL-7, produce IgM unmutated antibodies, and express AID, exhibiting a phenotype consistent with germinal center B cells. B220lo cells do not express GL7, produce IgG mutated antibodies, and express Blimp-1 and XBP. These are late post-germinal center B cells [15]. We have previously demonstrated that by day 17 after immunization, the tetramer-reactive B220lo population expresses RAG1 and RAG2 and initiates new light chain rearrangements [14]. This process requires BCR engagement, hence; it does not occur in mice treated with DNase to remove the BCR-binding autoantigen [15]. The process also requires IL-7; blocking antibody to the IL-7 receptor (IL-7R) leads to a marked decrease in RAG expression [15]. The role of receptor editing in these antigen-activated B cells is to reduce the number of autoreactive B cells; as interfering with the process leads to increased titers of autoantibodies [15]. Interestingly, the RAG expressing B cells are located in extrafollicular regions of the spleen [15].
It has been reported that re-expression of RAG can be triggered in mature human B cells by exposure to IL-6 [18]. Studies of ex vivo cultures of human peripheral blood B cells have shown that RAG expression can be induced by IL-6 exposure and blocking with anti-IL-6 receptor (IL-6R) antibodies inhibits RAG production in peripheral blood B cells of healthy individuals [18, 19]. Based on this activity of IL-6 in human B cells ex vivo we speculated that IL-6 contributes to the in vivo regulation of receptor editing in post germinal center B cells.
In this study, we show that IL-6 functions to upregulate the expression of IL-7R on tetramer-reactive B220lo B cells. When IL-6 signaling is blocked, either by neutralizing antibody or by genetic deletion, receptor editing is diminished and autoantibody titers increase in mice immunized with the DWEYS peptide. In this process, IL-6 seems to function in an autocrine fashion because a B cell specific decrease in IL-6 production abrogates receptor editing. Moreover, the production of IL-6 is regulated by estrogen; estradiol treatment downregulates IL-6 and, thereby, decreases receptor editing. These observations identify a novel role for IL-6 in tolerance induction in antigen-activated B cells, and further suggest that hormonal milieu contributes to the control of autoreactivity.
2. Materials and Methods
2.1 Mice and Immunizations
3-month-old female wild type (WT) BALB/c mice or BALB/c mice heterozygous for IL-6 expression (IL-6+/−) (The Jackson Laboratory, Bar Harbor, ME) were housed according to AAALAC regulations. Female μMT mice on a (C57BL/6 × 129/Sv) background were obtained from K. Rajewsky (Cologne, Germany)[20] and backcrossed to the BALB/c strain for greater than ten generations. Mice were immunized with 100 μg of DWEYSVWLSN peptide on a poly-lysine backbone (DWEYS-MAP, AnaSpec, San Jose, CA) intraperitoneally on day 0 in complete Freund’s adjuvant (CFA, Difco Laboratories, Detroit, MI). On day 7, mice were boosted intraperitoneally with 100 μg DWEYS-MAP in incomplete Freund’s adjuvant (IFA, Difco Laboratories). Rat anti-mouse-IL-6 (BD Bioscience, San Jose, CA) or isotype control antibody (purified rat IgG1, BD Bioscience) was administrated intravenously (i.v.) on day 8 and day 14. Mice were sacrificed on day 17 for analysis.
2.2 Adoptive Transfer
B cells from WT BALB/c or IL-6+/− mice were purified using the mouse B cell isolation kit (Miltenyi Biotec, Auburn, CA). Purified splenic B cells (2–5 × 107 cells/mouse) were injected i.v. into μMT mice. Three weeks later, recipient μMT mice were immunized with DWEYS-MAP as described above. Spleens from μMT mice with WT B cells or IL-6+/− B cells were harvested on day 17 following the initial immunization.
2.3 Tetramer Generation
DWEYSVWLSN-streptavidin-allophycocyanin (DWEYS-APC) tetramers were generated as previously described [21]. Biotinylated peptide was synthesized by AnaSpec. Streptavidin-APC was purchased from Invitrogen (Carlsbad, CA).
2.4 Cell Sorting
To sort the tetramer binding populations, splenocytes were harvested from 3–5 mice at day 17 after the first immunization and placed in cold Hank’s Balanced Salt Solution (HBSS, Invitrogen) supplemented with 5% fetal calf serum (FCS, Invitrogen) and 10mM HEPES (Invitrogen). PerCP-anti-B220 antibody (clone RA3–6B2, BD Bioscience) and APC-DWEYS tetramer were used to detect antigen-binding B cells. 4′-6-Diamidino-2-phenylindole (DAPI) was added before flow cytometry to exclude dead cells. Immediately after isolation, tetramer-reactive B cells were resuspended in Trizol (Invitrogen) and frozen at −80°C until RNA isolation. Cell sorting was performed on a FACSAria Flow Cytometer (BD Bioscience). Data were analyzed by using FlowJo software (Tree Star Inc., Ashland, OR).
