

Abstract
Shigella flexneri, a facultative intracellular pathogen, is exposed to a variety of environments inside and outside of the human host. Some of these environments may contain significant oxidative stress. S. flexneri mutants were generated with deletions in the major oxidative stress regulators oxyR and/or soxRS to test their importance in Shigella biology. Strains that contained a deletion of oxyR had reduced growth and survival during aerobic growth, but not microaerobic growth. The mutants were also defective in surviving exposure to oxidative stress: oxyR mutants were sensitive to hydrogen peroxide, while soxRS mutants were sensitive to superoxide. Although the ΔsoxRS, ΔoxyR, and ΔoxyR/ΔsoxRS mutant Shigellae survived similarly to the parental strains within macrophages, the mutants formed plaques on Henle cell monolayers that were slightly smaller than the plaques formed by the wildtype strain.
Keywords: Shigella, oxidative stress, OxyR, SoxRS
1. Introduction
The facultative intracellular pathogens from the genus Shigella are the causative agents of shigellosis. Shigella invades the epithelium of the human colon and spreads from cell-to-cell, resulting in a characteristic intestinal tract aggravation and subsequent dysentery-like symptoms caused by inflammation in response to Shigella invasion (Takeuchi et al., 1965). In healthy adults, shigellosis is self-limiting, and the infection is likely cleared by neutrophils (Mandic-Mulec et al., 1997). Of the four species, S. flexneri is the most common cause of endemic shigellosis in the developing world(Bennish and Wojtyniak, 1991).
Shigella initially crosses the colonic epithelium through the M cells of the follicular-associated epithelium and is then phagocytosed by resident macrophages; however, Shigella rapidly escapes the phagocytotic vacuole, initiates apoptosis in the macrophage, and is released by the apoptotic macrophage into the basolateral space of the colonic epithelium. Shigella can then attach to and invade colonic epithelial cells. Within these epithelial cells, the phagocytic vacuole is lysed and Shigella is released into the cytoplasm. Unlike in macrophages, Shigella-infected epithelial cells do not undergo rapid cell death, but instead serve as a replicative niche where the bacteria multiply and eventually spread to adjacent cells via actin based motility, with limited exposure to the extracellular environment (for review see (Sansonetti et al., 2001)).
Shigella is exposed to a variety of environments, both inside and outside of the human host. Some of these environments may contain significant oxidative stress. Microarray analysis has shown that expression of several oxidative stress response genes (oxyR, soxR, soxS and grxA) is induced when Shigella is within the epithelial or macrophage cytosol (Lucchini et al., 2005). Furthermore, normal flora in the colon may produce reactive oxygen species, and bile may cause oxidative stress through the generation of free radicals(Begley et al., 2005). Additionally, in aerobic environments, Shigella must cope with the oxidative stress associated with autooxidation during normal aerobic growth(Dukan and Nystrom, 1999).
Two of the primary mediators of oxidative stress are the reactive oxygen species (ROS) superoxide (O2−) and hydrogen peroxide (H2O2). Arising primarily from the transfer of electrons to oxygen molecules, O2− and H2O2 damage numerous biomolecules within cells, and thus disrupt many cellular processes(Imlay, 2003). In all aerobic organisms, the principal oxidative stress defense mechanisms are enzymes that either repair damage caused by ROS or neutralize the damaging ROS(Storz and Imlay, 1999; Imlay, 2008). Sensing of oxidative stress signals and subsequent activation of these defense mechanisms is mediated by the OxyR and SoxRS transcriptional regulators in E. coli (for review, (Zheng and Storz, 2000)). S. flexneri contains homologues of these proteins.
The regulatory action of SoxRS is mediated via a two step process. First, SoxR is activated when the [2Fe-2S] centers it contains are oxidized either directly or indirectly by the superoxide anion (Hidalgo et al., 1997). Upon activation, SoxR enhances the transcription of its only known target in E. coli: soxS. In turn, SoxS activates the expression of the SoxRS regulon (Nunoshiba et al., 1992; Wu and Weiss, 1992). This regulon contains numerous genes such as the manganese superoxide dismutase (sodA), DNA repair enzyme endonuclease IV (nfo), superoxide-resistant isozymes of fumarase (fumC) and aconitase (acnA), and glucose-6-phosphate dehydrodgenase (zwf) (Storz and Imlay, 1999).
Activation of OxyR occurs via direct oxidization of disulfide bonds by hydrogen peroxide (Zheng et al., 1998). Once activated, OxyR induces expression of genes in the OxyR regulon including: a stress response DNA binding protein (dps), hydroperoxidase I (katG), a chaperone protein (ibpA), thiorodoxin 2 (trxC), an Fe-S cluster repair protein (sufA), two subunits of an alkyl hydroperoxide reductase (ahpCF), ferredoxin NADP+ reducatase (fpr), glutaredoxin 1 (grxA), glutathione reductase (gorA), and cysteine synthase (cysK)(Zheng et al., 2001).
Prior to this study, aS. flexneri strain deficient in oxyR (UR021) was constructed for examining the regulation of the suf and isc operons (Runyen-Janecky et al., 2008). During these studies, it was noted that UR021 did not reach the same optical densities as the parent S. flexneri strain even after prolonged aerobic incubation. It was hypothesized that the oxyR-deficient mutant was not surviving extended growth under aerobic conditions as well as wildtype Shigella due to an inability to combat the oxidative stress from autooxidation. The study described here extends this initial work by examining the effect of mutations in oxyR and/or soxRS on Shigella growth in survival in both extra- and intra-cellular environments.
2. Material and methods
2.1. Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this work are listed in Table 1. E. coli strains were routinely grown in Luria-Bertani broth (LB) or Luria agar (L agar). S. flexneri strains were grown in LB or on tryptic soy broth (TSB) agar plus 0.01% Congo red dye (CR agar) at 37°C unless otherwise indicated. All strains were stored as freezer stocks at -80°C in TSB + 20% glycerol. Antibiotics were used at the following concentrations (per milliliter), unless otherwise noted: 125 μg carbenicillin, 10 μg chloramphenicol, and 12.5 μg tetracycline.
Table 1.
