

Abstract
In recent years, colorectal cancer (CRC) incidence has been increasing to become a major cause of morbidity and mortality worldwide from cancers, with high rates in westernized societies and increasing rates in developing countries. Epigenetic modifications including changes in DNA methylation, histone modifications, and non-coding RNAs play a critical role in carcinogenesis. Epidemiological data suggest that, in comparison to other cancers, these alterations are particularly common within the gastrointestinal tract. To explain these observations, environmental factors and especially diet were suggested to both prevent and induce CRC. Epigenetic alterations are, in contrast to genetic modifications, potentially reversible, making the use of dietary agents a promising approach in CRC for the development of chemopreventive strategies targeting epigenetic mechanisms. This review focuses on CRC-related epigenetic alterations as a rationale for various levels of prevention strategies and their potential modulation by natural dietary compounds.
Keywords: Colorectal cancer, Epigenetic, DNA methylation, Histone modification, Non-coding, microRNA, Cancer prevention, Early detection, Biomarker, Predictive marker, Prognostic marker, Chemoprevention, Molecular epidemiology
Introduction
Colon and rectal cancers (colorectal cancer, CRC) represent globally, in terms of frequency, the third leading cause of cancer-related mortality (ie, after lung and breast cancer). Nevertheless, CRC incidence and mortality rates vary over 10-fold worldwide. Lowest incidence rates are observed in Africa and Asia and highest ones are found in Australia/New Zealand, North America, and Western Europe with a mortality rate of approximately 30%. Although incidence rates in developed countries are stabilizing, they are severely increasing in both developing countries and several areas historically at low risk [1]. Since the 1970s, CRC incidence in USA has continuously increased in the African-American population to become more frequent in this population than in Caucasians or other ethnic groups [2]. Similarly, data from migration population studies revealed that some ethnic groups are showing increased CRC incidence rate while they are migrating from low-risk to high-risk areas, to finally reach rates comparable to the host country [3–5]. Despite genetic variation, these epidemiological data strongly suggest a role of environmental and lifestyle factors deeply contributing to the etiology of CRC.
Although it is well accepted that genetic factors and inflammatory bowel disease place certain individuals at increased risk [6], various modifiable lifestyle factors have been identified related to CRC pathogenesis. Significant lifestyle risk factors are represented by sedentarity and changes in dietary habits, from a moderate to a Western-like enriched diet associated with high consumption of unsaturated fats and red meat, high intake of alcohol, and smoking.
Epigenetic mechanisms are fundamental to tightly regulated cellular processes. Epigenetic aberrations governing tumor suppressor gene (TSG) inactivation, oncogene activation, and chromosomal instability play a fundamental role in tumorigenesis including CRC. Epigenetic events are involved in all critical pathways and steps of carcinogenesis including tumor initiation, and some events are usually detectable before neoplastic transformation [7, 8, 9•, 10]. Nonetheless, it is well accepted that environmental and dietary factors greatly influence epigenetic events. Moreover, the reversibility of epigenetic alterations stimulates the development of novel therapeutic approaches with an open field for development in cancer chemoprevention. Taking together, these observations suggest that improved early detection and dietary intervention are preventive approaches of choice to decrease CRC incidence.
In this review, we focus on epigenetic alterations associated with CRC, which offer promising novel biomarkers for early detection, with an emphasis on how these alterations can potentially be modulated by dietary compounds for preventive interventions.
Colorectal Carcinogenesis and Cancer Prevention
The vast majority of CRCs are a multistep-associated adenoma-carcinoma progression associated with successive clinico-histopathological stages. This transformation initially starts with a premalignant lesion, called aberrant crypt foci (ACF), rising from normal colonic mucosa, progressing to a premalignant lesion (ie, adenoma), and finally evolving to invasive adenocarcinoma and metastasis (Fig. 1). The tumor-node-metastasis (TNM) system stages CRCs depending on the extent of invasion of the intestinal wall (T), the degree of lymphatic node involvement (N), and whether there is presence of metastasis (M). Based on this system, CRC is ranked from 0 (in situ tumor confined to mucosa) to stage IV (presence of metastasis). Thus, an increasing ranking correlates to a more advanced cancer and likely a worse outcome [11].
Fig. 1.
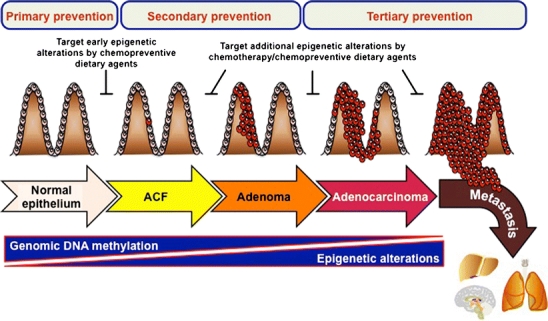
Colorectal cancer (CRC) progression as a model for epigenetic alteration cascade and prevention strategies. CRC development is initially starting by a premalignant lesion, called aberrant crypt foci (ACF), rising from normal colonic mucosa, progressing to a premalignant lesion (adenoma) and then to invasive adenocarcinoma, and finally evolving to metastatic adenocarcinoma. Epigenetic alterations are largely contributing to CRC initiation and adenoma-carcinoma progression. These alterations are characterized by global genomic DNA hypomethylation leading to genomic instability and oncogene activation concomitantly to an increase of CpG island promoter hypermethylation-mediated silencing of tumor suppressor genes. These changes are accompanied by an increase of aberrant histone modification profiles and miRNA signatures reinforcing oncogenic activation and tumor suppressor loss associated with CRC progression. Consequently, epigenetic alterations represent promising targets for CRC prevention. Early epigenetic aberrations represent interesting targets for primary prevention, especially through chemoprevention by dietary epigenetic modulators, as well as for secondary prevention as early biomarkers of CRC initiation. Modifications occurring at later stages may be targeted by chemotherapeutic interventions as well as chemopreventive agents to limit or block disease progression (secondary and tertiary prevention activities)
Although most CRCs occur sporadically, the importance of inheritance associated with a family history of the disease is evaluated to approximately 25% [12]. Some well-defined syndromes associated with CRC pathobiology have been identified: hereditary non-polyposis CRC (HNPCC), familial adenomatous polyposis (FAP), and MUTYH-associated polyposis (MAP), which are caused by germline mutations in DNA mismatch repair (MMR) genes, adenomatous polyposis coli (APC) TSG, and MUTYH gene, respectively; plus a number of relatively rare polyposis syndromes [13].
Mechanisms underlying the adenoma-carcinoma sequence have been identified for their contribution to CRC pathogenesis in relation to alterations of TSG and oncogene functions. Among these mechanisms, genomic instability represented by two “genotypic” subtypes pathways associated with somatic mutations are frequently identified: chromosomal instability (CIN) and microsatellite instability (MSI) [9•, 13]. Although CIN is observed in approximately 85% of CRC cases, the initiating mechanism is still poorly understood. The most common cytogenic abnormalities observed in CIN are mutations of APC gene, which occur in the majority of sporadic CRCs and also very early in adenoma development, and chromosome aberrations such as loss of heterozygosity of 5, 17p, and 18q. The latter contains the deleted in colorectal cancer (DCC) TSG. MSI is characterized by the inactivation of genes implicated in mismatch repair (MMR) system leading to subsequent mutations in the microsatellite repeat sequences of genes linked to tumor progression [13]. In addition to somatic mutations, epigenetic alterations are also particularly common in CRC. Epigenetics is defined as the heritable changes in gene expression patterns that occur without a change in the primary DNA sequence. This field encompasses DNA methylation, histone modifications and chromatin remodeling, and non-coding RNA-mediated interference [9•, 10, 13].
After years of research it appears that the best way to avoid the burden of cancer might be prevention. Under the general concept of prevention, several levels of approaches are encompassed [14]. Avoiding exposure to potential carcinogens or life risk factors is associated with the primary level of prevention. However, preemptive behavior prevention is not limited to this aspect. Indeed, chemoprevention, ie, the use of natural agents in healthy individuals without signs of premalignancy, falls also in this category. Secondary prevention corresponds to early detection of tumor-related abnormal changes aiming to prevent cancer development. Screening tests are included in this category, which require robust clinical biomarkers for early diagnosis. Finally, tertiary prevention consists to control cancer development to a more advanced-stage or reoccurrence after treatment and reduce adverse health effects.
Given the fact that epimutations are potentially reversible, the major field of applications regarding epigenetics might be cancer prevention. Accordingly, epimutations represent secondary prevention biomarkers by their precocity in carcinogenesis processes (ie, before neoplastic transformation). Primary to tertiary prevention may be achieved through chemoprevention, with dietary agents controlling epigenetic (re)programming, to either prevent or reverse premalignant stem cell phenotypes (Fig. 1).
Epimutations in CRC: Biomarkers and Targets for Prevention
DNA Methylation in CRC
In humans, DNA methylation occurs at the 5′ position of the pyrimidine ring of the cytosine residues within CpG dinucleotides through addition of a methyl moiety to form 5-methylcytosines. This process is catalyzed by three DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) using the cofactor S-adenosyl-methionine (SAM). Although CpG dinucleotides represent approximately 1% of the human genome, they are unequally distributed across the genome and are clustered in small DNA stretches. These CpG-rich regions, called CpG islands (CGIs), are usually present near promoters and exogenic regions. CGIs are usually unmethylated in normal differentiated cells, whereas CpGs located in intergenic regions are methylated [8, 15].