2.5 RNA isolation and qPCR
RNA was extracted from the isolated cells using TriZol reagent (Invitrogen) following the manufacturer’s protocol. cDNA synthesis was performed on 15 μl RNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) in a final volume of 20 μl. qPCR was performed on a LightCycler®480 Real-Time PCR System (Roche, Indianapolis, IN) and analyzed using LightCycler®480 Software (Roche). ABI TaqMan Gene Expression Assays sets were used, and the reactions were performed using LightCycler®480 Probes Master Mix (Roche) in a final volume of 10 μl. All primer sets spanned an intron/exon border. ABI (Applied Biosystems, Carlsbad, CA) Primer IDs: RAG1= Mm01270936_m1, RAG2= Mm00501300_m1, IL-7R= Mm00434295_m1, IL-6= Mm99999064_m1, polr2a= Mm00839493_m1.
2.6 Histology
Spleens were collected on day 17 after the initial immunization and frozen in Tissue-Tek OCT Compound (Miles, Elkhart, IN) by immersion in a 2-methylbutane bath on dry ice. Sections were cut to 5-micron slices using a Leica CM3050S cryostat. Prior to staining, sections were warmed to room temperature and rehydrated in PBS. Slides were then fixed in ice-cold acetone for 10 minutes and blocked with PBS, containing 2% BSA, 5% goat serum, and 0.2% Triton X-100 (blocking buffer), for 30 minutes at room temperature. Staining was performed with antibodies in blocking buffer for 1.5 hour at room temperature. Slides were washed in PBS and mounted in Aqua-Poly/Mount (Polysciences, Inc, Warrington, PA). Fluorescence microscopy was performed on an AxioCam II microscope (Carl Zeiss Microimaging, Thornwood, NY). Image acquisition was performed with a Hamamatu ORCA-ER camera using the Openlab imaging software (PerkinElmer, Inc., Waltham, MA). The following antibodies were used: anti-RAG1 or anti-RAG2 (rabbit polyclonal, BD Biosicence), PE-anti-B220 (RA3-6B2, BD Bioscience), Streptavidin-Alexa-Fluor-350 (Invitrogen) for tetramer labeling. Anti-RAG antibodies were detected using Zenon Alexa-Fluor-488 anti-rabbit IgG labeling kit (Invitrogen). Isotype control for RAG was rabbit IgG (blocking reagent from the kit).
2.7 ELISA
ELISA plates (Costar, Corning, NY) were coated with 15 μg/ml DWEYS-MAP or 100 μg/ml sonicated, filtered calf-thymus DNA (Calbiochem, San Diego, CA). Plates were blocked with phosphate buffered saline with 1% bovine serum albumin (PBS/BSA). Serum antibodies were diluted in PBS/0.5%BSA and incubated for one hour at room temperature for the peptide ELISA or 37°C for DNA ELISA. After washing the plates in PBS-Tween20, anti-mouse-IgG-AP (goat polyclonal, Southern Biotech, Birmingham, AL) in PBS/0.5% BSA was added at 1 μg/ml. Plates were washed and 1mg/ml PNPP (Sigma, St Louis, MO) was added for detection. The assay was read at 405nm on a 1430 Multilabel Counter Spectrometer (PerkinElmer, Inc., Waltham, MA).
2.8 Ovariectomy
3-month-old female mice were ovariectomized. Sixty-day time-release pellets containing 17β estradiol (E2, 0.18 mg/pellet) or placebo pellets (vehicle control) (Innovative Research of America, Sarasota, FL) were implanted subcutaneously. The E2 implant elevates serum E2 levels to 75–100 pg/ml [22]. Mice were then rested for 7–10 days before immunization.
2.9 Data Analysis
Data were analyzed in Microsoft Excel (Microsoft, Redmond, WA) or Graphpad Prism v4.02 (Graphpad Software, San Diego, CA).
3. Results
3.1 Anti-IL-6 antibody treatment diminishes the expression of RAG and IL-7R in DWEYS-MAP immunized BALB/c mice
In vitro studies showed that IL-6 regulates the expression of RAG in mature human B cells [18]. We were interested in determining whether IL-6 also participates in the regulation of RAG and receptor editing in antigen-activated post-germinal center B cells in vivo. We, therefore, immunized BALB/c mice with MAP-peptide as previously described, to induce a cross-reactive anti-peptide, anti-DNA response. On day 8 and 14 following immunization, we administered either anti-IL-6 or isotype control antibody. Three days later, we harvested spleen and isolated B220hi and B220lo tetramer-reactive B cells by flow cytometry.