Bacterial strains and plasmids.
| Strain or Plasmid | Characteristics | Reference or source |
|---|---|---|
| E. coli strains | ||
| DH5α | endA1 hsdR17 supE44 thi-1 recA1 gyrA relA1 Δ(lacZYA-argF)U169 deoR (Φ80dlacΔ(lacZ)M15) | (Sambrook et al., 1989) |
| SM10λpir | pirR6K | (Taylor et al., 1989) |
| S. flexneri strains | ||
| SA100 | S. flexneri wild-type serotype 2a | (Payne et al., 1983) |
| SM100 | SA100 StrR | S. Seliger |
| UR021 | SM100 ΔoxyR::cam | (Runyen-Janecky et al., 2008) |
| UR023 | SM100 soxRS::cam | This study |
| UR024 | UR021 ΔoxyR:: Δcam | This study |
| UR025 | UR024 ΔoxyR:: Δcam soxRS::cam | This study |
| Plasmids | ||
| pKD3 | Contains cam resistance gene | (Datsenko and Wanner, 2000) |
| pKM208 | Phage lambda Red recombinase genes under the control of an inducible promoter on temperature sensitive plasmid | (Murphy and Campellone, 2003) |
| pCP20 | FLP recombinase on temperature sensitive plasmid | (Datsenko and Wanner, 2000) |
| pWKS30 | Low-copy-number cloning vector | (Wang and Kushner, 1991) |
| pCW1 | S. flexneri oxyR in pWKS30 | This study |
| pRJ3 | S. flexneri soxRS in pWKS30 | This study |
2.2. DNA isolation and plasmid construction
Plasmid and chromosomal DNA were isolated using the QIAprep Spin Miniprep kit or the DNeasy Tissue Kit (Qiagen, Santa Clarita, CA), respectively. Isolation of DNA fragments from agarose gels was performed using the QIAquick Gel Extraction kit (Qiagen). All standard PCR reactions were carried out using either GoTaq (Promega, Madison, WI) or Pfu polymerase (Stratagene Cloning Systems, La Jolla, CA) according to the manufacturer’s instructions. To clone the oxyRgene, the gene was amplified from Shigella DNA by PCR with primers UR029 (5′CCGGAATTCTCTGGCGTCAATTAT ACCG) & UR084 (5′ACCACCTTTAACTACCCGACG) and Pfu polymerase. The oxyR fragment was digested with EcoRI and ligated with pWKS30 digested with EcoRI and HincII to generate pCW1. To clone the soxRS genes, the genes were amplified from Shigella DNA by PCR with primers UR027 (5′CCGGAATTCTGACTGAGCATGCATTTCTTG) & UR028 (5′CGCGGATCCGCTTTAGTTTTGTGTTTGC) and Pfu polymerase. The soxRS fragment and pWKS30 was digested with EcoRI and BamHI and ligated with digested with EcoRI and BamHI to generate pRJ3.
2.3. Construction of gene mutations in Shigella
The oxyR and soxRS mutants were constructed using a modified one step inactivation of chromosomal genes protocol(Datsenko and Wanner, 2000). The oxyR mutant UR021 was made previously(Runyen-Janecky et al., 2008). To construct the soxRS mutant allele, a PCR product for allelic exchange that contained approximately 50 bp of the beginning the soxRS locus, a chloramphenicol resistance gene (cam), and approximately 50 bp at the end of the soxRS locus was generated. The plasmid pKD3, which contains a chloramphenicol resistance gene (cam), was the template for the PCR using Pfu polymerase. The primers used to construct the soxRS mutant UR023 were UR152 (5′CGCGCGGGAGTTAACGCGCGGGCAATAAAACTACAGGCGGTGGCGATAATTGTG TAGGCTGGAGCTGCTTCG) and UR153 (5′ATAGTTAACGATGAGTCAGCTTGTCCTTGTGGCGCTTTAGTTTTGTTCATCTTCCA GCAACATATGAATATCCTCCTTAG). These primers contained 50 nucleotide overhangs that were homologous to either end of the soxRS locus as well as a sequence that hybridized to the cam gene on pKD3. The soxRS::cam PCR fragment was electroporated into Shigella SM100 containing the plasmid pKM208 (Murphy and Campellone, 2003), which harbors the phage lambda Red recombinase genes under the control of an IPTG-inducible promoter as described in (Runyen-Janecky et al., 2008). Transformants were selected on Congo Red agar containing 5–10 μg chloramphenicol per ml. Growth of the cultures at 42°C eliminated pKM208 due to its temperature sensitive origin of replication.
Construction of the oxyR/soxRS double mutant, UR025, was accomplished in two steps. First, UR024, an oxyR-deficient strain that had cam removed, was constructed as an intermediate strain. UR024 was constructed by removing the FLP recombination target (FRT) flanked cam gene from UR021 by incubating the strain at 30°C for 2 hr following transformation with pCP20, which contains contains the FLP recombinase gene (Datsenko and Wanner, 2000). Single-colony purified transformants were streaked on CR plates and incubated at 42°C to select for the loss of pCP20. UR025 was then constructed by P1 transduction (Miller, 1972) of the soxRS:cam mutation from UR023 into UR024. PCR using primers that flanked the gene of interest was employed to verify the disruption of the appropriate genes for all strains constructed.
2.4. Aerobic and microaerobic growth assays
Inoculums from Shigella freezer stocks were streaked on CR agar plates. Following overnight growth at 37°C, a minimum of five colonies were used to start 1 mL LB cultures grown at 37°C 17 hr in 13×100 mm tubes with aeration for aerobic conditions or in microcentrifuge tubes in a BD GasPak EZ Campy Pouch (Becton-Dickenson, Franklin Lakes, NJ) for microaerobic conditions. 1–2 mL LB subcultures were then started from these overnight cultures at equal optical denisites and incubated as described in each figure. Growth was measured via optical density at 650 nm (OD650) or by spreading serial dilutions of each culture on CRagar plates with or without 40 units of bovine catalase per plate (USB Corp, Cleveland, OH) to determine viable bacterial colony forming units (CFU) per ml.
2.5. Colony size assays
Subcultures of Shigella strains were grown as described above aerobic conditions. For those strains containing plasmids, all plates and broth included carbenicillin. After 24 hours, serial dilutions of each culture were plated on CRagar plates. Plates were incubated at 37°C for 22 hours, and then the colony diameter of at least ten well-isolated colonies was measured using a dissecting microscope from at least three replicate experiments.