In cancer, promoter CGI of numerous TSGs are found to be densely methylated, which results in transcriptional silencing. Interestingly, these epimutations may be cancer type-specific and tumor stage-specific. Thus, methylation patterns can be considered as biomarkers for diagnosis, prognosis, as well as prediction and monitoring of therapy response [8, 10, 15, 16]. Therefore, the identification of these cancer-associated methylation signatures is really critical for cancer prevention purposes.
Recent studies show that CRC is strongly associated with aberrant DNA methylation profiles, which has been linked to the origin and progression of the disease. The list of epimutations is growing quickly with the use of developing technologies allowing genome studies. To date, a long list of TSGs involved in numerous signalization pathways and cellular processes were found frequently methylated in CRC (Table 1). Noteworthy, a widespread contribution of DNA methylation participates in the disruption of β-catenin–dependent Wnt signaling pathway, which plays an important role in colorectal tumor development [9•, 13]. Moreover, methylation can affect coding and non-coding genes (eg, microRNA, miRNA) participating in loss of tumor suppressor functions. Remarkably, most of these methylated genes are investigated as potential biomarkers for preventive or therapeutic purposes. However, the methylation prevalence varies substantively depending on the considered genes as well as between studies regarding a same gene. The discrepancy between studies, like the case of CDKN2A (p16), for which methylation ranged from 10% to 58% depending on reports [17–19], could be explained by the phenotype of patients constituting these cohorts as well as how clinical parameters were included in these analyses. Indeed, certain genes are found frequently methylated in specific CRC subgroups, such as AXIN2 found preferentially associated with MSI tumors [20].
Table 1.
Epimutations associated with colorectal cancer based on experimental data from patientsa
| Epigenetic event | Name | Locus | Function/targets | Noteb | Comments |
|---|---|---|---|---|---|
| Hypermethylation | ADAMTS5 | 21q22.1-q22 | Protease | NA | Increase of methylation level in CRC |
| ADHFE1 | 8q12.3 | Alcohol dehydrogenase | NA | Increase of methylation level in CRC | |
| ALX4 | 11p11.2 | Homeobox gene | 85/64 | Adenoma vs CRC | |
| APBA1 | 9q13-q21 | Intracellular signaling | 16–28 | ||
| APBA2 | 15q11-q12 | Intracellular signaling | 22/26 | Stage I + III vs IV | |
| APC | 5q22.2 | Wnt signaling | 21 | ||
| APC2 | 19p13.3 | Wnt signaling | 100 | ||
| AXIN2 | 17q24.1 | Wnt signaling | 29 | Associated with MSI tumors | |
| B4GALNT1 | 12q13.3 | Lipid metabolism | 100 | ||
| B4GALNT2 | 17q21.3 | Lipid metabolism | 50 | Correlated with EBV-associated gastric carcinomas | |
| BARX1 | 9q12 | Homeobox gene | 56 | ||
| BMP3 | 4q21.21 | Bone and cartilage formation | 72/60 | Adenoma vs CRC | |
| BNIP3 | 10q26.3 | Apoptosis | 66 | ||
| BOLL | 2q33.1 | Development | NA | Increase of methylation level in CRC | |
| CACNA1G | 17q22 | Calcium metabolism | 39 | ||
| CASR | 3q21.1 | Calcium metabolism | 9/69/90 | Adenoma vs CRC vs lymph node metastatic tissues | |
| CCNA1 | 13q13.3 | Cell cycle | 100 | ||
| CD109 | 6q13 | Complement system | 33 | ||
| CDH1 | 16q22.1 | Cell adhesion | 51 | ||
| CDH13 | 16q23.3 | Cell adhesion | 32–66 | Poor prognosis | |
| CDH2 | 18q12.1 | Cell adhesion | 45 | ||
| CDH4 | 20q13.3 | Cell adhesion | 78 | ||
| CDKN2A (p14) | 9p21.3 | Cell cycle | 34 | ||
| CDKN2A (p16) | 9p21.3 | Cell cycle | 10–58 | ||
| CDKN2B (p15) | 9p21.3 | Cell cycle | 68 | ||
| CDX1 | 5q33.1 | Homeobox gene | 100 | ||
| CHFR | 12q24.33 | Cell cycle | 26–63 | Associated with disease recurrence | |
| CNRIP1 | 2p14 | G protein-coupled receptor | 91/94 | Adenoma vs CRC | |
| CNTFR | 9p13.3 | Cytokine signaling | 22 | ||
| CPAMD8 | 19p13.12 | Innate immunity | 90 | ||
| CXCL12 | 10q11.21 | Cytokine signaling | 62 | ||
| DAPK1 | 9q21.33 | Apoptosis | 43 | ||
| DCC | 18q21.2 | Putative TSG | 81/83 | Adenoma vs CRC (20% in normal) | |
| DFNA5 | 7p15.3 | Unknown | 65 | ||
| DKK1 | 10q21.1 | Wnt signaling | 13–35 | Associated with MSI tumors | |
| DKK2 | 4q25 | Wnt signaling | 65 | ||
| DKK3 | 11p15.3 | Wnt signaling | 35 | ||
| DKK4 | 8p11.2-p11.1 | Wnt signaling | 20 | ||
| DLC1-i4 | 8p22 | Putative TSG | 100 | ||
| DLEC1 | 3p22.2 | Putative TSG | 38 | Poor prognosis | |
| EFEMP1 | 2p16.1 | Cell migration | 39 | Poor prognosis | |
| EGFR | 7p11.2 | Cytokine signaling | 58 | Poor prognosis | |
| EN1 | 2q13-q21 | Homeobox gene | 33 | ||
| EphA1 | 7q32-q34 | Intercellular signaling | 49 | Poor prognosis | |
| EphA5 | 4q13.1 | Intercellular signaling | 53 | ||
| EphA7 | 6q16.1 | Intercellular signaling | 49 | More frequent in moderately differentiated adenocarcinomas | |
| EPHB2 | 1p36.12 | Intercellular signaling | 53 | ||
| ESR1 | 6q25.1 | Hormonal signaling | 31 | ||
| EVL | 14q32.32 | Cell migration | 60 | ||
| EYA2 | 20q13.1 | Development | 44/51 | Adenoma vs CRC | |
| EYA4 | 6q23 | Development | 70 | ||
| FAM127A | Xq26 | Unknown | 58 | ||
| FBN1 | 15q21.1 | ECM component | 69/79 | Adenoma vs CRC | |
| FBN2 | 5q23.3 | ECM component | 90 | ||
| FLNC | 7q32.1 | Cell migration | 30 | ||
| FOXL2 | 3q23 | Transcription factor | 50 | ||
| GAS7 | 17p13.1 | Development | NA | Increase of methylation level in CRC | |
| GATA4 | 8p23.1 | Transcription factor | 70 | Independent of clinicopathologic features | |
| GATA5 | 20q13.33 | Transcription factor | 79 | Independent of clinicopathologic features | |
| GPNMB | 7p15 | Development | 100 | ||
| GPR101 | Xq25-q27.1 | G protein-coupled receptor | 40 | ||
| GRID1 | 10q22 | Glutamate receptor | 60 | ||
| GRIN2A | 16p13.2 | Glutamate receptor | 82 | ||
| GSPT2 | Xp11.22 | GTPase | 21 | ||
| GUCY1A2 | 11q21-q22 | Intercellular signaling | 50 | ||
| HACE1 | 6q16.3 | Stress response | 28 | ||
| HIC1 | 17p13.3 | Transcriptional repressor | 35/42 | Adenoma vs CRC | |
| HLTF | 3q24 | Transcription factor | 18–34 | ||
| HOXB13 | 17q21.32 | Homeobox gene | 40 | ||
| HRK | 12q24.23 | Apoptosis | 36 | ||
| HUS1 | 7p12.3 | Cell cycle | 22 | ||
| ID4 | 6p22.3 | Transcription factor | 46 | ||
| IGF2 | 11p15.5 | Development | 22 | ||
| IGFBP3 | 7p12.3 | Hormonal signaling | 25 | ||
| IGFBP7 | 4q12 | Hormonal signaling | 18/23 | Adenoma vs CRC | |
| IKZF1 | 7p12.2 | Transcriptional activator | 30–82 | % increase with Duke’s stages | |
| INA | 10q24.33 | Development | 35/65 | Adenoma vs CRC | |
| INHBB | 2q14.2 | Inhibin | 30 | ||
| IRF8 | 16q24.1 | Transcription factor | 43 | ||
| ITGA4 | 2q31.3 | Cell adhesion | 75/92 | Adenoma vs CRC | |
| KCNK12 | 2p16.3 | Potential potassium channel | 41 | ||
| KLF4 | 9q31.2 | Transcription factor | 25 | ||
| LAMA1 | 18p11.31 | Cell migration | 100 | ||
| LRRC3B | 3p24.1 | Putative TSG | 77 | ||
| MAL | 2q11.1 | Proteolipids | 84/91 | Adenoma vs CRC | |
| MGMT | 10q26.3 | DNA repair | 20–60 | ||
| miR-1-1 | 20q13.33 | Translational repression | 50 | ||
| miR-9-1 | 1q22 | Translational repression | 50 | Associated with the presence of lymph node metastasis | |
| miR-34a | 1p36.22 | Translational repression | 74 | ||
| miR-34b/c | 11q23.1 | Translational repression | 99 | ||
| miR-124-1 | 8p23.1 | Translational repression | 75 | ||
| miR-129-2 | 11p11.2 | Translational repression | 83 | ||