We first examined the transcription of RAG by qPCR and observed that anti-IL-6-treated mice displayed a reduction in RAG expression in the antigen-specific (DWEYS-tetramer binding, Figure 1A and 1D/1E) B220lo B cells compared to mice treated with isotype control antibody (Figure 1B). This observation was confirmed by immunohistochemistry of the spleen showing a decrease in RAG protein in anti-IL-6-treated mice (Figure 1B and supplementary Figure 1). To gain insight into how RAG is downregulated by the IL-6 blocking antibody, we next measured the expression of IL-7R, as previous data from our laboratory showed that the B220lo antigen-specific B cells upregulate IL-7R and receptor editing in this population requires IL-7R signaling [15]. qPCR analysis showed that the expression of IL-7R in the B220lo antigen-specific B cells is reduced in mice given anti-IL-6 antibody compared to control group (Figure 1C). These data suggest that IL-6 is required for re-induction of RAG in antigen-activated B cells and that IL-6 may act through regulating IL-7/IL-7R signaling.
Figure 1. Reduced RAG/IL-7R expression after anti-IL-6 treatment in DWEYS-MAP immunized BALB/c mice.


BALB/c mice (3-month-old) were immunized with DWEYS-MAP and boosted a week later (see Material and Methods). Anti-IL-6 antibody or isotype control (3–5 mice each group) was administrated to mice intravenously at day 8 and day 14 following the initial immunization. Spleens were collected at day 17. Antigen specific B cells were sorted as shown in (A) and RNA was extracted for qPCR to assess RAG (B) and IL-7R (C) mRNA. Spleen sections were frozen on the same day and immunohistochemistry was performed to detect RAG (left column, staining in green) and DWEYS peptide reactivity (mid column, tetramer staining in blue) levels (D and E).
3.2 Anti-IL-6 treatment elevates serum autoantibody titers in DWEYS-MAP immunized BALB/c mice
Next, we wanted to examine the impact of diminished RAG expression in mice treated with anti-IL-6 antibody on the production of autoantibodies. On week 6 after the initial immunization, we collected serum from anti-IL-6-treated or isotype control-treated mice. The serum level of anti-dsDNA antibody was significantly higher in the anti-IL-6 treated mice, while the anti-DWEYS peptide level was not affected by anti-IL-6 administration (Figure 2). These data indicates IL-6 contributes to the regulation of autoimmunity, and that the anti-IL-6 antibody, that treatment did not affect the immune response to foreign antigens.
Figure 2. Elevated serum anti-dsDNA IgG titer after anti-IL-6 treatment in DWEYS-MAP immunized BALB/c mice.
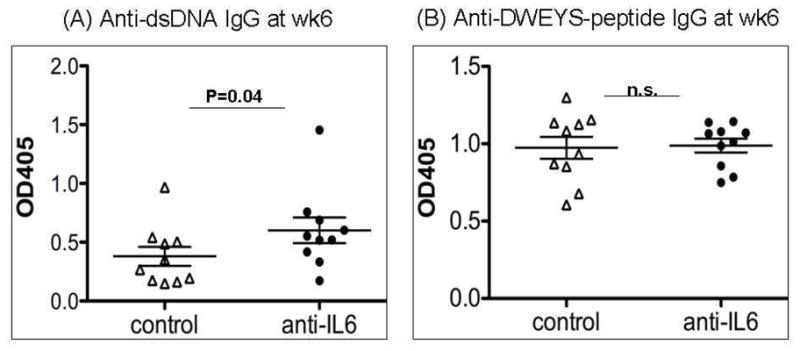
BALB/c mice (3-month-old) were immunized with DWEYS-MAP and boosted a week later (see Material and Methods). Anti-IL-6 antibody or isotype control (10 mice each group) was injected to mice intravenously at day 8 and day 14 following the initial immunization. Serum was collected at week 6 post initial immunization. ELISA was performed to assess IgG titer of anti-dsDNA autoantibody (A) and anti-DWEYS-peptide antibody levels (B). The experiment was performed twice. Data are from a representative experiment.
3.3 IL-6 haplo-deficiency diminishes RAG/IL-7R expression in DWEYS-MAP immunized BALB/c mice
To confirm the requirement for IL-6 to initiate receptor editing, we repeated the study in mice that were heterozygous for a deletion of the IL-6 gene. BALB/c mice heterozygous for IL-6 (IL-6+/−) were immunized with DWEYS-MAP as described above. On day 17, we harvested spleen cells and isolated B220hi and B220lo cells. The IL-6+/− mice had fewer RAG-expressing B cells in their extrafollicular region compared to wild-type BALB/c mice (Figure 3A and 3B). Consistent with the data generated in mice treated with anti-IL6 antibody, IL-7R was also reduced in this subset.
Figure 3. Reduced RAG/IL-7R expression in DWEYS-MAP immunized IL-6+/− mice.

IL-6 heterozygous (IL-6+/−) mice (3-month old) and age-matched wild type BALB/c mice were immunized with DWEYS-MAP and boosted a week later (see Material and Methods). Antigen specific B cells were sorted on day 17 following the initial immunization and RNA was extracted for qPCR to check RAG and IL-7R mRNA (A). Spleen sections were frozen on the same day of sorting and immunohistochemistry was performed to detect RAG protein levels (B).