2.6. Oxidative stress disk diffusion assays
Shigella cultures were started directly from freezer stocks in 2 mL LB containing carbenicillin and were incubated under aerobic conditions for 12–16 hr at 37°C. The SM100 and UR023 cultures were diluted 1:100 in saline and 100 μL plated on 20 mL LB + carbenicillin plates; the UR021 and UR025 cultures were diluted 1:25 and 150 μL was plated. A 6 mm BBL disk was then placed in the center of each plate. To this disk, 10 μL of the oxidizing agent (either 1M H2O2 or 50mM PMS) was added. The plates then incubated overnight at 37°C, and then the diameters of the zones of growth inhibition were measured.
2.7. Bile sensitivity assays
Inoculums from Shigella freezer stocks were streaked on CR agar plates. Following overnight growth at 37°C, a minimum of five colonies were used to start 2 mL LB cultures, which were grown at 30°C overnight with aeration. Approximately 5×105 bacteria were added to LB containing either 0, 1, or 7.5% ox bile (Oxgall) (US Biologicals, Swampscott, MA) and incubated at 37°C for 3 hours. Survival was assessed by spreading serial dilutions of each sample on CRagar plates with 40 units of bovine catalase to determine viable bacterial colony forming units (CFU) per ml.
2.8. Cell culture assays
Monolayers of Henle cells (intestine 407 cells, American Type Culture Collection, Manassas, VA) were maintained in Minimum Essential Medium (Invitrogen, Carlsbad, CA) supplemented with 1X MEM non-essential amino acid solution (Invitrogen) and 10% fetal bovine serum (Invitrogen). Plaque assays on Henle cells were done as described previously (Oaks et al., 1985)using the modifications described in Hong et al. (Hong et al., 1998), and plaques were scored after three days.
RAW 264.7 murine macrophages (ATCC TIB-71) were maintained and passaged in DMEM (American Type Culture Collection) containing 10% heat-inactivated FBS (Invitrogen). For the experiments described herein, macrophages were between passage 3 and 20 from the original ATCC stock. Shigella infections were performed as described elsewhere (Barnoy et al., 2010). Briefly, macrophages were seeded into 24-well plates in DMEM/10% FBS at ~ 106 cells/well and incubated overnight at 37°C in a humidified, 5% CO2 atmosphere for infection the following day. When activated macrophages were used, the cells were seeded as above in the presence of 200 U/ml recombinant mouse interferon-γ (R&D Systems). Cultures of Shigella were grown aerobically at 37°C in LB Miller broth overnight (12–16 hr), then sub-cultured until an OD600 of 0.3–0.6 was reached. Macrophages were infected at an MOI of 10 by adding 107 Shigella directly to appropriate wells, then centrifuging the plates at ~ 250×g for 5 min at ambient temperature to synchronize cell contact. The plates were then incubated at 37°C in a humidified 5% CO2 atmosphere for 25 min. Subsequently, all wells were washed three times with pre-warmed PBS to remove extracellular bacteria, then incubated as above with fresh, pre-warmed DMEM/10% FBS containing 100 μg/ml gentamicin to inhibit re-infection; this was defined as the start of infection. 30 min later, the medium was replaced with fresh, pre-warmed DMEM/10% FBS containing 10 μg/ml gentamicin to reduce bactericidal effects on intracellular bacteria. For CFU enumeration, macrophages were lysed in 1% Triton X-100 in PBS at time points from 0 to 6 hours post-infection, and lysates were diluted and plated on LB agar containing 40 U catalase.
Shigella-induced cytotoxicity on RAW 264.7 macrophages was assessed using the Cytotox 96 Non-Radioactive Cytotoxicity Assay kit (Promega Corp, Madison, WI) to quantify lactate dehydrogenase (LDH) activity in culture supernatants to ensure that any Shigella-induced cytotoxicity was similar for wildtype and mutant Shigellae. The assay was performed according to the manufacturer’s protocol.
2.9. Statistical analysis
Statistical analyses of the data were performed using the single factor analysis of variance statistics package in Microsoft Excel 2010 (Microsoft Corporation, Redmond, WA).
3. Results
3.1. Growth of oxidative stress regulator-deficient mutants in aerobic conditions
In previous studies, we noticed that an aerobically-grown overnight culture of a mutant S. flexneri strain lacking oxyR did not achieve the same turbidity as wildtype cultures(Runyen-Janecky et al., 2008). Thus, to investigate further the importance of oxidative stress regulators in Shigella growth, we generated a series of mutants with deletions in the oxyR and/or soxRS oxidative stress regulators and tested these mutants in a variety of growth assays. During growth in LB, the soxRS deletion mutant UR023 and the parental, wildtype strain SM100 had similar optical densities (Fig. 1). However, strains that contained a deletion of oxyR, UR021 (ΔoxyR) and UR025 (ΔoxyR ΔsoxRS), exhibited an approximately 2-fold reduction in optical density as compared to SM100 (Fig. 1).
Figure 1. The S. flexnerioxyR mutants show reduced growth in LB under aerobic growth conditions.
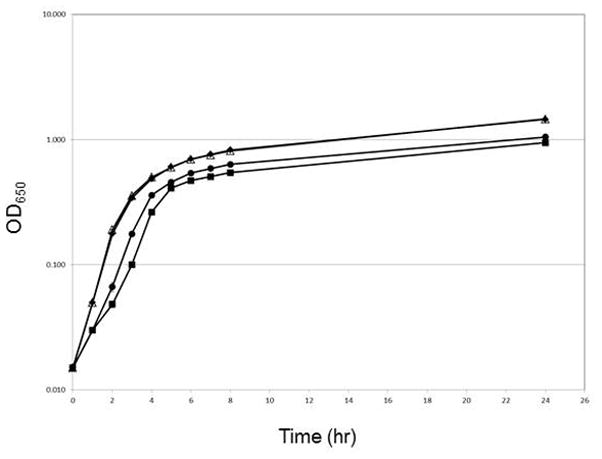
Overnight cultures of the each strain [SM100, ◆; UR021, ■; UR23,Δ; and UR025, ●] were diluted to an OD650 of 0.015 in LB and grown at 37°C under aerobic conditions. Growth was measured via optical density at 650 nm (OD650). The data presented are the means of three experiments, and the standard deviations of the means are indicated.