| miR-137 | 1p21.3 | Translational repression | 100 | ||
| miR-148 | NA | Translational repression | 65 | ||
| miR-342 | 14q32.2 | Translational repression | 67/86 | Adenoma vs CRC | |
| miR-345 | 14q32.2 | Translational repression | 87 | ||
| miR-373 | 19q13.42 | Translational repression | 88 | ||
| MLH1 | 3p22.2 | DNA repair | 18–22 | Poor prognosis | |
| MMP2 | 16q12.2 | Protease | 95 | ||
| MYOD1 | 11p15.1 | Transcription factor | 69 | ||
| NDRG2 | 14q11.2 | Putative TSG | 27 | ||
| NDRG4 | 16q21 | Putative TSG | 70–86 | ||
| NEURL | 10q25.1 | Putative TSG | 31 | ||
| NEUROG1 | 5q31.1 | Putative TSG | 36 | ||
| NPY | 7p15.1 | Putative TSG | NA | Increase of methylation level in CRC | |
| NRCAM | 7q31.1 | Cell adhesion | 50 | ||
| NTNG1 | 1p13.3 | Development | 70 | ||
| NTRK2 | 9q21.33 | Differentiation | 100 | ||
| OSMR | 5p13.1 | Cytokine signaling | 55/89/90 | Mucosa adjacent to CRC vs colorectal polyps vs carcinoma | |
| PAPSS2 | 10q23.2 | Development | 100 | ||
| PDLIM4 | 5q31.1 | Development | 85/70 | Adenoma vs CRC | |
| PPM1E | 17q23.2 | Phosphatase | 55 | ||
| PRKD1 | 14q12 | Kinase | 20 | ||
| PROM1 | 4p15.32 | Putative TSG | 62 | ||
| PTGIS | 20q13.1-q13.3 | Prostaglandin signaling | 30/44 | Adenoma vs CRC | |
| PTGS2 | 1p25.2-3 | Inflammation | 72 | ||
| PTPRD | 9p23 | Phosphatase | 50 | ||
| RAB32 | 6q24.3 | Ras signaling | 56 | MSI tumors | |
| RARβ | 3p24.2 | Hormonal signaling | 33–85 | ||
| RASSF1A | 3p21.2 | Ras signaling | 41/57 | Stage I/III vs IV | |
| RASSF2 | 20p13 | Ras signaling | 42 | ||
| RASSF5 | 1q32.1 | Ras signaling | NA | Increase of methylation level in CRC | |
| RECK | 9p13.3 | Putative TSG | 44 | ||
| RUNX3 | 1p36.11 | Transcription factor | 27–63 | Poor prognosis | |
| SCTR | 2q14.1 | G protein-coupled receptor | 81 | ||
| SFRP1 | 8p11.21 | Wnt signaling | 95–100 | ||
| SFRP4 | 7p14.1 | Wnt signaling | 100 | ||
| SH3TC1 | 4p16.1 | Putative TSG | 40 | ||
| SLC5A8 | 12q23.1 | Solute carrier | 80 | ||
| SLC6A15 | 12q21.31 | Solute carrier | NA | Increase of methylation level in CRC | |
| SLIT2 | 4p15.2 | Cell migration | 72 | ||
| SMO | 7q32.1 | G protein-coupled receptor | 21 | ||
| SNCA | 4q21.3-q22 | Dopamine signaling | 53/66 | Adenoma vs CRC | |
| SOCS1 | 16p13.13 | Cytokine signaling | 22 | ||
| SOX17 | 8q11.23 | Transcription factor | 86/89–100 | Adenoma vs CRC | |
| SPARC | 5q33.1 | ECM component | 100 | ||
| SPG20 | 13q13.3 | Putative TSG | 78/89 | Adenoma vs CRC | |
| SST | 3q28 | Hormonal signaling | 90 | ||
| ST3GAL6 | 3q12.2 | Putative TSG | 44 | Correlated with EBV-associated gastric carcinomas | |
| STARD8 | Xq13.1 | Putative TSG | 55 | ||
| SYNE1 | 6q25.2 | Putative TSG | 95 | ||
| SYT6 | 1p13.2 | Calcium metabolism | 64 | ||
| TAC1 | 7q21.3 | Hormonal signaling | 95 | ||
| TCERG1L | 10q26.3 | Putative TSG | 100 | ||
| TFPI2 | 7q22 | ECM component | NA | Increase of methylation level in CRC | |
| TIMP3 | 22q12–13 | ECM component | 23 | ||
| TMEFF2 | 2q32.3-q33 | Cell proliferation | 77 | ||
| TP73 | 1p36.33 | Cell cycle control (G1-S) | 63 | ||
| TUBG2 | 17q21 | Cell migration | 71 | ||
| TUSC3 | 8p22 | Putative TSG | 66 | Associated with ulcerative colitis | |
| TWIST1 | 7p21.1 | Transcription factor | NA | Increase of methylation level in CRC | |
| UNC5C | 4q22.3 | Development | 64/76 | Adenoma vs CRC | |
| VIM | 10p13 | Cell migration | 91/77 | Adenoma vs CRC | |
| WIF-1 | 12q13.13 | WIF-1 | 100 | Very limited number of samples | |
| WNT5a | 3p14.3 | Wnt signaling | 20 | Associated with MSI and BRAF V600E mutation | |
| WRN | 8p12 | DNA repair | 29 | ||
| WT1 | 11p13 | Transcription factor | 58 | ||
| ZNF569 | 19q13.12 | Transcription factor | 40 | ||
| Hypomethylation | C7orf50 | 7p22.3 | Unknown | NA | |
| CARD14 | 17q25.3 | NF-κB signaling | NA | ||
| CCDC116 | 22q11.21 | Transcriptional regulator | NA | ||
| CDH3 | 16q22.1 | Cell adhesion | 77 | ||
| CSRP1 | 1q32.1 | Development | NA | ||
| EPHX4 | 1p22.1 | Cell detoxification | NA | ||
| GPR109A | 12q24.31 | G protein-coupled receptor | NA | ||
| GPSM1 | 9q34.3 | G protein signaling | NA | ||
| GRAP | 17p11.2 | Intracellular signaling | NA | ||
| H19 | 11p15.5 | Putative TSG | 18 | ||
| HIST1H2BO | 6p22.1 | Histone | NA | ||
| IGF2 | 11p15.5 | Development | 35 | Poor prognosis | |
| L1CAM | Xq28 | Cell adhesion | NA | ||
| LAMB1 | 7q22 | ECM component | NA | ||
| LILRA4 | 19q13.4 | Cytokine signaling | NA | ||
| LINE1 | NA | Retrotransposon | NA | Associated with MSI and CIMP tumors | |
| MAEL | 1q24.1 | piRNA system | NA | ||
| MIRLET7BHG | 22q13.31 | Long non-coding RNA | NA | ||
| NRXN1 | 2p16.3 | Cell adhesion | NA | ||
| NUP50 | 22q13.3 | Macromolecule trafficking | NA | ||
| S100A4 | 1q21.3 | Cell cycle | NA | ||
| S1PR4 | 19p13.3 | G protein-coupled receptor | NA | ||
| SFT2D3 | 2q14.3 | Transport and trafficking | NA | ||
| SLC39A4 | 8q24.3 | Solute carrier | NA | ||
| SLC6A18 | 5p15.33 | Solute carrier | NA | ||
| SLC6A6 | 3p25.1 | Solute carrier | NA | ||
| TIAM1 | 21q22.1 | Cell migration | NA | Associated with metastasis | |
| miRNA | let-7 family | NA | DLD-1, c-Myc, K-RAS | − | Poor prognosis |
| miR-1-1 | 20q13.33 | TAGLN2 | − | ||
| miR-9-1 | 1q22 | − | |||
| miR-10b | 2q31.1 | − | |||
| miR-15b | 3q25.33 | + | |||
| miR-16 | NA | Wip1 | − | ||
| miR-17 | 13q31.3 | E2F1 | + | Poor prognosis, MSS tumors | |
| miR-18a | 13q31.3 | K-RAS | + | Without lymph node metastasis | |
| miR-18b | Xq26.2 | + | Without lymph node metastasis | ||
| miR-19a | 13q31.3 | PTEN | + | Without lymph node metastasis | |
| miR-19b | NA | + | |||
| miR-20a | 13q31.3 | BNIP2 | + | MSI | |
| miR-21 | 17q23.1 | Cdc25A, MSH2, PTEN, RECK, TIMP3 | + | Poor prognosis, decrease of chemotherapy response, MSI tumors | |
| mir-24 | NA | DHFR | − | ||
| miR-25 | 7q22.1 | + | |||
| miR-26b | 2q35 | EphA2 | − | ||
| miR-29a | 7q32.3 | + | |||
| miR-29b | NA | + | |||
| miR-30a | 6q13 | Beclin 1 | − | ||
| miR-30c | NA | − | |||
| miR-31 | 9p21.3 | FIH-1 | + | Poor prognosis | |
| miR-32 | 9q31.3 | + | |||
| miR-33a | 22q13.2 | + | |||
| miR-34a | 1p36.22 | Bcl2, CDK4/6, E2F3, MET, SIRT1 | − | ||
| miR-34b/c | 11q23.1 | Tp53 | − | ||
| miR-92a | NA | + | MSS tumors | ||
| miR-93 | 7q22.1 | + | |||
| miR-95 | 4 | SNX1 | + | ||
| miR-96 | 7q32.2 | + | |||
| miR-99a | 21q21.1 | − | |||
| miR-101 | NA | COX-2 | − | MSI tumors | |
| miR-106a | Xq26.2 | E2F1 | + | ||
| miR-106b | 7q22.1 | CDKN1A (p21) | + | Without lymph node metastasis | |
| miR-124-1 | 8p23.1 | − | |||
| miR-125a | 19q13.41 | − | |||
| miR-125b | NA | + | Poor prognosis | ||
| miR-126 | 9q34.3 | p85β | − | Associated with metastasis | |
| miR-127 | 14q32.2 | − | |||
| miR-129-2 | 11p11.2 | − | |||
| miR-103b | NA | + | |||
| miR-133a | NA | − | |||
| miR-133b | 6p12.2 | c-Met | + | ||
| miR-135a | NA | APC | + | ||
| miR-135b | 1q32.1 | APC | + | Without lymph node metastasis | |
| miR-137 | 1p21.3 | Cdc42, LSD-1 | − | ||
| miR-139 | 11q13.4 | β–Catenin | − | ||
| miR-140 | 16q22.1 | HDAC4 | − | ||
| miR-141 | 12p13.31 | TGF-β1 | + | ||
| miR-142 | 17q22 | − | MSS tumors | ||
| miR-143 | 5q32 | DNMT3A, Erk5, K-RAS | − | Decrease of chemotherapy response, associated with metastasis | |
| miR-145 | 5q32 | FLI1, IRS1, STAT1, YES | − | MSI tumors | |
| miR-146b | 10q24.32 | − | MSS tumors | ||
| miR-155 | 21q21.3 | MLH1, MSH2, MSH6 | + | With lymph node metastasis | |
| miR-181b | NA | + | Decrease of chemotherapy response | ||
| miR-182 | 7q32.2 | + | |||