3.4 B cell production of IL-6 is required for re-induction of receptor editing in activated B cells
According to Jamin and colleagues, the IL-6 that contributes to RAG expression is made by B cells and functions in an autocrine fashion [19]. We first showed that B cells undergoing receptor editing produce IL-6 (Figure 4A). The antigen-activated B cells themselves are a source of IL-6. We further explored this paradigm by transferring splenic B cells from wild type or IL-6+/− mice to BALB/c μMT mice. We then immunized the recipient mice with DWEYS-MAP as described above. Because all the B cells in the recipient mice were of donor origin, we were able to address the requirement for B cell-derived IL-6 in this process. As shown in Figure 4B, both RAG and IL-7R transcripts levels were decreased in the μMT mice reconstituted with IL-6+/− B cells. RAG protein levels were reduced in the spleens of mice reconstituted with B cells from IL-6 heterozygous (Figure 4C). In composite, these observations indicate that the dependence on IL-6 to initiate receptor editing is B cell intrinsic.
Figure 4. B cell production of IL-6 is required for re-induction of receptor editing. BALB/c mice (3-month-old) were immunized with DWEYS-MAP and boosted a week later. RNA was extracted from splenocytes at day 17 for qPCR to assess IL-6 mRNA level.

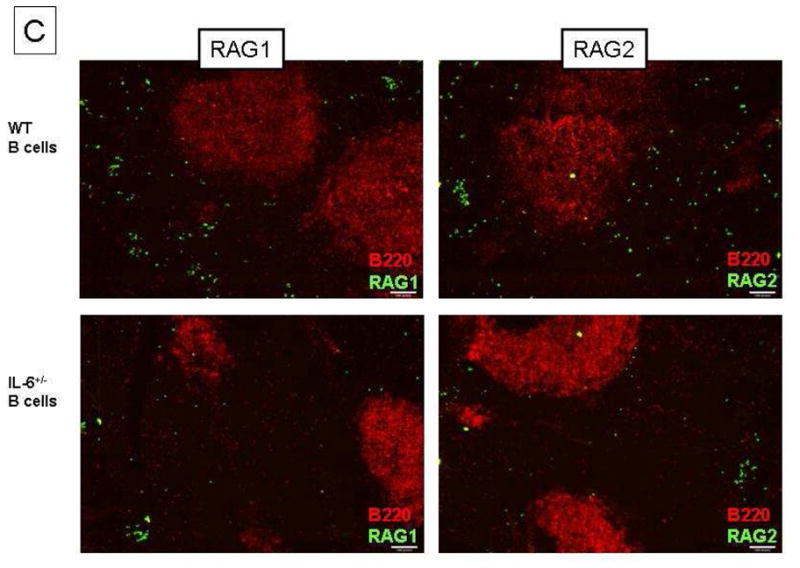
(A). Splenic B cells from 3-month-old IL-6+/− mice or age-matched wild type BALB/c mice were purified and adoptively transferred to μMT mice by intravenous injections. 3 weeks later, the recipient mice were immunized with DWEYS-MAP and boosted a week later (see Material and Methods). Spleens were collected at day 17. Antigen specific B cells were isolated and RNA was extracted for qPCR to assess RAG and IL-7R mRNA (B). Spleen sections were frozen on the same day and immunohistochemistry was performed to detect RAG protein levels (C).
3.5 Estrogen regulates receptor editing in activated B cells through IL-6/IL-7R
Previous studies from our laboratory have shown that hormones can modulate B cell development and central tolerance. In particular, estrogen increases the signaling threshold for B cell apoptosis and abrogates naïve B cell tolerance at both the immature to T1 junction and the T2 to naïve junction [22, 23]. Thus, mice transgenic for the R4A heavy chain of an anti-dsDNA antibody exhibit an expanded marginal zone compartment that is enriched with immunocompetent DNA-specific autoreactive B cells when treated with estradiol [23]. Interestingly, RAG expression is negatively regulated by estrogen [24]. We, therefore, investigated the regulatory role of estrogen on receptor editing in antigen-activated B cells.
To circumvent the influence of the endogenous estrus cycle on estrogen concentrations, ovariectomy was performed on BALB/c mice 7–10 days prior to immunization with DWEYS-MAP. Mice were implanted with placebo or 17β estradiol (E2) pellets (60-day release) subcutaneously. The E2 pellet generates a continuous serum concentration of 75–100pg/ml, which represents peak levels during the estrus cycle [22].