Since optical density does not always correlate with cell viability, we plated samples from 8 hour LB cultures on Congo Red (CR) agar plates to determine the number of viable cells in each culture. We found that the soxRS deletion mutant UR023 and the parental strain SM100 had similar viable cell counts (7.4×108± 6.4×107 and 7.0×108± 3.8×107, respectively). However, the oxyR mutant strains UR021 and UR025 exhibited severe defects in the ability to recover reproducible numbers of viable cells from the liquid cultures on CRagar plates. The calculated number of viable oxyR mutant bacteria per ml was always at least five-fold less than that of the wildtype strain SM100, but varied by a factor of over 10-fold from experiment to experiment. This defect in the ability to recover reproducible numbers of viable cells from aerobic liquid cultures could be suppressed by plating the oxyR mutant strains UR021 and UR025 on CRagar plates containing the anti-oxidant catalase (Fig. 2). In these experiments, the number of viable oxyR mutant bacteria per ml was consistently two-fold less than that of the wildtype strain SM100 (P<0.05).
Figure 2. The S. flexnerioxyR mutants show reduced viability in LB under aerobic growth conditions.
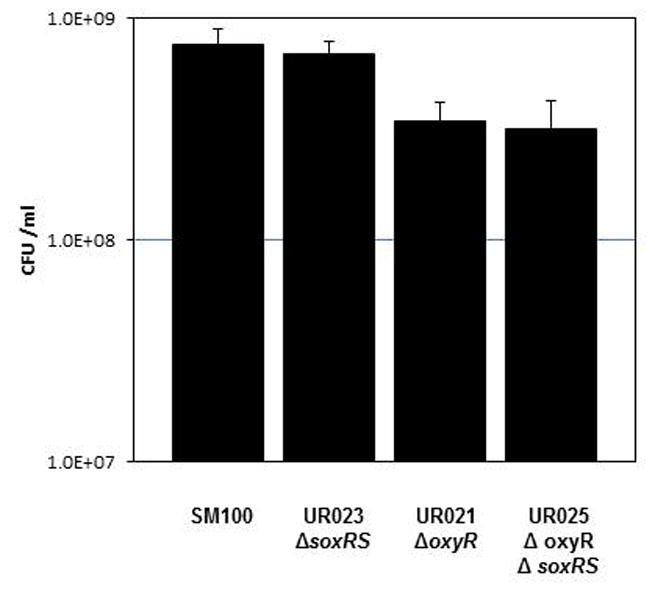
Overnight cultures of the each strain were diluted to an OD650 of 0.015 in LB and grown at 37°C under aerobic conditions for 8 hours. Serial dilutions of each culture were spotted on Congo Red plates containing 40 units of catalase per plate to determine viable bacterial colony forming units (CFU) per ml. The data presented are the means of three experiments, and the standard deviations of the means are indicated. The difference between SM100 and UR021 and between SM100 and UR025 was significant (p<0.05).
Colony size on agar plates was examined as an alternative means to investigate the relative growth rate of each strain. For this assay, after 24 hr of aerobic growth in LB, Shigella strains were plated at a low density on CRagar and colony diameter was measured after 22 hr growth at 37°C. Colonies were assigned to 4 classes: normal (>2.0 mm), medium (1.1 – 2 mm), small (0.5 – 1 mm), and very small (<0.5 mm). Both of the oxyR mutant strains UR021 and UR025 containing a vector control plasmid pWKS30 showed a greater percentage of small/very small colonies (19% and 61%, respectively) relative to the wildtype strain SM100 carrying pWKS30, which had no small/very small colonies (Fig. 3). Addition of pCW1 (which carries S. flexneri oxyR) to the oxyR mutants significantly increased the proportion of colonies with relatively larger sizes, although not entirely to the level of the parent strain (Fig. 3). UR023 had colony sizes (85% >2 mm) that were similar to SM100 (95% >2 mm). Addition of catalase to the agar plates restored the colony size of the oxyR mutants to that of SM100 (data not shown).
Figure 3. S. flexneri oxyR mutants show reduced colony size on agar plates.
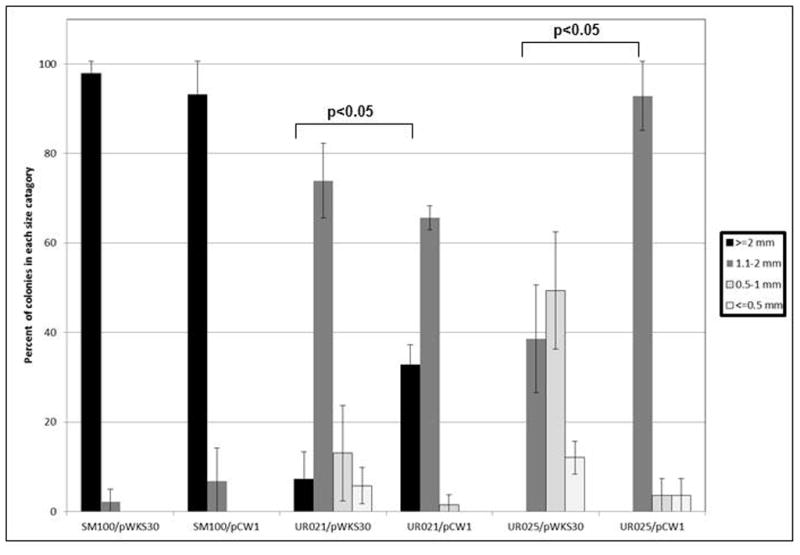
Subcultures of each strain were started from 17 hr overnight cultures (1:100 for wildtype and 1:50 for mutants) and grown under aerobic growth conditions at 37°C for 24 hr. Serial dilutions of each culture were then spread on Congo Red plates. Colony diameter was measured for each strain 22 hr after plating. The percentage of total colonies falling in each size category is reported as indicated in the accompanying legend. The data presented are the means of at least three replicate experiments. pCW1 carries the S. flexneri oxyR gene on the vector pWKS30.
3.2. Growth in microaerobic conditions suppresses the growth/survival defects of oxyR mutants
Because oxyR is known to be involved in the response to oxidative stress in many bacterial species(Storz and Imlay, 1999), we hypothesized that the growth and survival defects observed in the oxyR mutants UR021 and UR025 were due to their inability to cope with the oxidative stress associated with rapid aerobic growth. To test this hypothesis, we grew the strains microaerobically in an attempt to suppress these defects. It was predicted that the growth and survival defects of the oxyR-mutants would be diminished when the cultures were grown microaerobically. Both oxyR mutants UR021 and UR025 showed growth and survival comparable to wildtype strain SM100 when grown microaerobically (Fig. 4), suggesting that the defects seen during aerobic growth of the oxyR mutants is due to the inability to effectively combat oxidative stress.