| miR-183 | 7q32.2 | Klf4, Sox2, BMI1 | + | ||
| miR-191 | 3p21.31 | − | |||
| miR-192 | 11q13.1 | DHFR, TS, TYMS | − | Decrease of chemotherapy response | |
| miR-195 | 17p13.1 | Bcl-2 | − | ||
| mir-196a | NA | AKT | − | Increase metastasis potential | |
| mir-196b | 7p15.2 | + | Without lymph node metastasis | ||
| miR-200a | 1p36.33 | ZEB1, ZEB2, MLH1, MSH2 | + | Associated with metastasis | |
| miR-200b | 1p36.33 | MLH1, MSH2 | + | Associated with metastasis | |
| miR-200c | 12p13.31 | TGF-β2, ZEB1, ZEB2, BMI1, PTPN12 | + | Poor prognosis, associated with metastasis | |
| miR-203 | 14q32.33 | Klf4, Sox2, BMI1 | + | ||
| miR-212 | 17p13.3 | − | MSS | ||
| miR-215 | 1q41 | DHFR, TS, TYMS | − | Decrease of chemotherapy response | |
| miR-217 | 2p16.1 | − | MSS | ||
| miR-223 | Xq12 | + | |||
| miR-224 | Xq28 | + | Without lymph node metastasis | ||
| miR-301b | 22q11.21 | + | Without lymph node metastasis | ||
| miR-320 | 8p21.3 | − | Poor prognosis | ||
| miR-328 | 16q22.1 | − | |||
| miR-335 | 7q32.2 | + | Without lymph node metastasis | ||
| miR-342 | 14q32.2 | DNMT1 | − | ||
| miR-345 | 14q32.2 | BAG3 | − | ||
| miR-373 | 19q13.42 | LATS2, CD44, RAB22A | − | ||
| miR-374a | Xq13.2 | + | Without lymph node metastasis | ||
| miR-378 | 5q32 | − | Without lymph node metastasis | ||
| miR-378* | 5q32 | − | Without lymph node metastasis | ||
| miR-422a | 15q22.31 | − | |||
| miR-424 | Xq26.3 | + | Without lymph node metastasis | ||
| miR-432* | 14q32.2 | + | MSI tumors | ||
| miR-451 | 17q11.2 | MIF | − | Poor prognosis | |
| miR-455 | 9q32 | − | MSI tumors | ||
| miR-484 | 16p13.11 | − | MSI tumors | ||
| miR-486 | 8p11.21 | − | |||
| miR-492 | 12q22 | + | MSI tumors | ||
| miR-497 | 17p13.1 | − | |||
| miR-498 | 19q13.42 | − | Poor prognosis | ||
| miR-510 | Xq27.3 | + | MSS tumors | ||
| miR-513 | NA | + | MSS tumors | ||
| miR-542 | Xq26.3 | + | |||
| miR-552 | 1p34.3 | + | |||
| miR-592 | 7q31.33 | + | MSS tumors | ||
| miR-675 | 11p15.5 | Rb | + |
CIMP, CpG island methylator phenotype; ECM, extracellular matrix; MSI, microsatellite instability; MSS, microsatellite stable; TSG, tumor suppressor gene.
aOnly hypermethylated genes with methylation prevalence ≥ 20% in CRC patients and ≤ 10% in normal mucosa were reported. Gene symbols and chromosome location are in accordance with www.genecards.org.
bFor DNA hypermethylation/hypomethylation, number represent prevalence (%) in CRC; for miRNAs, - and + mean down-regulated and up-regulated in CRC compared to normal mucosa, respectively; NA means “not applicable.”
Besides its diagnostic potential, methylated genes are associated with a number of clinical features correlated with poor prognosis (DLEC1, EFEMP1, EphA1, EGFR, MLH1, CDH13) [19, 21–24], Epstein-Barr virus–associated gastric carcinomas (B4GALNT2, ST3GAL6) [25]. In contrast, some methylated genes (GATA4, GATA5) are found methylated independently of clinicopathologic features [26].
Some genes are not, at least alone, good biomarkers for CRC since they are frequently methylated in other cancer types such CDKN2A (p16), found methylated across various tumors [10, 16]. In contrast, APC2, B4GALNT1, CCNA1, CDX1, GPNMB, LAMA1, NTRK2, PAPSS2, TCERG1L, and SFRP4 genes are found methylated near 100% of patients tested [19, 27–29]. Therefore, these genes could represent promising CRC biomarkers, similarly to the methylation of detoxification enzyme GSTP1, which is a hallmark of prostate cancer, even though data suggest it may also occur in other cancers [16, 30]. Nevertheless, it confirms that epigenetic silencing is far more common than mutations (see for review of mutation frequencies [13]). Interestingly, numerous genes are gradually methylated during colorectal carcinogenesis. By example, CASR is found methylated at 9%, 69%, and 90% in adenoma, carcinoma, and lymph node metastatic tissues, respectively [31]. Intriguingly, some CRC patients accumulate methylation abnormalities in a large number of genes. This CRC subset is defined with CpG island methylator (CIMP) phenotype characterized by clinicopathological and genetic (chromosomal instability) features, which are the consequence of hypermethylation-mediated TSG silencing involved in the malignant transformation of colonic tissue [32]. In sporadic MSI tumors, hypermethylation-mediated silencing of MMR genes such as MLH1 is common [19, 23, 24].
Concomitant with DNA promoter CGI hypermethylation-mediated silencing, global genomic hypomethylation is observed in CRC. This hypomethylation is usually associated with oncogene activation and genetic instability. Accordingly, an increasing list of genes were identified as hypomethylated in CRC patients, such as CCDC116, SFT2D3, MAEL, and H19/IGF2, which could also be used as biomarkers to reinforce CRC detection [33••, 34]. Furthermore, a recent study suggests that long interspersed nuclear element-1 (LINE-1) hypomethylation could be used as a predictive biomarker of chemotherapy response to fluoropyrimidines in CRC patients [35].
Finally, DNMT expression might also be used as a marker, since overexpression of DNMT1 mRNA was reported in 42% of CRC [36].
All together, these events may represent powerful biomarkers for secondary prevention and risk stratification in CRC. Accordingly, these markers represent promising targets for therapeutic/chemopreventive interventions.
miRNA in CRC
MiRNA pathway is an additional epigenetic mechanism implicated in the regulation of tightly regulated biological processes. MiRNAs are endogenous short non-coding RNAs (~22 nucleotides) that post-transcriptionally regulate mRNA expression levels in a sequence-specific manner. MiRNAs bind sequences located essentially in 5′ and 3′ untranslated regions of target genes degrading mRNA or blocking translation. Increasing amount of evidence reveals that miRNA expression signature dysregulations are associated with carcinogenesis, suggesting miRNAs might act as a novel class of oncogenes or TSGs [8, 10, 37].
An increasing number of reports indicate that miRNA dysregulations are important in colorectal carcinogenesis. Table 1 summarizes these alterations based on experimental data from patients. MiRNome signatures revealed that miRNA affected many tumor-suppressive and oncogenic pathways implicated in CRC pathobiology, including β-catenin/Wnt signaling (miR-135a, -135b, -139, -145) [38•, 39, 40], apoptosis (miR-34a, -133b, -195) [38•, 41], differentiation (miR-141, -200c) [42–44], p53 signaling (miR-34b/c) [45], proliferation (K-RAS signaling: let7 family, miR-18a, -143, -200c) [38•, 41, 46], cell cycle control (miR-34a, -192, -215, -675) [38•, 41, 47], and migration, invasion, and metastasis (miR-126, -143, -196a, -200a, -200b, -200c, -373, -520c) [38•, 41, 44]. MiRNA pathway may also modulate DNA methylation (miR-143, -342) [48, 49].
In addition, miRNA alterations are correlated to a number of clinicopathologic features and outcomes related to CRC pathogenesis. MiR-21 is a representative example, since high levels of expression are associated with lymph node positivity, increased metastasis propensity and advanced tumor stages associated with worse overall survival [50, 51]. Additional miRNAs, including miR-17, -31, -125b, -126, -143, -196a, -200c, -320, -451, and -498, were identified as associated to an increase of metastasis potential, a decrease of disease-free survival, and a poor prognosis [38•, 40–42, 44, 46, 52, 53].
