

Abstract
Interleukin-23 (IL-23) is a member of the IL-12 family of cytokines with pro-inflammatory properties. Its ability to potently enhance the expansion of T helper type 17 (Th17) cells indicates the responsibility for many of the inflammatory autoimmune responses. Emerging data demonstrate that IL-23 is a key participant in central regulation of the cellular mechanisms involved in inflammation. Both IL-23 and IL-17 form a new axis through Th17 cells, which has evolved in response to human diseases associated with immunoactivation and immunopathogeny, including bacterial or viral infections and chronic inflammation. Targeting of IL-23 or the IL-23 receptor or IL-23 axis is a potential therapeutic approach for autoimmune diseases including psoriasis, inflammatory bowel disease, rheumatoid arthritis and multiple sclerosis. The current review focuses on the immunobiology of IL-23 and summarizes the most recent findings on the role of IL-23 in the pre-clinical and ongoing clinical studies.
Keywords: autoimmune inflammation, briakinumab, inflammatory bowel disease, interleukin-23, psoriasis, rheumatoid arthritis, ustekinumab
Introduction
Interleukin-23 (IL-23), a member of the IL-12 cytokine family, is a heterodimeric cytokine composed of the IL-12p40 subunit, and with a novel p19 subunit.1 Interleukin-23 is mainly secreted by activated macrophages and dendritic cells (DCs) located in peripheral tissues (skin, intestinal mucosa and lung)2 as a disulphide-linked complex with the polypeptide p19 binding protein p40.3,4
Interleukin-23 has been implicated in several autoimmune inflammatory disorders such as colitis, gastritis, psoriasis and arthritis, 5–7 and as a novel pro-inflammatory cytokine, with close resemblance to IL-12.5 Although similar to IL-12 both structurally and in the ability of memory T cells to increase interferon-γ (IFN-γ) production and proliferation, the ability of IL-23 to induce IL-17 provides a unique role compared with that of IL-12 in both the development and the maintenance of autoimmune inflammation.5,8 The balance of IL-12 and IL-23 production by DCs was controlled by prostaglandin E2, which promotes inflammatory responses.9,10 However, in contrast to IL-12, which is important for the differentiation of naive CD4+ T cells,11 whereas it is crucial for the proliferation and development of CD4+ CD45RO+ memory T cells in humans and CD4+ CD45RBlow in mice,5 IL-23 amplifies and stabilizes the proliferation of IL-17-secreting CD4+ memory T cells [T helper type 17 (Th17 cells)],12–14 which produce IL-17, a pro-inflammatory cytokine that stimulates the production of molecules such as IL-1, IL-6, tumour necrosis factor-α (TNF-α), nitric oixide synthase-2 and chemokines responsible for inflammation,2,15–18 whereas IL-12 induce Th1 cell differentiation, which has protective effects on autoimmune responses in certain conditions.19–22 Consequently, although IL-12 and IL-23, members of IL-12 family, have similar structure, the roles of these two cytokines in the differentiation of Th cells are totally different.23,24 The production of both IL-12 and IL-23 requires nuclear factor-κB (NF-κB) achieved by different signalling to produce two cytokines, reflecting that these two cytokines initially trigger immune responses leading to Th1 or Th17 cell-mediated immunity.25 The Th17 cells differentiate in the mouse from naive T cells under the influence of transforming growth factor-β and IL-6,26 and are maintained and expanded primarily by IL-23. An enormous increase in IL-23 release and Th17 priming by human DCs has been identified,27 which are vital mediators of autoimmune28 and inflammatory pathology.15
Research has demonstrated that IL-1β and IL-23 stimulate IL-17 production and expression.29 Moreover, an enhancing effect of IL-17 on TNF-α is mediated IL-23 p19 expression.30
Recent data indicate that IL-23, not IL-12, is a dominant cytokine controlling inflammation in peripheral tissues and joints, based on the finding of p19 and p40, but not p35 mRNA.31,32 Some findings are consistent with the abundant expression of p19 protein in rheumatoid arthritis tissues, despite the apparent absence of heterodimeric IL-23.30,33 The possibility exists that additional by-products of the inflammatory milieu may be required for the generation and secretion of bioactive IL-23. The expressed p19 protein may be playing an independent role in modulating inflammation and may be up-regulated in a human disease.30,31 Interestingly, while p40 over-expression promotes IL-23 formation, a secreted p40 is able to inhibit IL-23-mediated immune responses,34,35 which means that p40 could be another novel target.