Figure 4. Growth in microaerobic conditions suppresses the growth and survival defects of oxyR mutants.
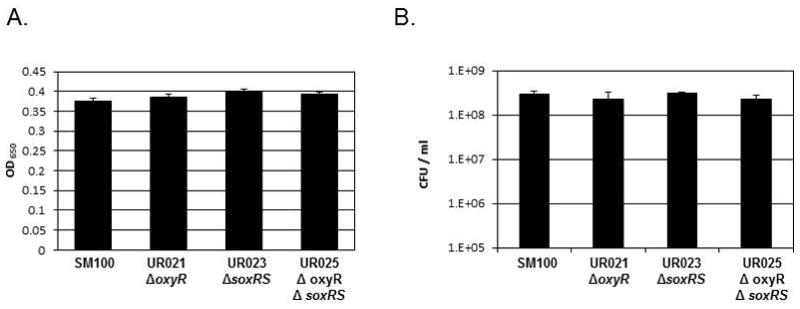
Overnight cultures of the each strain were subcultured into L broth and grown at 37°C under microaerobic conditions in a BD GasPak EZ Campy Pouch for 24 hours. (A) Growth was measured via optical density at 650 nm (OD650). (B) Serial dilutions of each culture were spotted on Congo Red plates containing 40 units of catalase to determine viable bacterial colony forming units (CFU) per ml. The data presented are the means of three experiments, and the standard deviations of the means are indicated.
3.3. Contribution of OxyR and SoxRS to oxidative stress survival in Shigella
To provide further support for the hypothesis that oxidative stress was the underlying cause of the observed growth/survival deficiencies in the oxyR-mutants, the sensitivity of the Shigella strains to hydrogen peroxide and the superoxide generator phenazine methosulfate (PMS)was examined using a disk diffusion assay.
The oxyR-deficient mutant UR021 carrying the vector control pWKS30 had significantly larger zones of growth inhibition by hydrogen peroxide as compared to the OxyR+ strain SM100/pWKS30(p<0.0001)(Fig. 5A). Addition of the oxyR-containing plasmid pCW1 to UR021 resulted in a significant decrease in the zone of growth inhibition by hydrogen peroxideas compared to the strain with the vector pWKS30(p<0.0001)(Fig. 5A). In comparison, the soxRS mutant UR023/pWKS30 had a zone of growth inhibition by hydrogen peroxide that was similar in size to SM100/pWKS30 (Fig. 5A).
Figure 5. Diffusion Assays with Oxidative Stressors.
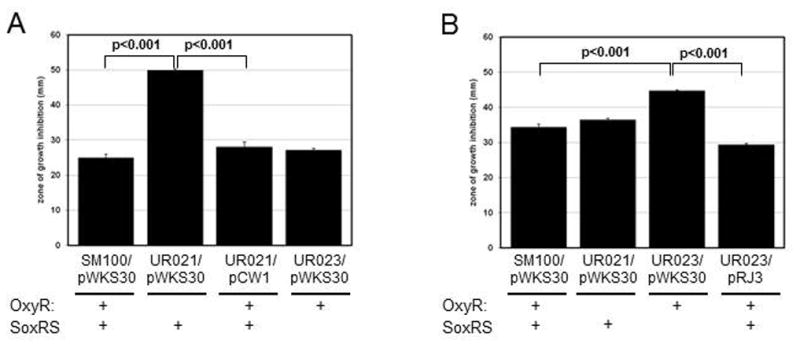
Approximately 1–6 x106 cells were spread on 20 mL LB + carbenicillin plates. Next, either 10 μl of 1M H2O2 (Panel A) or 50 mM PMS (Panel B) was added to a 6 mm BBL disk in the center of the plate. Following overnight growth, the diameters of the areas of clearing surrounding the discs were measured. The data presented are the means of three experiments, and the standard deviations of the means are indicated. pWKS30 is the vector control; pCW1 carries oxyR on pWKS30, and pRJ3 carries soxRS on pWKS30
In contrast to sensitivity to hydrogen peroxide, the oxyR mutant UR021 containing pWKS30 had a sensitivity to PMS similar to that of the OxyR+ strain SM100/pWKS30(Fig. 5B). ThesoxRS-deficient mutant UR023/pWKS30, however, had a significantly larger zone of growth inhibition by PMS relative to SM100/pWKS30(p<0.001)(Fig. 5B). Addition of the soxRS-containing plasmid pRJ3 to UR023 resulted in a significant decrease in the zone of growth inhibition by PMS as compared to the strain with pWKS30(p<0.001)(Fig. 5B).
Taken together, these data suggest that in Shigella OxyR is more important for mediating hydrogen peroxide stress survival and SoxRS is more important for mediating superoxide stress survival.
3.4. Contribution of the OxyR and SoxRS to Shigella survival in bile
Bile may cause oxidative stress through the generation of free radicals(Begley et al., 2005). In fact, Henneqin et al. (Hennequin and Forestier, 2009)showed that OxyR enhances resistance to bile in Klebsiella pseumoniae. Since Shigella encounters bile during transit through the gastrointestinal track, we tested whether any of the oxidative stress regulatory mutants had an increased sensitivity to bile. After three hour incubation in 10% ox-bile, the mutants survived as well as wildtype Shigella (data not shown).
3.5. Contribution of the OxyR and SoxRS to Shigella survival in eukaryotic cells
Because oxidative stress is a primary mechanism of macrophage antibacterial activity, we tested the ability of the Shigella mutants to survive in RAW 264.7 macrophages using a gentamicin-protection assay. Over a time course from 0 to 6 hr, the intramacrophage survival curves of the ΔoxyR/ΔsoxRS mutants were indistinguishable from the parental wild-type. To ensure that the Shigella mutants were being exposed to oxidative stress in the assay, we activated the macrophages with interferon-γ prior to infection. We tested the ΔoxyR/ΔsoxRS mutant UR025 and found that both the mutant and wild-type were killed more rapidly in the activated macrophages, but the intramacrophage survival curves for the mutant remained indistinguishable from the wild-type (Fig. 6). Collectively, these data suggest that neither OxyR nor SoxRS contribute significantly to intramacrophage survival.