Several studies have identified miRNA expression signatures associated with MSS or MSI CRC phenotypes. These include miR-17, -92, -142, -146b, -212, -217, -510, -513, and -592 associated with MSS, whereas miR-20a, -101, -145, -432*, -455, -484, -492, and -625 were higher in MSI-H tumors [39, 52, 54]. Furthermore, four miRNAs (miR-31, -224, -552, and -592) were identified as able to discriminate between MMR-proficient and MMR-defective adenocarcinomas [40].
All together, these data suggest that CRC-specific miRNA expression signatures are common events, which are representative of CRC-related genetic instability and may be a key event for tumor onset and development. Accordingly, miRNA expression signatures have great and valuable potential for diagnostic and prognostic purposes.
For the past decades, 5-fluorouracil (5-FU) has been and still is the most commonly used chemotherapeutic agent in CRC treatments. However, a significant fraction of patients are refractory or become resistant to 5-FU–based chemotherapies. A growing body of evidence is revealing the importance of miRNA alterations in the modulation of tumor response to 5-FU treatments. For instance, miR-92, -143, and -215, by impairing 5-FU–induced apoptosis [55], could be implicated in the resistance to 5-FU developed by CRC patients presenting low level of expression of miR-92, -143, and -215 [38•, 41]. In addition, miR-21, which plays a central role in colon cancer pathogenesis by targeting many TSGs with elevated expression in advanced tumor stages, was described as an independent predictive marker associated with poor survival and for which overexpression predicts a poor response to therapy [50, 51]. Finally, a recent study suggests that miRNA SNPs rs7372209 and rs1834306 in miR-26-a-1 and miR-100 genes, respectively, affect the clinical outcome of 5-FU–treated CRC patients [56]. These data suggest that miRNA signatures have a potential as marker to predict chemotherapy response.
It has been suggested that, in addition to DNA hypermethylation-mediated silencing of miRNAs (Table 1) [45, 53, 57, 58••, 59–61], alterations of proteins involved in miRNA processing is observed in CRC. Indeed, Papachristou et al. [62] reported that the nuclear ribonuclease Drosha and the cytoplasmic ribonucleases Dicer and Ago2 are possibly implicated in colorectal carcinogenesis and that Dicer could influence tumor progression to advanced stages.
Taken together, these findings demonstrate that miRNome alterations represent promising candidates to develop specific and sensitive biomarkers in CRC pathology with opportunities for primary to tertiary prevention levels.
Histones and Histone-Modifying Enzymes in CRC
An additional layer of epigenetic regulation of gene expression is represented by histone tail post-translational covalent modifications. Core histone (H2A, H2B, H3, H4) N-termini are modified by phosphorylation, acetylation, methylation, ubiquitylation, sumoylation, citrullination, β-N-acetylglucosamination, deimination, and ADP-ribosylation. Altogether, these dynamic and reversible modifications establish a “histone code” regulating chromatin structure and activity. The better understood modifications are acetylation of lysine and methylation of arginine and lysine residues. The acetylation/deacetylation reactions are catalyzed by histone acetyl transferases (HATs) and histone deacetylases (HDACs), respectively. Similarly, methylation/demethylation processes are driven by histone methyltransferase (HMTs) and histone demethylases (HDM). While acetylation occurs as a single addition, methylation exists at various levels on the same residue (ie, mono-, di-, and tri-methylation) [63, 64].
There is now clear evidence that aberrant histone modification profiles are closely connected to tumorigenesis. Indeed, dysregulated activity or expression of histone-modifying enzymes as well as their aberrant recruitment by cytogenetic alterations (eg, leukemia-associated fusion proteins) participate in cancer development by inducing aberrant regulation of oncogenes and/or TSGs, and affecting genome stability and/or chromosome segregation [10, 64, 65]. Although our knowledge about histone code and histone-modifying enzymes is incomplete, some data suggest their implications in CRC. A study from Weichert et al. [66] revealed that HDAC1, HDAC2, and HDAC3 are overexpressed in 36.4%, 57.9%, and 72.9% of CRC cases, respectively. Interestingly, the expression was significantly enhanced in strongly proliferating and poorly differentiated tumors. Thus, high HDAC expression levels are associated with reduced patient survival, with in addition, HDAC2 expression being a prognostic factor for survival [66]. HDAC2 overexpression is accompanied by H4K12 and H3K18 acetylation and correlates with adenoma-carcinoma progression [67]. HDAC1 increase was confirmed in another study reporting an upregulation of two HATs: K(lysine) acetyltransferase 2B (KAT2B, CBP) and p300. KAT2B overexpression was associated with long-term survival, whereas p300 overexpression was correlated with a poor prognosis [68]. Interestingly, the class III HDAC sirtuin 1 is overexpressed in 37% of CRC patients and is mainly associated with MSI and CIMP-high phenotypes [69•]. Finally, it was demonstrated that the expression of the cell-cycle regulator p21 is lower in CRC associated with widespread histone H3 hypo-acetylation in chromatin. These observations were connected to the development and progression of CRC but not with tumor biological behaviors [70].
Dysregulation of enzymes involved in histone methylation is also observed in CRC. Indeed, the HMT suppressor of variegation 3–9 homolog 1 (SUV39H1) is overexpressed in 25% of CRC patients and its expression is significantly associated with DNMT1 expression [36]. Furthermore, the histone H3 lysine 4-specific HMT suppressor of variegation, enhancer of zeste, and trithorax (SET)1 is over-expressed in colon tumor cells, where its expression promotes cell proliferation and survival [71]. Moreover, the multiple myeloma SET domain (MMSET) HMT and putative oncoprotein is overexpressed in CRC patients with a worse 5-year survival. Recently, MMSET expression was associated with a good prognostic value in colon cancer and is more pronounced in early stages of colon carcinogenesis (dysplasia) than in adenocarcinomas [72]. Noteworthy, the histone H3 lysine 9-specific HDM, Jumonji domain containing 1A (JMJD1A) was reported as a useful biomarker for hypoxic tumor cells [73]. In humans, enhancer of zeste homolog 2 (EZH2) overexpression-mediated gene silencing has been identified in numerous tumor types associated with H3K27me3 widespread high levels in chromatin. Recent evidence demonstrated that EZH2 overexpression is a common feature of CRC (observed in 87% of cases) [74]. Finally, it was suggested that oncogenic RAS pathways could modulate histone modifications to influence the expression of target genes involved in the regulation of cell proliferation [75]. Accordingly, overexpression of the HMT SET and MYND domain-containing protein 3 (SMYD3) has been reported in mutated K-RAS CRC patients [76].
Taken together these data suggest that histone modification profiles and histone-modifying enzymes could be used as marker as well as therapeutic/chemopreventive targets in CRC and therefore play a role in CRC prevention.
Chemoprevention, Epigenetics, and CRC
Epigenetic mechanisms by their potential reversibility represent interesting targets in CRC for chemopreventive approaches using dietary agents. Accumulating evidence suggests that natural molecules/nutrients present in our diet might modulate epigenetic events in humans. Table 2 summarizes compounds identified in various in vitro and in vivo tumor models that may exert their chemopreventive potential by targeting epigenetic mechanism(s). The current knowledge about some naturally occurring compounds, which may play a significant role in CRC chemoprevention related to epigenetic modulation, is discussed below.
Table 2.
Compounds present in diet acting as epigenetic modulators
| Dietary agent | Food source | Potential epigenetic target |
|---|---|---|
| 3,3′-diindolylmethane | Broccoli, cauliflower (indole-3-carbinol metabolite) | Histone modifications, miRNAs |
| 6-methoxy-2E,9E-humuladien-8-one | Ginger | Histone modifications |
| Allicin | Garlic | Histone modifications |
| Allyl mercaptan | Garlic | Histone modifications |
| Anacardic acid | Cashew nuts | Histone modifications |
| Apigenin | Parsley, celery | DNA methylation |
| Biochanin A | Soy | Histone modifications |
| Butein | Toxicodendron vernicifluum | Histone modifications |
| Butyrate | Gut flora–mediated fermentation of dietary fiber | Histone modifications |
| Caffeic acid | Coffea | Histone modifications |
| Catechin | Green tea | Histone modifications |
| Chlorogenic acid | Coffea | Histone modifications |
| Cinnamic acid | Cinnamon | Histone modifications |
| Coumaric acid | Cinnamon | Histone modifications |
| Curcumin (diferuloylmethane) | Turmeric | Histone modifications, miRNAs |
| Daidzein | Soy | Histone modifications |
| Delphinidin | Cranberries, Concord grapes, pomegranates | Histone modifications |
| Diallyl disulfide | Garlic | Histone modifications |
| Dihydrocoumarin | Sweet clover (Meliotus officinalis) | Histone modifications |
| (-)-Epigallocatechin gallate | Green tea | DNA methylation, histone modifications, miRNAs |
| Equol | Soy | Histone modifications |
| Fisetin | Strawberries | DNA methylation |
| Flavone | Mandarin | Histone modifications |
| Folate | Leafy vegetables, beans, peas, lentils, eggs, liver | DNA methylation, histone modifications |
| Garcinol, isogarcinol | Garcinia indica | Histone modifications |
| Genistein | Soybean | DNA methylation, histone modifications, miRNAs |
| Hesperidin | Citrus | DNA methylation |
| Isoliquiritigenin | Licorice | Histone modifications |
| Isothiocyanates | Broccoli | Histone modifications, miRNAs |
| Kaempferol | Apples, nuts, tea, onions | Histone modifications |
| Luteolin | Celery, parsley | Histone modifications |
| Lycopene | Tomatoes and various fruits | DNA methylation |
| MCP30 | Bitter melon | Histone modifications |
| Myricetin | Walnuts and various berries, fruits, and vegetables | DNA methylation |
| Naringenin | Citrus | DNA methylation |
| Phloretin | Apples | DNA methylation |
| Piceatannol | Grapes (resveratrol metabolite) | Histone modifications |
| Polyphenon B | Green and black tea | Histone modifications |
| Pomiferin | Maclura pomifera | Histone modifications |
| Protocatechuric acid | Olives | DNA methylation |
| Quercetin | Apples, tea, onion, nuts, berries | DNA methylation, histone modifications |
| Resveratrol | Grapes | Histone modifications |
| Rosmarinic acid | Rosemary | DNA methylation |
| S-allylmercaptocysteine | Garlic | Histone modifications |
| Sanguinarine | Opium poppy | Histone modifications |
| Silibinin | Milk thistle | Histone modifications |
| Sinapinic acid | Sinapis (mustard) | DNA methylation |
| Sulforaphane | Broccoli | DNA methylation, histone modifications |
| Syringic acid | Red grapes | DNA methylation |
| Theophylline | Green and black tea | Histone modifications |
| Ursolic acid | Basil | Histone modifications |
| Selenium | Nuts, cereals, meat, fish, eggs, kidney | DNA methylation, histone modifications |
Curcumin is well recognized for its chemopreventive and therapeutic properties in vitro and in vivo against many tumor types. Curcumin decreases inflammation cell proliferation, invasion, and angiogenesis, triggers apoptosis, and sensitizes tumor cells to cancer therapies [77–79]. These protective properties could be, at least partially, mediated by a modulation of epigenetic events. While no study was performed in colon cells, curcumin is a well-known inhibitor of p300/ KAT2B HAT activity [80]. Furthermore, it was shown that curcumin modulates the miRNA pathway. Specifically, curcumin inhibits miR-21 expression via AP-1 leading to a decreased proliferation and metastasis potential in CRC [81].