On day 17 following the initial immunization with MAP-peptide it was evident that RAG transcripts in antigen-specific B cells were decreased by estradiol treatment (Figure 5). Estradiol-treated mice displayed lower expression of IL-7R on B220lo tetramer-reactive B cells than placebo-treated mice. These changes are likely due to an estradiol-mediated reduction in IL-6, as IL-6 transcripts were also lower in estradiol-treated mice in the antigen-specific B220lo population that is the B cell source of IL-6 (Figure 5). Moreover, estradiol treated mice had significantly higher levels of anti-dsDNA autoantibodies in their sera than placebo-treated mice (Figure 6). Thus, in addition to interfering with tolerance induction in immature B cells, estrogen also perturbs tolerance maintenance in antigen-activated mature B cells by inhibiting receptor editing. The observed drop in IL-6 production is consistent with a model in which the effects of estrogen on editing are mediated through its effects on IL-6.
Figure 5. Estrogen downregulates RAG expression through IL-6/IL-7R.

BALB/c mice (3-month-old) were ovariectomized and implanted with either estradiol (E2) or placebo pellets subcutaneously. Mice were rested 7–10 days and then immunized with DWEYS-MAP as described in Material and Methods. On day 17, antigen-specific B cells were isolated and RNA was extracted for qPCR to assess mRNA levels of RAG, IL-7R and IL-6.
Figure 6. Estrogen increases serum anti-dsDNA IgG titer in DWEYS-immunized BALB/c mice.

BALB/c mice (3-month-old) were ovariectomized and implanted with either estradiol (E2) or placebo pellets subcutaneously. Mice were rested 7–10 days and then immunized with DWEYS-MAP as described in Material and Methods. On week 6 following the initial immunization, serum was collected to detect anti-dsDNA autoantibody (A) and anti-DWEYS-peptide antibody (B) levels by ELISA.
4. Discussion
In this study, we have identified a novel function of IL-6 in the maintenance of B cell tolerance. IL-6, a cytokine produced by a variety of cells, mediates a broad range of biological activities including immune regulation, inflammation, hematopoiesis and oncogenesis [25, 26]. It was first discovered as a T-cell-derived B cell stimulation factor that induces B cells to produce IgG antibodies [26]. The role of IL-6 in promoting B cell terminal differentiation into antibody-secreting plasma cell has been well demonstrated [25]. Studies in myeloma cells and B cell lymphomas have shown that IL-6 functions in an autocrine fashion to promote cell proliferation and survival but paracrine signaling also occurs [27].
The importance of IL-6 in immune homeostasis has been highlighted in studies using IL-6 deficient mice and IL-6 overexpressing mice. IL-6 deficient mice display impaired GC formation with a decrease in both size and number of GCs [28]. The mice also exhibit defects in T cell growth and function, failure of an acute phase response and impaired self-renewal of uncommitted progenitor cells [29]. Mice overexpressing the human form of IL-6 have increased IgG production, an increased number of plasma cells, and autoantibody production [30].
We investigated the role of IL-6 in the regulation of receptor editing after antigen activation when autoreactive BCRs have been generated through somatic hypermutation during the GC reaction. We were led to consider the role of IL-6 by previous studies by Jamin and colleagues who demonstrated that human mature peripheral blood B cells upregulate RAG expression upon BCR stimulation and that abrogation of IL-6 signaling with a blocking antibody to the IL-6 receptor (IL-6R) inhibits the expression of RAG genes [18, 19]. In the present study, blocking IL-6 activity by anti-IL-6 antibody not only reduced RAG expression but also decreased B cell expression of IL-7R. Previous studies from our laboratory have demonstrated the importance of signaling through the IL-7R in the initiation of receptor editing in post GC B cells [15]. These observations suggest that IL-6 regulates receptor editing in activated B cells through the control of IL-7/IL-7R signaling. It is important to note that receptor editing alters autoantibody response without altering the antibody response to foreign antigen; thus, it does not lead to impairment in protective immunity. It is possible that the most critical function of the receptor editing process is to purge the B cells that enter the memory compartment of autoreactivity as we have previously shown [15].
The dependence of light chain rearrangement on IL-7 signaling remains controversial. Early studies demonstrated that withdrawing IL-7 allowed pre-B cells to mature and generate an intact light chain gene [31]. Recent studies in a pre-B cell line have been cited to confirm that attenuation of IL-7 signaling promotes RAG expression [32]. In contrast, others have argued that these data reflect an in vitro culture system in which cells lack exposure to some vital factors like hepatocyte growth factor β that forms a chimeric molecule, pre-pro-B cell growth stimulating factor (PPBSF), which is required to upregulate IL-7R, thereby enabling the B cell to respond to IL-7 [33]. In fact, others have shown directly that IL-7 does not prevent light chain rearrangement in pre-B cells [34]. Rather, withdrawal of IL-7 from the culture only causes the death of a subset of IgM-negative pro-B cells but does not change the number of cells that differentiate to the surface IgM-positive stage. Our recent studies, together with those described here, further support the notion that IL-7R does not repress light-chain rearrangement, but rather, is positively correlated with RAG expression in post-germinal center B cells [15]. Moreover, inhibition of the IL-7R abrogates RAG expression in these cells and leads to greater autoantibody production [15].