Figure 6. Survival of the S. flexnerioxyR/soxRS mutant in activated macrophage cells.
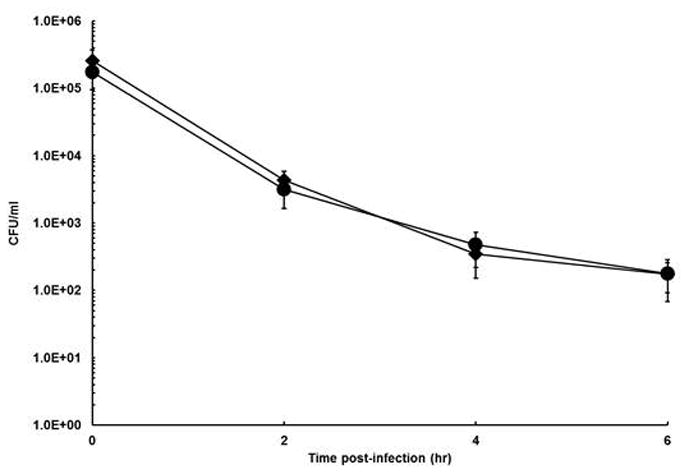
RAW264.1 macrophages, activated by mouse interferon-γ, were infected with Shigella (SM100, ◆ or UR025, ●) at an MOI of 10. Gentamicin was added to kill all extracellular bacteria, and the infected monolayers were allowed to incubate for up to 6 hours. At each time point, macrophages were lysed with Triton X-100, and lysates were diluted and plated on LB agar containing 40 U catalase. The data presented are the means of at least three experiments, and the standard deviations of the means are indicated.
Microarray analysis of Shigella gene expression in HeLa epithelial cells suggested that oxyR, soxR and soxS transcription increased in the intracellular environment, which suggested that these genes might be important for survival within the intracellular environment (Lucchini et al., 2005). Thus, we tested the S. flexneri oxidative stress regulator mutants for growth in the Henle cell intracellular environment by examining the ability of the mutants to form plaques on Henle cell monolayers. All of the mutants formed similar numbers of plaques on Henle cell monolayers (data not shown), suggesting the ability of Shigella to invade Henle cells was not affected by lack of OxyR or SoxRS. However, the diameter of the plaques formed by the mutants was always slightly smaller than the plaques formed by the wildtype strain (Fig. 7) (p< 0.05). These data indicate that neither the OxyR nor SoxRS is absolutely required for growth-dependent plaque formation in epithelial cells, but that the presence of either or both of the genes does enhance plaque formation slightly.
Figure 7. Plaque assay with S. flexnerioxyR and soxRS mutants.
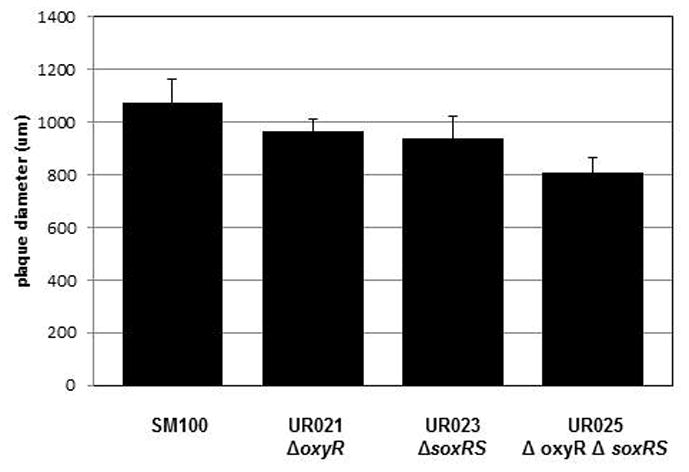
Confluent Henle cell monolayers were infected with 104 bacteria per 35-mm diameter plate and the diameter of the plaques were measured after 3 days. The data presented are the means of at least three experiments, and the standard deviations of the means are indicated. The difference between SM100 and UR025 was significant (p<0.05).
4. Discussion
Previously, we observed that a S. flexneri mutant lacking the oxidative stress responsive transcriptional regulator OxyR grew to lower cell densities than wildtype during aerobic growth(Runyen-Janecky et al., 2008). We have now examined the growth phenotypes of mutants lacking oxyR or soxRS, another important regulator of the oxidative stress response, as well as a third mutant lacking both oxyR and soxRS. In congruence with the initial observation, oxyR-deficient strains had a lower density of viable cells than the wildtype strain following aerobic growth. In addition, in both the strains lacking oxyR, colonies were characteristically smaller than wildtype colonies. Support for the poor growth phenotypes being due to the absence of OxyR was provided by the ability of plasmid-borne oxyR to complement the colony size defect of both oxyR-deficient strains. Although complementation was not complete, possibly because the plasmid was not maintained at 100% in the cells on agar plates, there was a statistically significant shift to relatively larger colony sizes when plasmid-borne oxyR was added to any of our oxyR mutants. Our results are consistent with other studies of oxyR mutants that show the absence of OxyR alters the growth and survival of bacterial species including E. coli, Pseudomonas aeruginosa, Klebsiella pneumonia, and Moraxella catarrhalis (Dukan and Nystrom, 1999; Hassett et al., 2000; Johnson et al., 2006; Hennequin and Forestier, 2009; Hoopman et al., 2010).
Because OxyR is a transcriptional activator of the oxidative stress response in many enterobacteria, it was hypothesized that the observed growth phenotypes were due to inability of the oxyR-deficient Shigella strains to survive the oxidative stress produced during normal aerobic growth. Several pieces of evidence support this hypothesis. In microaerobic environments with presumably less oxidative stress, the oxyR-deficient strains demonstrated improved growth phenotypes that were much more similar to wildtype. Secondly, the oxyR mutant was unable to survive the same concentration of H2O2 as either wildtype or the soxRS-deficient strain in disc diffusion assays. This phenotype was complemented with the addition of plasmid-borne oxyR. Finally, the difficultly in reproducible recovery of viable Shigella in plate count assays could be suppressed by the addition of catalase or by microaerobic growth conditions. Other groups have reported similar phenomena in P. aeruginosa, K. pneumonia, and M. catarrhalis which they refer to as an “aerobic dilution defect” or an “inoculum effect” (Hassett et al., 2000; Hennequin and Forestier, 2009; Hoopman et al., 2010). Consistent with our results, these defects were suppressed under anaerobic/microaerobic conditions or in the presence of anti-oxidants like catalase. It is possible that a burst of new aerobic growth may kills the cells in the absence of an appropriate level of antioxidant (Dukan and Nystrom, 1999).