Butyrate is an essential short-chained fatty acid (SCFA) for the colon epithelia formed from bacteria-fermented dietary fibers. Butyrate triggers growth arrest, differentiation, and/or apoptosis in many in vitro and in vivo precancerous and tumor cell models including CRC cell lines [82–84]. These biological effects leading to carcinogenesis suppression have been proposed to account for the chemopreventive properties of butyrate and to be mediated by HDAC inhibition–induced histone hyperacetylation [83, 84]. Furthermore, butyrate was identified as the most potent HDAC inhibitor among various SCFAs tested in colon carcinoma cells. In the same study, cinnamic acid, coumaric acid, and caffeic acid also showed HDAC inhibitory activities [85].
(-)-Epigallocatechin gallate (EGCG), the major polyphenol in green tea, has been extensively studied both in vitro and in animal models of carcinogenesis and is well recognized for its chemopreventive properties. EGCG seems to have DNA-demethylating properties since it can induce the reactivation of some methylation-silenced TSGs in various tumor models including human colon cancer cells, limiting their proliferation and invasiveness [86, 87].
Isothiocyanates such as sulforaphane (SFN) are sulfur phytonutrients abundant in broccoli reported to present chemopreventive properties in CRC. SFN has been initially found to inhibit in vitro HDAC activity in human colon cancer cells and then in numerous other models [88, 89]. In vivo, a study demonstrated that APCmin/+ mice with SFN-enriched diet have reduced tumor development associated with an increased histone acetylation and p21 expression [90]. Remarkably, in humans, consumption of 68 g broccoli resulted in a significant inhibition of blood HDAC activity 3 h following intake [91]. Furthermore, prolonged exposure to SFN induces a decrease of various class I and selected class II HDAC proteins and especially HDAC3 [92].
3,3′-diindolylmethane (DIM) is a digestive metabolite of indole-3-carbinol, which is found in vegetables such as broccoli or cauliflower. DIM strongly decreases the expression of the anti-apoptotic protein survivin and enhances the effect of butyrate on both apoptosis in colon cancer cells and prevention of FAP in APCmin/+ mice. These effects were accompanied by a drastic decrease of HDAC1, HDAC2, and HDAC3 expression [93], which could be explained by selective DIM-induced proteasomal degradation of class I HDACs (HDAC1–3, and 8), leading to p21 and p27 overexpression. These data may account for DIM’s capability to trigger G2-cell cycle arrest and apoptosis [94].
Garlic-derived sulfur compounds such as diallyl disulphide (DADS) or allyl mercaptan (AM) are known for their HDAC inhibitory potential. Thus, these compounds induce total histone hyperacetylation in colon cancer cells as well as CDKN1A promoter-associated histone hyperacetylation, which is responsible for p21 overexpression and correlated with a G2/M-cell cycle arrest [89, 95]. Remarkably, epidemiological data suggest that garlic consumption decreases risks of CRC. Thus, it is believed that the effect of these sulfur compounds on HDAC account for their anticarcinogenic and chemopreventive properties.
Quercetin has been shown to activate the class III HDAC sirtuin 1 (SIRT1) and to be a potent antitumor agent by decreasing proliferation, and triggering G2/M-cell cycle arrest and apoptosis in cancer cells [96, 97]. In addition, a study revealed that quercetin demethylates CDKN2A promoter in colon cells [98]. Therefore, quercetin might present protective properties against CRC.
Finally, folate and selenium are common nutrients reported to influence epigenetic events. Epidemiological studies support the link between low folate concentrations and increased CRC risk [99]. Folate is the main source of methyl group necessary for the production of SAM, a universal cofactor in methylation reactions. Thus, defects in folate metabolism or intake lead to hypomethylation of genomic DNA or proto-oncogene and alterations of histone methylation patterns associated with genomic instability in colon cells [83]. Selenium has also been reported to alter epigenetic mechanisms, providing a rationale for its potential chemopreventive efficacy. Indeed, it was shown that colon DNA from rats fed a selenium-rich diet was hypomethylated, whereas low-selenium diet increases DNA methylation of the TSG von Hippel-Lindau [100]. These data were linked to selenium propensity to inhibit DNMT1 activity and protein expression in colon cells [101]. Furthermore, organoselenium metabolites of Se-methyl-L-selenocysteine and L-selenomethionine methylselenopyruvate induce HDAC inhibition–dependent histone H3 acetylation in colon cancer cells associated with an induction of p21 expression, which could account for G2/M cell cycle arrest and apoptosis [102]. Therefore, unbalanced and improper consumption of these nutrients might have an injurious impact on colorectal carcinogenesis.
Conclusions and Perspectives
Since epigenetic alterations are reversible, they were initially considered as interesting targets for chemotherapy using DNMT and HDAC inhibitors such as 5-aza-2′-deoxycytidine (decitabine) and suberoylanilide hydroxamic acid (SAHA, vorinostat), respectively. These compounds induce pleiotropic biological effects including regulation of cell growth, differentiation, autophagy, senescence, and apoptosis. Additionally, they sensitize cells to classical chemotherapeutic agents and they mostly act synergistically as antitumor agents against cancer cells [10, 63, 103, 104]. Nonetheless, the use of such pharmacological epigenetic modulators is associated with some dose-limiting toxicities such as neutropenia and thrombocytopenia observed with SAHA or nonspecific cytotoxic effects observed with nucleoside analogues DNA demethylating agents inherent to their incorporation into DNA. In the perspective to reduce these drawbacks, natural compounds might represent a good alternative to identify safer epigenetic modulators. Accordingly, increasing evidence about the impact of environment on epigenetics as well as early occurrence of epimutations in carcinogenesis make us reconsider epigenetic events as promising preventive targets. However, to reach these attractive perspectives, we need to improve our current knowledge of CRC-associated early epigenetic changes, for early detection and to define promising epigenetic targets for chemoprevention. In addition, a clear impact of such chemopreventive strategies is needed, which requires a better rationale of studies to determine detail mechanisms, and assess safety and efficient doses for humans. Nevertheless, epigenetics and chemoprevention by dietary modulators is well associated with targeted therapy and personalized oncology and should ultimately aid to decrease CRC incidence and mortality rate.