Although the present study shows that IL-6 is required for receptor editing in antigen-activated B cells and controls titers of serum autoantibody, the pleiotropic IL-6 also plays a pivotal, but contradictory, role in pathogenesis of many autoimmune and inflammatory diseases [25]. Elevated IL-6 levels have been found in serum of individuals with systemic lupus erythematosus (SLE), Sjogren’s syndrome (SS) and rheumatoid arthritis (RA) patients [25]. A humanized anti-IL-6 antibody, tocilizumab, has demonstrated efficacy in the treatment of RA and has been approved for clinical use [35]. IL-6 directly contributes to the production of autoantibodies since anti-IL-6R antibody diminishes anti-dsDNA production by B cells of SLE patients [36] and anti-aquaporin production by B cells of patients with neuromyelitis optica [37]. B cells from NZB/W F1 mice that spontaneously develop SLE show hyper-responsiveness to IL-6 and produce anti-DNA antibody following exposure to IL-6 [38]. IL-6R blockade ablates the onset of autoimmune kidney disease in these mice [38]. In another SLE-like autoimmune mouse model, the Lyn-deficient mice, Lyn-null B cells produce high levels of IL-6 and genetic deletion of IL-6 ameliorates the autoimmune disease in these mice [39]. B cell-derived IL-6 directly facilitates activation of B cells, splenomegaly and generation of pathogenic autoantibodies in the Lyn-deficient mice [39]. The constitutive expression of IL-6R on SLE B cells and an auto-stimulatory function of IL-6 in proliferation and differentiation of these cells has been reported [40]. IL-6 helps modify autoreactive B cell receptors after the GC reaction, but we speculate that if any such cells pass through that checkpoint and become cells that are responsive to IL-6’s autocrine, proliferative effects, autoimmunity is thus promoted by IL-6.
The role of estrogen in SLE is somewhat controversial but a substantial amount of data suggests that sex hormones contribute to disease expression. The incidence of SLE in human is 9 times greater in woman than in men, but in individuals with disease onset prior to puberty or after menopause the female to male ratio is 3:1 [41]. The timing of lupus onset and disease activity suggests that female sex hormones may be regulators of lupus activity [42]. Studies of animal models provide clear insight into the hormonal modulation of autoimmune diseases. In the lupus-prone NZB/W mouse, there is a greater incidence of disease in females than in males and administration of exogenous estrogen leads to earlier onset and increased severity of disease [43]. In contrast, ovariectomy of female mice reduces disease incidence and severity and castration of male mice increases their disease incidence [43]. Estrogen also exacerbates disease in another spontaneous mouse model of lupus, the MRL/lpr mouse, although to a lesser extent [44].
Studies from our laboratory indicate that estrogen can regulate B cell development and tolerance induction in mice. Estrogen increases the signaling threshold for BCR-mediated apoptosis of transitional B cells and impairs naïve B cell tolerance [22, 23]. Because estrogen has been shown to down-regulate the expression of RAG [24] and IL-6 [45, 46] in B cell precursors and osteoblasts, respectively, we asked whether it also plays a similar role in mature post-GC B cells. Indeed, our data confirm the suppressive effect of estrogen on RAG and IL-6 expression in antigen-activated B cells. In our DWEYS-induced autoimmunity model, BCR engagement by autoantigen, along with autocrine IL-6 production and IL-7R signaling leads to induction of RAG expression and subsequent light-chain editing to convert the cell to an innocuous specificity. This whole process is inhibited by estrogen, while we did not determine whether estrogen regulates RAG expression directly or indirectly, we surmise an indirect effect through IL-6 production, although it may be that there are both direct and indirect effects of estrogen on RAG gene expression.
Two signals are involved in mediating RAG expression in lymphoid progenitor cells [47]. Interestingly, both our model of receptor editing in antigen-activated in B cells and the model of receptor editing in cultured mature human B cells also require two signals. Our model requires BCR engagement and IL-7 signaling; human B cells require BAFF as well as IL-6 [48]. Whether the same downstream pathways are involved in each model remains to be determined. An increased number of RAG-expressing B cells are observed in both the peripheral blood of SLE patients and in the synovial tissues of RA patients [40]. This may reflect high serum levels of IL-6, which are present in both diseases.
In the present study, we elucidated a novel function of IL-6 in the modulation of tolerance induction in antigen-experienced B cells. This study suggests a need for careful monitoring of autoreactive B cells as anti-IL-6 antibody is taken to the clinic in SLE treatment. More importantly, it demonstrates that IL-6 has functions that are pro-inflammatory, but it also functions to control autoimmunity. Similarly, IL-10 also functions in homeostatic regulation. It has also been suggested to act in an autocrine fashion to promote B cell survival and autoantibody secretion [49] and yet B cell production of IL-10 can suppress immune responses [50]. Herein, we identify an analogous quality in IL-6 function in which balanced IL-6 secretion is required for a properly behaved immune system; homeostasis is maintained through a coordination of discrete and seeming opposite functions of key cytokines.