Of the eight sequenced strains of Shigella, all possess oxyR, suggesting selective pressure for maintaining this gene. OxyR is necessary for cells to cope with the oxidative stress, such as hydrogen peroxide, which is an unavoidable byproduct of aerobic respiration. OxyR activates genes involved in detoxification of hydrogen peroxide including katG (encoding hydroperoxidase I or catalase G) and ahpCF(encoding alkyl hydroperoxide reductase)(Zheng et al., 2001). Most Shigella strains, with the exception of S. dysenteriae type 1 strains, are catalase positive (Karas et al., 2007). The sequenced S. dysenteriae type 1 strain Sd197 has a frame shift in katG and lacks katEF, but all of the other sequenced strains have intact katG genes. Interestingly though, Franzon et al. (Franzon et al., 1990) showed that catalase negative S. flexneri did not survive inside peritoneal mouse macrophages as well as wildtype S. flexneri. However, the catalase mutant they used was generated by P1 transduction and so it is possible that there are other mutations that were transduced into the strain at the same time. Alternatively, the experimental design of Franzon et al. (Franzon et al., 1990)utilized opsonized bacteria. Since an oxidative burst is not generated in Type 2 phagocytosis (opsonin-dependent, complement receptor-mediated), catalase may not have been required for survival in these experiments. All sequenced Shigella strains do have an intact ahpCF gene. These data suggest that alkyl hydroperoxide reductase is likely sufficient to scavenge hydrogen peroxide in Shigella and is consistent with reports in E. coli (Seaver and Imlay, 2001).
The S. flexneri soxRS strain UR023 demonstrated no detrimental growth phenotype in vitro. This was initially unexpected because superoxide is generated during normal cellular metabolism and since SoxRS was demonstrated to be important in the response of Shigella to superoxide generation. The Shigella strains lacking soxRS were more susceptible to the superoxide generator phenazine methosulfate (PMS) as demonstrated in disc diffusion assays as compared with the parent strains. This suggests that soxRS is primarily responsible for mediating superoxide stress in Shigella. Most likely under normal aerobic growth, the SoxRS-independent superoxide dismutase SodB detoxifies endogenous superoxide(Fee, 1991); however, with elevated levels of superoxide from PMS, the SoxRS-dependent SodA is required. Interestingly, S. sonnei Ss046 lack a functional soxR gene due to an insertion sequence that eliminated the first 141 nucleotides of the gene, and S. boydii lacks the soxRS locus entirely. These data suggest that soxRS is dispensable for virulence in Shigella.
Our plaque data provided preliminary evidence that OxyR and SoxRS may have a very small role in enhancing growth in Henle cells. Although these genes are induced when Shigella is within the epithelial cytosol, most, but not all, of the genes they regulate are not induced at the transcriptional level(Lucchini et al., 2005); thus, our plaque assay result was a bit surprising. The cytosolis thought to be reducing because the ratio of reduced glutathione to oxidized glutathione is at least 30:1; thus, a high level of oxidative stress in epithelial cells is not likely (Hwang et al., 1992). Also, previous work showed that a deletion of oxyR did not affect expression of the OxyR-regulated sufA gene in Henle cells, suggesting that the environment is not sufficiently oxidizing to allow OxyR enhancement of gene expression(Runyen-Janecky et al., 2008). At this time, the significance of the increased expression of these genes or small role for the genes in intracellular growth is unknown. Since only a few bacterial species replicate within the cytoplasm, and not membrane compartments in the host eukaryotic cell, it is difficult to make large comparison of Shigella with other intracellular, cytoplasmic pathogens. In Listeria monocytogenes, precise regulation of the PerR regulon, which includes genes involved in oxidative stress survival, is required for normal virulence in mice; PerR mutants were less virulent than the wild type(Rea et al., 2004).
The human host employs ROS as a defense mechanism against pathogens. Most notably, immune cells such as resident tissue macrophage, infiltrating monocyctes, and neutrophils can utilize the reactive oxygen burst in the phagolysome to kill pathogens. Our data suggest that even though OxyR and SoxRS are required for efficient in vitro survival in the presence of hydrogen peroxide and the superoxide generator PMS, these genes are not needed for Shigella survivalin activated RAW264.7 macrophages. There are several formal possibilities for why this is so. First, expression of the genes for ROS scavenging enzymes may be activated by OxyR and SoxR-independent mechanisms in macrophage cells. Second, other OxyR and SoxRS-independent ROS scavenging enzymes may be used for survival within the RAW264.7 macrophage. For example, expression of the katEF –encoded catalase and the Fe-containing superoxide dismutase SodB are not under OxyR or SoxRS control(Fee, 1991; Zheng et al., 2001). However, the most likely explanation is that Shigella rapidly escapes the phagocytotic vacuole and so that it experiences relatively little (or very transient) ROS stress. This is consistent with microarray data that suggests most genes in the oxyR and soxS regulons (i.e. katG, aphC, cysK, sodA) are not expressed when Shigella is in the U937 macrophage (Lucchini et al., 2005).
In conclusion, this study demonstrated that the oxyR gene in Shigella is important for the bacteria to survive in aerobic conditions. It is likely that these conditions are most relevant when Shigella is between hosts in the environment. Thus, the ability to combat oxidative stress may be an environmental survival factor for Shigella species.