Acknowledgments
Research at the Laboratoire de Biologie Moléculaire et Cellulaire du Cancer (LBMCC) is financially supported by the “Recherche Cancer et Sang” foundation, “Recherches Scientifiques Luxembourg,” the “Een Häerz fir Kriibskrank Kanner” association, the Action Lions “Vaincre le Cancer” Luxembourg, and Televie Luxembourg. MS is recipient of a Télévie Luxembourg fellowship. Editing and print costs were covered by the Fonds National de la Recherche (FNR), Luxembourg.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Dimou A, Syrigos KN, Saif MW. Disparities in colorectal cancer in African-Americans vs Whites: before and after diagnosis. World J Gastroenterol. 2009;15(30):3734–3743. doi: 10.3748/wjg.15.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grulich AE, McCredie M, Coates M. Cancer incidence in Asian migrants to New South Wales. Australia: Br J Cancer. 1995;71(2):400–408. doi: 10.1038/bjc.1995.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee J, Demissie K, Lu SE, Rhoads GG. Cancer incidence among Korean-American immigrants in the United States and native Koreans in South Korea. Cancer Control. 2007;14(1):78–85. doi: 10.1177/107327480701400111. [DOI] [PubMed] [Google Scholar]
- 5.Flood DM, Weiss NS, Cook LS, et al. Colorectal cancer incidence in Asian migrants to the United States and their descendants. Cancer Causes Control. 2000;11(5):403–411. doi: 10.1023/A:1008955722425. [DOI] [PubMed] [Google Scholar]
- 6.MacFarlane AJ, Stover PJ. Convergence of genetic, nutritional and inflammatory factors in gastrointestinal cancers. Nutr Rev. 2007;65(12 Pt 2):S157–S166. doi: 10.1301/nr.2007.dec.S157-S166. [DOI] [PubMed] [Google Scholar]
- 7.Carmona FJ, Esteller M. Epigenomics of human colon cancer. Mutat Res. 2010;693(1–2):53–60. doi: 10.1016/j.mrfmmm.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60(6):376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 9.• Migliore L, Migheli F, Spisni R, Coppede F. Genetics, cytogenetics, and epigenetics of colorectal cancer: J Biomed Biotechnol 2011,2011:792362. Detailed and well-written review about the genetics and epigenetics of colorectal cancer pathobiology. [DOI] [PMC free article] [PubMed]
- 10.Florean C, Schnekenburger M, Grandjenette C, et al. Epigenomics of leukemia: from mechanisms to therapeutic applications. Epigenomics. 2011;3(5):581–609. doi: 10.2217/epi.11.73. [DOI] [PubMed] [Google Scholar]
- 11.Scoazec JY, Sabourin JC. The seventh edition of the TNM classification. Ann Pathol. 2010;30(1):2–6. doi: 10.1016/j.annpat.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138(6):2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 14.Blackburn EH, Tlsty TD, Lippman SM. Unprecedented opportunities and promise for cancer prevention research. Cancer Prev Res (Phila) 2010;3(4):394–402. doi: 10.1158/1940-6207.CAPR-10-0051. [DOI] [PubMed] [Google Scholar]
- 15.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16(Spec No 1):R50–R59. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- 16.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61(8):3225–3229. [PubMed] [Google Scholar]
- 17.Ebert MP, Model F, Mooney S, et al. Aristaless-like homeobox-4 gene methylation is a potential marker for colorectal adenocarcinomas. Gastroenterology. 2006;131(5):1418–1430. doi: 10.1053/j.gastro.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 18.Ju HX, An B, Okamoto Y, et al. Distinct profiles of epigenetic evolution between colorectal cancers with and without metastasis. Am J Pathol. 2011;178(4):1835–1846. doi: 10.1016/j.ajpath.2010.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu XL, Yu J, Zhang HY, et al. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J Gastroenterol. 2004;10(23):3441–3454. doi: 10.3748/wjg.v10.i23.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koinuma K, Yamashita Y, Liu W, et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene. 2006;25(1):139–146. doi: 10.1038/sj.onc.1209009. [DOI] [PubMed] [Google Scholar]
- 21.Herath NI, Doecke J, Spanevello MD, et al. Epigenetic silencing of EphA1 expression in colorectal cancer is correlated with poor survival. Br J Cancer. 2009;100(7):1095–1102. doi: 10.1038/sj.bjc.6604970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scartozzi M, Bearzi I, Mandolesi A, et al. Epidermal growth factor receptor (EGFR) gene promoter methylation and cetuximab treatment in colorectal cancer patients. Br J Cancer. 2011;104(11):1786–1790. doi: 10.1038/bjc.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Yuan X, Jiao N et al. CDH13 and FLBN3 Gene Methylation are Associated with Poor Prognosis in Colorectal Cancer: Pathol Oncol Res 2011. [DOI] [PubMed]
- 24.Deng G, Kakar S, Okudiara K, et al. Unique methylation pattern of oncostatin m receptor gene in cancers of colorectum and other digestive organs. Clin Cancer Res. 2009;15(5):1519–1526. doi: 10.1158/1078-0432.CCR-08-1778. [DOI] [PubMed] [Google Scholar]
- 25.Kawamura YI, Toyota M, Kawashima R, et al. DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology. 2008;135(1):142–51 e3. doi: 10.1053/j.gastro.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 26.Hellebrekers DM, Lentjes MH, van den Bosch SM, et al. GATA4 and GATA5 are potential tumor suppressors and biomarkers in colorectal cancer. Clin Cancer Res. 2009;15(12):3990–3997. doi: 10.1158/1078-0432.CCR-09-0055. [DOI] [PubMed] [Google Scholar]
- 27.Yi JM, Dhir M, Van Neste L, et al. Genomic and epigenomic integration identifies a prognostic signature in colon cancer. Clin Cancer Res. 2011;17(6):1535–1545. doi: 10.1158/1078-0432.CCR-10-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuebel KE, Chen W, Cope L, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3(9):1709–1723. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MS, Louwagie J, Carvalho B, et al. Promoter DNA methylation of oncostatin m receptor-beta as a novel diagnostic and therapeutic marker in colon cancer. PLoS One. 2009;4(8):e6555. doi: 10.1371/journal.pone.0006555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karius T, Schnekenburger M, Ghelfi J, et al. Reversible epigenetic fingerprint-mediated glutathione-S-transferase P1 gene silencing in human leukemia cell lines. Biochem Pharmacol. 2011;81(11):1329–1342. doi: 10.1016/j.bcp.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 31.Hizaki K, Yamamoto H, Taniguchi H, et al. Epigenetic inactivation of calcium-sensing receptor in colorectal carcinogenesis. Mod Pathol. 2011;24(6):876–884. doi: 10.1038/modpathol.2011.10. [DOI] [PubMed] [Google Scholar]
- 32.Curtin K, Slattery ML, Samowitz WS. CpG island methylation in colorectal cancer: past, present and future: Patholog Res Int 2011,2011:902674. [DOI] [PMC free article] [PubMed]
- 33.Kim YH, Lee HC, Kim SY, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol. 2011;18(8):2338–2347. doi: 10.1245/s10434-011-1573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng YW, Idrees K, Shattock R, et al. Loss of imprinting and marked gene elevation are 2 forms of aberrant IGF2 expression in colorectal cancer. Int J Cancer. 2010;127(3):568–577. doi: 10.1002/ijc.25086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawakami K, Matsunoki A, Kaneko M, et al. Long interspersed nuclear element-1 hypomethylation is a potential biomarker for the prediction of response to oral fluoropyrimidines in microsatellite stable and CpG island methylator phenotype-negative colorectal cancer. Cancer Sci. 2011;102(1):166–174. doi: 10.1111/j.1349-7006.2010.01776.x. [DOI] [PubMed] [Google Scholar]
- 36.Kang MY, Lee BB, Kim YH, et al. Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J Cancer. 2007;121(10):2192–2197. doi: 10.1002/ijc.22953. [DOI] [PubMed] [Google Scholar]
- 37.Winter J, Diederichs S. MicroRNA biogenesis and cancer. Methods Mol Biol. 2011;676:3–22. doi: 10.1007/978-1-60761-863-8_1. [DOI] [PubMed] [Google Scholar]
- 38.Arndt GM, Dossey L, Cullen LM, et al. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer. 2009;9:374. doi: 10.1186/1471-2407-9-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarver AL, French AJ, Borralho PM, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;9:401. doi: 10.1186/1471-2407-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oberg AL, French AJ, Sarver AL, et al. miRNA expression in colon polyps provides evidence for a multihit model of colon cancer. PLoS One. 2011;6(6):e20465. doi: 10.1371/journal.pone.0020465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Earle JS, Luthra R, Romans A, et al. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J Mol Diagn. 2010;12(4):433–440. doi: 10.2353/jmoldx.2010.090154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xi Y, Formentini A, Chien M, et al. Prognostic Values of microRNAs in Colorectal Cancer. Biomark Insights. 2006;2:113–121. [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng H, Zhang L, Cogdell DE, et al. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS One. 2011;6(3):e17745. doi: 10.1371/journal.pone.0017745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stratmann J, Wang CJ, Gnosa S, et al. Dicer and miRNA in relation to clinicopathological variables in colorectal cancer patients. BMC Cancer. 2011;11(1):345. doi: 10.1186/1471-2407-11-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogt M, Munding J, Gruner M, et al. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011;458(3):313–322. doi: 10.1007/s00428-010-1030-5. [DOI] [PubMed] [Google Scholar]
- 46.Motoyama K, Inoue H, Takatsuno Y, et al. Over- and under-expressed microRNAs in human colorectal cancer. Int J Oncol. 2009;34(4):1069–1075. doi: 10.3892/ijo_00000233. [DOI] [PubMed] [Google Scholar]
- 47.Tsang WP, Ng EK, Ng SS, et al. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis. 2010;31(3):350–358. doi: 10.1093/carcin/bgp181. [DOI] [PubMed] [Google Scholar]
- 48.Ng EK, Tsang WP, Ng SS, et al. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br J Cancer. 2009;101(4):699–706. doi: 10.1038/sj.bjc.6605195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H, Wu J, Meng X, et al. MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis. 2011;32(7):1033–1042. doi: 10.1093/carcin/bgr081. [DOI] [PubMed] [Google Scholar]
- 50.Slaby O, Svoboda M, Fabian P, et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72(5–6):397–402. doi: 10.1159/000113489. [DOI] [PubMed] [Google Scholar]
- 51.Schetter AJ, Leung SY, Sohn JJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. Jama. 2008;299(4):425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schepeler T, Reinert JT, Ostenfeld MS, et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68(15):6416–6424. doi: 10.1158/0008-5472.CAN-07-6110. [DOI] [PubMed] [Google Scholar]