Supplementary Material
IL-6 is required for post germinal center receptor editing that diminishes autoreactivity.
IL-6 is regulated by estrogen with estrogen decreasing IL-6 production by B cells.
These data suggest one mechanism for the increased humoral autoimmunity in women.
Acknowledgments
The authors gratefully acknowledge Dr. Laurel Eckhardt (Hunter College, The City University of New York) for helpful discussions and critical reading of the manuscript. We thank Ms. Sylvia Jones for the help of preparing the manuscript and Flow Cytometry Facility of Feinstein Institute for expert help with cell sorting.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 2.Basten A, Silveira PA. B-cell tolerance: mechanisms and implications. Curr Opin Immunol. 2010;22:566–74. doi: 10.1016/j.coi.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B-cell anergy. Immunol Rev. 2010;237:249–63. doi: 10.1111/j.1600-065X.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–20. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat Immunol. 2004;5:645–50. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 7.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9:845–57. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 9.Kuo P, Alban A, Gebhard D, Diamond B. Overexpression of bcl-2 alters usage of mutational hot spots in germinal center B cells. Mol Immunol. 1997;34:1011–8. doi: 10.1016/s0161-5890(97)00117-x. [DOI] [PubMed] [Google Scholar]
- 10.Diamond B, Scharff MD. Somatic mutation of the T15 heavy chain gives rise to an antibody with autoantibody specificity. Proc Natl Acad Sci U S A. 1984;81:5841–4. doi: 10.1073/pnas.81.18.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Putterman C, Limpanasithikul W, Edelman M, Diamond B. The double edged sword of the immune response: mutational analysis of a murine anti-pneumococcal, anti-DNA antibody. J Clin Invest. 1996;97:2251–9. doi: 10.1172/JCI118666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Putterman C, Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med. 1998;188:29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. 2007;26:205–13. doi: 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice JS, Newman J, Wang C, Michael DJ, Diamond B. Receptor editing in peripheral B cell tolerance. Proc Natl Acad Sci U S A. 2005;102:1608–13. doi: 10.1073/pnas.0409217102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang YH, Diamond B. B cell receptor revision diminishes the autoreactive B cell response after antigen activation in mice. J Clin Invest. 2008;118:2896–907. doi: 10.1172/JCI35618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–93. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 17.Wang C, Khalil M, Ravetch J, Diamond B. The naive B cell repertoire predisposes to antigen-induced systemic lupus erythematosus. J Immunol. 2003;170:4826–32. doi: 10.4049/jimmunol.170.9.4826. [DOI] [PubMed] [Google Scholar]
- 18.Hillion S, Dueymes M, Youinou P, Jamin C. IL-6 contributes to the expression of RAGs in human mature B cells. J Immunol. 2007;179:6790–8. doi: 10.4049/jimmunol.179.10.6790. [DOI] [PubMed] [Google Scholar]
- 19.Hillion S, Garaud S, Devauchelle V, Bordron A, Berthou C, Youinou P, Jamin C. Interleukin-6 is responsible for aberrant B-cell receptor-mediated regulation of RAG expression in systemic lupus erythematosus. Immunology. 2007;122:371–80. doi: 10.1111/j.1365-2567.2007.02649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–6. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 21.Newman J, Rice JS, Wang C, Harris SL, Diamond B. Identification of an antigen-specific B cell population. J Immunol Methods. 2003;272:177–87. doi: 10.1016/s0022-1759(02)00499-4. [DOI] [PubMed] [Google Scholar]
- 22.Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci U S A. 2000;97:2703–8. doi: 10.1073/pnas.040577497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. 2002;109:1625–33. doi: 10.1172/JCI14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medina KL, Strasser A, Kincade PW. Estrogen influences the differentiation, proliferation, and survival of early B-lineage precursors. Blood. 2000;95:2059–67. [PubMed] [Google Scholar]
- 25.Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011;22:83–9. doi: 10.1016/j.cytogfr.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73–6. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 27.Weidle UH, Klostermann S, Eggle D, Kruger A. Interleukin 6/interleukin 6 receptor interaction and its role as a therapeutic target for treatment of cachexia and cancer. Cancer Genomics Proteomics. 2010;7:287–302. [PubMed] [Google Scholar]
- 28.Kishimoto T. IL-6: from laboratory to bedside. Clin Rev Allergy Immunol. 2005;28:177–86. doi: 10.1385/CRIAI:28:3:177. [DOI] [PubMed] [Google Scholar]