Acknowledgments
This work was supported by Public Health Service Grants AI075330 awarded to L. R-J and by funding from the University of Richmond School of Arts and Sciences and the University of Richmond Ethyl Science Scholarship to A.D. We would like to thank Dr. Malabi M. Venkatesan for support and guidance. The content of this publication does not necessarily reflect the views or policies of the U.S. Department of the Army, or the U.S. Department of Defense, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnoy S, Jeong KI, Helm RF, Suvarnapunya AE, Ranallo RT, Tzipori S, Venkatesan MM. Characterization of WRSs2 and WRSs3, new second-generation virG(icsA)-based Shigella sonnei vaccine candidates with the potential for reduced reactogenicity. Vaccine. 2010;28(6):1642–1654. doi: 10.1016/j.vaccine.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29(4):625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Bennish ML, Wojtyniak BJ. Mortality due to shigellosis: community and hospital data. Rev Infect Dis. 1991;13 (Suppl 4):S245–251. doi: 10.1093/clinids/13.supplement_4.s245. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukan S, Nystrom T. Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J Biol Chem. 1999;274(37):26027–32. doi: 10.1074/jbc.274.37.26027. [DOI] [PubMed] [Google Scholar]
- Fee JA. Regulation of Sod genes in Escherichia coli: relevance to superoxide dismutase function. Mol Microbiol. 1991;5(11):2599–2610. doi: 10.1111/j.1365-2958.1991.tb01968.x. [DOI] [PubMed] [Google Scholar]
- Franzon VL, Arondel J, Sansonetti PJ. Contribution of superoxide dismutase and catalase activities to Shigella flexneri pathogenesis. Infect Immun. 1990;58(2):529–35. doi: 10.1128/iai.58.2.529-535.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett DJ, Alsabbagh E, Parvatiyar K, Howell ML, Wilmott RW, Ochsner UA. A protease-resistant catalase, KatA, released upon cell lysis during stationary phase is essential for aerobic survival of a Pseudomonas aeruginosa oxyR mutant at low cell densities. J Bacteriol. 2000;182(16):4557–563. doi: 10.1128/jb.182.16.4557-4563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennequin C, Forestier C. OxyR, a LysR-type regulator involved in Klebsiella pneumoniae mucosal and abiotic colonization. Infect Immun. 2009;77(12):5449–5457. doi: 10.1128/IAI.00837-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo E, Ding H, Demple B. Redox signal transduction: mutations shifting [2Fe-2S] centers of the SoxR sensor-regulator to the oxidized form. Cell. 1997;88(1):121–129. doi: 10.1016/s0092-8674(00)81864-4. [DOI] [PubMed] [Google Scholar]
- Hong M, Gleason Y, Wyckoff EE, Payne SM. Identification of two Shigella flexneri chromosomal loci involved in intercellular spreading. Infect Immun. 1998;66(10):4700–4710. doi: 10.1128/iai.66.10.4700-4710.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopman TC, Liu W, Joslin SN, Pybus C, Brautigam CA, Hansen EJ. Identification of Gene Products Involved in the Oxidative Stress Response of Moraxella catarrhalis. Infect Immun. 2010:745–755. doi: 10.1128/IAI.01060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JR, Clabots C, Rosen H. Effect of inactivation of the global oxidative stress regulator oxyR on the colonization ability of Escherichia coli O1:K1:H7 in a mouse model of ascending urinary tract infection. Infect Immun. 2006;74(1):461–8. doi: 10.1128/IAI.74.1.461-468.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas JA, Pillay DG, Sturm AW. The catalase reaction of Shigella species and its use in rapid screening for epidemic Shigella dysenteriae type 1. Ann Trop Med Parasitol. 2007;101(1):79–84. doi: 10.1179/136485907154575. [DOI] [PubMed] [Google Scholar]
- Lucchini S, Liu H, Jin Q, Hinton JC, Yu J. Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect Immun. 2005;73(1):88–102. doi: 10.1128/IAI.73.1.88-102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic-Mulec I, Weiss J, Zychlinsky A. Shigella flexneri is trapped in polymorphonuclear leukocyte vacuoles and efficiently killed. Infect Immun. 1997;65(1):110–115. doi: 10.1128/iai.65.1.110-115.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol. 2003:4. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoshiba T, Hidalgo E, Amabile Cuevas CF, Demple B. Two-stage control of an oxidative stress regulon: the Escherichia coli SoxR protein triggers redox-inducible expression of the soxS regulatory gene. J Bacteriol. 1992;174(19):6054–6060. doi: 10.1128/jb.174.19.6054-6060.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oaks EV, Wingfield ME, Formal SB. Plaque formation by virulent Shigella flexneri. Infect Immun. 1985;48(1):124–129. doi: 10.1128/iai.48.1.124-129.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SM, Niesel DW, Peixotto SS, Lawlor KM. Expression of hydroxamate and phenolate siderophores by Shigella flexneri. J Bacteriol. 1983;155(3):949–955. doi: 10.1128/jb.155.3.949-955.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea RB, Gahan CG, Hill C. Disruption of putative regulatory loci in Listeria monocytogenes demonstrates a significant role for Fur and PerR in virulence. Infect Immun. 2004;72(2):717–727. doi: 10.1128/IAI.72.2.717-727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyen-Janecky L, Daugherty A, Lloyd B, Wellington C, Eskandarian H, Sagransky M. Role and regulation of iron-sulfur cluster biosynthesis genes in Shigella flexneri virulence. Infect Immun. 2008;76(3):1083–1092. doi: 10.1128/IAI.01211-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sansonetti PJ, Egile C, Wenneras C. Shigellosis: from disease symptoms to molecular and cellular pathogenesis. In: Groisman E, editor. Principles of bacterial pathogenesis. New York, N.Y: Academic Press; 2001. pp. 335–385. [Google Scholar]
- Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J Bacteriol. 2001;183(24):7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Imlay JA. Oxidative stress. Curr Opin Microbiol. 1999;2(2):188–194. doi: 10.1016/s1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Sprinz H, LaBrec EH, Formal SB. Experimental bacillary dysentery. An electron microscopic study of the response of the intestinal mucosa to bacterial invasion. Am J Pathol. 1965;47(6):1011–44. [PMC free article] [PubMed] [Google Scholar]
- Taylor RK, Manoil C, Mekalanos JJ. Broad-host-range vectors for delivery of TnphoA: use in genetic analysis of secreted virulence determinants of Vibrio cholerae. J Bacteriol. 1989;171:1870–1878. doi: 10.1128/jb.171.4.1870-1878.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- Wu J, Weiss B. Two-stage induction of the soxRS (superoxide response) regulon of Escherichia coli. J Bacteriol. 1992;174(12):3915–3920. doi: 10.1128/jb.174.12.3915-3920.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Storz G. Redox sensing by prokaryotic transcription factors. Biochem Pharmacol. 2000;59(1):1–6. doi: 10.1016/s0006-2952(99)00289-0. [DOI] [PubMed] [Google Scholar]
- Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279(5357):1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol. 2001;183(15):4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]