- 53.Bandres E, Agirre X, Bitarte N, et al. Epigenetic regulation of microRNA expression in colorectal cancer. Int J Cancer. 2009;125(11):2737–2743. doi: 10.1002/ijc.24638. [DOI] [PubMed] [Google Scholar]
- 54.Lanza G, Ferracin M, Gafa R, et al. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Mol Cancer. 2007;6:54. doi: 10.1186/1476-4598-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borralho PM, Kren BT, Castro RE, et al. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J. 2009;276(22):6689–6700. doi: 10.1111/j.1742-4658.2009.07383.x. [DOI] [PubMed] [Google Scholar]
- 56.Boni V, Zarate R, Villa JC et al. Role of primary miRNA polymorphic variants in metastatic colon cancer patients treated with 5-fluorouracil and irinotecan: Pharmacogenomics J 2010. [DOI] [PubMed]
- 57.Grady WM, Parkin RK, Mitchell PS, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene. 2008;27(27):3880–3888. doi: 10.1038/onc.2008.10. [DOI] [PubMed] [Google Scholar]
- 58.•• Suzuki H, Takatsuka S, Akashi H et al. Genome-wide profiling of chromatin signatures reveals epigenetic regulation of microRNA genes in colorectal cancer: Cancer Res 2011. Study investigating the role of chromatin signatures in miRNA dysregulation in colorectal cancer. [DOI] [PubMed]
- 59.Kalimutho M, Di Cecilia S, Del Vecchio Blanco G, et al. Epigenetically silenced miR-34b/c as a novel faecal-based screening marker for colorectal cancer. Br J Cancer. 2011;104(11):1770–1778. doi: 10.1038/bjc.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang JT, Wang JL, Du W, et al. MicroRNA 345, a methylation-sensitive microRNA is involved in cell proliferation and invasion in human colorectal cancer. Carcinogenesis. 2011;32(8):1207–1215. doi: 10.1093/carcin/bgr114. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka T, Arai M, Wu S et al. Epigenetic silencing of microRNA-373 plays an important role in regulating cell proliferation in colon cancer: Oncol Rep 2011. [DOI] [PubMed]
- 62.Papachristou DJ, Korpetinou A, Giannopoulou E et al. Expression of the ribonucleases Drosha, Dicer, and Ago2 in colorectal carcinomas: Virchows Arch 2011. [DOI] [PubMed]
- 63.Folmer F, Orlikova B, Schnekenburger M, et al. Naturally occurring regulators of histone acetylation/deacetylation. Current Nutrition & Food Science. 2010;6:78–99. doi: 10.2174/157340110790909581. [DOI] [Google Scholar]
- 64.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mravinac B, Sullivan LL, Reeves JW, et al. Histone modifications within the human X centromere region. PLoS One. 2009;4(8):e6602. doi: 10.1371/journal.pone.0006602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weichert W, Roske A, Niesporek S, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14(6):1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 67.Ashktorab H, Belgrave K, Hosseinkhah F, et al. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig Dis Sci. 2009;54(10):2109–2117. doi: 10.1007/s10620-008-0601-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishihama K, Yamakawa M, Semba S, et al. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol. 2007;60(11):1205–1210. doi: 10.1136/jcp.2005.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nosho K, Shima K, Irahara N, et al. SIRT1 histone deacetylase expression is associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2009;22(7):922–932. doi: 10.1038/modpathol.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen YX, Fang JY, Lu R, Qiu DK. Expression of p21(WAF1) is related to acetylation of histone H3 in total chromatin in human colorectal cancer. World J Gastroenterol. 2007;13(15):2209–2213. doi: 10.3748/wjg.v13.i15.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yadav S, Singhal J, Singhal SS, Awasthi S. hSET1: a novel approach for colon cancer therapy. Biochem Pharmacol. 2009;77(10):1635–1641. doi: 10.1016/j.bcp.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hudlebusch HR, Santoni-Rugiu E, Simon R, et al. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res. 2011;17(9):2919–2933. doi: 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- 73.Uemura M, Yamamoto H, Takemasa I, et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin Cancer Res. 2010;16(18):4636–4646. doi: 10.1158/1078-0432.CCR-10-0407. [DOI] [PubMed] [Google Scholar]
- 74.Takawa M, Masuda K, Kunizaki M, et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011;102(7):1298–1305. doi: 10.1111/j.1349-7006.2011.01958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pelaez IM, Kalogeropoulou M, Ferraro A, et al. Oncogenic RAS alters the global and gene-specific histone modification pattern during epithelial-mesenchymal transition in colorectal carcinoma cells. Int J Biochem Cell Biol. 2010;42(6):911–920. doi: 10.1016/j.biocel.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 76.Gaedcke J, Grade M, Jung K, et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer. 2010;49(11):1024–1034. doi: 10.1002/gcc.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duvoix A, Morceau F, Delhalle S, et al. Induction of apoptosis by curcumin: mediation by glutathione S-transferase P1-1 inhibition. Biochem Pharmacol. 2003;66(8):1475–1483. doi: 10.1016/S0006-2952(03)00501-X. [DOI] [PubMed] [Google Scholar]
- 78.Duvoix A, Morceau F, Schnekenburger M, et al. Curcumin-induced cell death in two leukemia cell lines: K562 and Jurkat. Ann N Y Acad Sci. 2003;1010:389–392. doi: 10.1196/annals.1299.071. [DOI] [PubMed] [Google Scholar]
- 79.Duvoix A, Blasius R, Delhalle S, et al. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005;223(2):181–190. doi: 10.1016/j.canlet.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 80.Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279(49):51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 81.Mudduluru G, George-William JN, Muppala S, et al. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci Rep. 2011;31(3):185–197. doi: 10.1042/BSR20100065. [DOI] [PubMed] [Google Scholar]
- 82.Schnekenburger M, Morceau F, Henry E, et al. Transcriptional and post-transcriptional regulation of glutathione S-transferase P1 expression during butyric acid-induced differentiation of K562 cells. Leuk Res. 2006;30(5):561–568. doi: 10.1016/j.leukres.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 83.Lu R, Wang X, Sun DF, et al. Folic acid and sodium butyrate prevent tumorigenesis in a mouse model of colorectal cancer. Epigenetics. 2008;3(6):330–335. doi: 10.4161/epi.3.6.7125. [DOI] [PubMed] [Google Scholar]
- 84.Scharlau D, Borowicki A, Habermann N, et al. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat Res. 2009;682(1):39–53. doi: 10.1016/j.mrrev.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 85.Waldecker M, Kautenburger T, Daumann H, et al. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19(9):587–593. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 86.Fang MZ, Wang Y, Ai N, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63(22):7563–7570. [PubMed] [Google Scholar]
- 87.Larsen CA, Dashwood RH. (-)-Epigallocatechin-3-gallate inhibits Met signaling, proliferation, and invasiveness in human colon cancer cells. Arch Biochem Biophys. 2010;501(1):52–57. doi: 10.1016/j.abb.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64(16):5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 89.Nian H, Delage B, Ho E, Dashwood RH. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ Mol Mutagen. 2009;50(3):213–221. doi: 10.1002/em.20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Myzak MC, Dashwood WM, Orner GA, et al. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006;20(3):506–508. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Myzak MC, Tong P, Dashwood WM, et al. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232(2):227–234. [PMC free article] [PubMed] [Google Scholar]
- 92.Rajendran P, Delage B, Dashwood WM, et al. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. 2011;10:68. doi: 10.1186/1476-4598-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bhatnagar N, Li X, Chen Y, et al. 3,3′-diindolylmethane enhances the efficacy of butyrate in colon cancer prevention through down-regulation of survivin. Cancer Prev Res (Phila) 2009;2(6):581–589. doi: 10.1158/1940-6207.CAPR-08-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70(2):646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Druesne-Pecollo N, Chaumontet C, Latino-Martel P. Diallyl disulfide increases histone acetylation in colon cells in vitro and in vivo. Nutr Rev. 2008;66(Suppl 1):S39–S41. doi: 10.1111/j.1753-4887.2008.00066.x. [DOI] [PubMed] [Google Scholar]
- 96.Shan BE, Wang MX, Li RQ. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway. Cancer Invest. 2009;27(6):604–612. doi: 10.1080/07357900802337191. [DOI] [PubMed] [Google Scholar]
- 97.Kim HJ, Kim SK, Kim BS, et al. Apoptotic effect of quercetin on HT-29 colon cancer cells via the AMPK signaling pathway. J Agric Food Chem. 2010;58(15):8643–8650. doi: 10.1021/jf101510z. [DOI] [PubMed] [Google Scholar]
- 98.Tan S, Wang C, Lu C, et al. Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy. 2009;55(1):6–10. doi: 10.1159/000166383. [DOI] [PubMed] [Google Scholar]
- 99.Martinez ME, Henning SM, Alberts DS. Folate and colorectal neoplasia: relation between plasma and dietary markers of folate and adenoma recurrence. Am J Clin Nutr. 2004;79(4):691–697. doi: 10.1093/ajcn/79.4.691. [DOI] [PubMed] [Google Scholar]
- 100.Uthus E, Begaye A, Ross S, Zeng H. The von Hippel-Lindau (VHL) tumor-suppressor gene is down-regulated by selenium deficiency in Caco-2 cells and rat colon mucosa. Biol Trace Elem Res. 2011;142(2):223–231. doi: 10.1007/s12011-010-8764-4. [DOI] [PubMed] [Google Scholar]
- 101.Davis CD, Uthus EO, Finley JW. Dietary selenium and arsenic affect DNA methylation in vitro in Caco-2 cells and in vivo in rat liver and colon. J Nutr. 2000;130(12):2903–2909. doi: 10.1093/jn/130.12.2903. [DOI] [PubMed] [Google Scholar]
- 102.Pinto JT, Lee JI, Sinha R, et al. Chemopreventive mechanisms of alpha-keto acid metabolites of naturally occurring organoselenium compounds. Amino Acids. 2011;41(1):29–41. doi: 10.1007/s00726-010-0578-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107(4):600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schnekenburger M, Grandjenette C, Ghelfi J, et al. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem Pharmacol. 2011;81(3):364–378. doi: 10.1016/j.bcp.2010.10.013. [DOI] [PubMed] [Google Scholar]