- 29.Kopf M, Ramsay A, Brombacher F, Baumann H, Freer G, Galanos C, Gutierrez-Ramos JC, Kohler G. Pleiotropic defects of IL-6-deficient mice including early hematopoiesis, T and B cell function, and acute phase responses. Ann N Y Acad Sci. 1995;762:308–18. doi: 10.1111/j.1749-6632.1995.tb32335.x. [DOI] [PubMed] [Google Scholar]
- 30.Fattori E, Della Rocca C, Costa P, Giorgio M, Dente B, Pozzi L, Ciliberto G. Development of progressive kidney damage and myeloma kidney in interleukin-6 transgenic mice. Blood. 1994;83:2570–9. [PubMed] [Google Scholar]
- 31.Espeli M, Rossi B, Mancini SJ, Roche P, Gauthier L, Schiff C. Initiation of pre-B cell receptor signaling: common and distinctive features in human and mouse. Semin Immunol. 2006;18:56–66. doi: 10.1016/j.smim.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, Singh H. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–45. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 33.Wei C, Lai L, Goldschneider I. Pre-pro-B cell growth-stimulating factor (PPBSF) upregulates IL-7Ralpha chain expression and enables pro-B cells to respond to monomeric IL-7. J Interferon Cytokine Res. 2002;22:823–32. doi: 10.1089/107999002320271422. [DOI] [PubMed] [Google Scholar]
- 34.Milne CD, Fleming HE, Paige CJ. IL-7 does not prevent pro-B/pre-B cell maturation to the immature/sIgM(+) stage. Eur J Immunol. 2004;34:2647–55. doi: 10.1002/eji.200425400. [DOI] [PubMed] [Google Scholar]
- 35.Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22:347–52. doi: 10.1093/intimm/dxq030. [DOI] [PubMed] [Google Scholar]
- 36.Nagafuchi H, Suzuki N, Mizushima Y, Sakane T. Constitutive expression of IL-6 receptors and their role in the excessive B cell function in patients with systemic lupus erythematosus. J Immunol. 1993;151:6525–34. [PubMed] [Google Scholar]
- 37.Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011;108:3701–6. doi: 10.1073/pnas.1017385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mihara M, Takagi N, Takeda Y, Ohsugi Y. IL-6 receptor blockage inhibits the onset of autoimmune kidney disease in NZB/W F1 mice. Clinical and experimental immunology. 1998;112:397–402. doi: 10.1046/j.1365-2249.1998.00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML. Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J Immunol. 2010;184:1348–60. doi: 10.4049/jimmunol.0901878. [DOI] [PubMed] [Google Scholar]
- 40.Hillion S, Rochas C, Youinou P, Jamin C. Signaling pathways regulating RAG expression in B lymphocytes. Autoimmun Rev. 2009;8:599–604. doi: 10.1016/j.autrev.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–6. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 42.Walker SE. Estrogen and autoimmune disease. Clin Rev Allergy Immunol. 2010;40:60–5. doi: 10.1007/s12016-010-8199-x. [DOI] [PubMed] [Google Scholar]
- 43.Roubinian JR, Talal N, Greenspan JS, Goodman JR, Siiteri PK. Effect of castration and sex hormone treatment on survival, anti-nucleic acid antibodies, and glomerulonephritis in NZB/NZW F1 mice. J Exp Med. 1978;147:1568–83. doi: 10.1084/jem.147.6.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlsten H, Tarkowski A, Holmdahl R, Nilsson LA. Oestrogen is a potent disease accelerator in SLE-prone MRL lpr/lpr mice. Clin Exp Immunol. 1990;80:467–73. doi: 10.1111/j.1365-2249.1990.tb03311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stein B, Yang MX. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol Cell Biol. 1995;15:4971–9. doi: 10.1128/mcb.15.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galien R, Garcia T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. 1997;25:2424–9. doi: 10.1093/nar/25.12.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tagoh H, Kishi H, Okumura A, Kitagawa T, Nagata T, Mori K, Muraguchi A. Induction of recombination activating gene expression in a human lymphoid progenitor cell line: requirement of two separate signals from stromal cells and cytokines. Blood. 1996;88:4463–73. [PubMed] [Google Scholar]
- 48.Rochas C, Hillion S, Saraux A, Mageed RA, Youinou P, Jamin C, Devauchelle V. Transmembrane BAFF from rheumatoid synoviocytes requires interleukin-6 to induce the expression of recombination-activating gene in B lymphocytes. Arthritis Rheum. 2009;60:1261–71. doi: 10.1002/art.24498. [DOI] [PubMed] [Google Scholar]
- 49.Llorente L, Zou W, Levy Y, Richaud-Patin Y, Wijdenes J, Alcocer-Varela J, Morel-Fourrier B, Brouet JC, Alarcon-Segovia D, Galanaud P, et al. Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J Exp Med. 1995;181:839–44. doi: 10.1084/jem.181.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev. 2008;224:201–14. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.