IL-23 structure, receptor and signalling pathways
Interleukin-23 is a heterodimer with a 19 000 molecular weight fourfold helical core α subunit (IL-23p19), disulphide linked to an additional 40 000 molecular weight distinct β subunit (IL-12p40). The p19 expression is produced by antigen-presenting cells as well as by T cells and endothelial cells. The p40 is primarily restricted to antigen-presenting cells such as monocytes, macrophages and DCs. The formation of biologically active IL-23 requires the synthesis of both subunits p40 and p19 within the same cell.36,37 Both IL-12 and IL-23 bind to the β1 receptor of T cells and natural killer (NK) cells via their shared p40 subunit. 38
The p19 subunit is composed of four exons and three introns; its gene is located on chromosome 12q13.2. However, the p40 subunit consists of eight exons and seven introns, coded by a gene present on 11q1.3. Human p19 displays 70% structural homology with mouse p19, and shows homology with the p35 subunit of IL-12. Both p35 and p19 are most structurally related to IL-6 and granulocyte colony-stimulating factor, making them members of the gp130-class of long-chain cytokines.3,5 Moreover, the p40 subunit is composed of three domains, D1, D2 and D3, where the D1 domain is an S-type lg-fold and interacts with the D2 domain, and the D3 domain together with other domains represents the canonical cytokine binding homology region found in all class I cytokine receptors, such as non-signalling α receptors for IL-6 and ciliary neurotrophic factor.3,39,40
Interleukin-23 binds to IL-23 receptor (IL-23R) and IL-12Rβ1, but not to IL-12Rβ2. The IL-23R, which binds to IL-23p19, consisting of an extracellular N-terminal immunoglobulin-like domain and two cytokine receptor domains, is a member of the haematopoietin receptor family,41 whereas the IL-12Rβ1 subunit contains three membrane-proximal fibronectin type III domains and two cytokine receptor domains that interact with IL-12/23p40 (Fig. 1).36 The IL-23R chain is expressed mostly on activated memory T cells as well as on NK cells and monocytes/macrophage and DCs (at low levels), whereas the IL-12Rβ1 chain has been shown to be expressed on T cells, NK cells and DCs. Additionally, it has now been shown that the IL-12 p40 homodimer induced the production of lymphotoxin-α in microglia and macrophages via IL-12Rβ1, but not IL-12Rβ2.42 We hypothesize that IL-12Rβ1 is dominant in autoimmune inflammation rather than IL-12Rβ2. These facts suggest that IL-23 may induce an autocrine loop within the innate immune system, leading to the production of numerous mediators of inflammation.43
Figure 1.
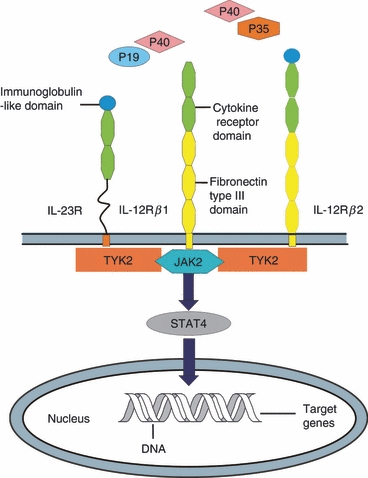
A schematic diagram of different components making up the interleukin-23 (IL-23) and IL-12 receptors and the common signal transduction and activator of transcription 4 (STAT4) activation pathway. IL-12 receptor β1 (IL-12Rβ1) and IL-12Rβ2 each consists of three fibronectin type III (yellow) domains and two cytokine receptor (green) domains with an additional immunoglobulin-like domain (blue) on the latter. IL-23R closely resembles IL-12Rβ2 but without the fibronectin type III domains.
The signalling pathway for IL-23 comprises two receptor chains and signalling proteins: Janus kinase 2 (Jak2), tyrosine kinase 2 (Tyk2), signal transducer and activator of transcription 3 (STAT3) and STAT4. Interleukin-23 uses Jak kinases (Jak2 and Tyk2) to phosphorylate and activate STAT3 and STAT4. Stimulation of the receptor complex activates Jak2 and Tyk2, resulting in the phosphorylation of the receptor complex, and in the formation of docking sites for STATs.2,44,45 The STATs are subsequently phosphorylated, dimerized and translocated into the nucleus, and then activate target genes. The phosphorylation of STAT4 is essential for increasing IFN-γ production and subsequent differentiation of Th1 cells, whereas STAT3 phosphorylation is essential for the development of Th17 cells.46,47 In particular, STAT3 is required for the expression of IL-17A, IL-17F and the transcription factor retinoid-related orphan receptor (ROR) γt in Th17 cultures.48,49 On the other hand, STAT4 is only partially required for the development of IL-23-primed Th17 cells, but it is essential for IL-17 secretion in response to IL-23 plus IL-18 stimulation (Fig. 2).48 Overall, this process organizes the cytokine cascade, activating the necessary immune cells to participate in the eradication of any pathogenic/antigenic challenge.
Figure 2.
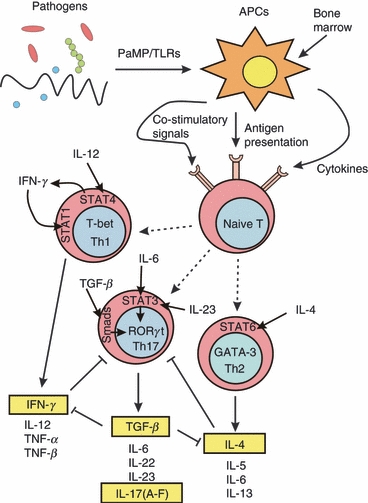
T helper type 17 (Th17) cell differentiation. Following T-cell receptor activation, naive CD4 T cells may differentiate into either Th1 cells in the presence of interleukin-12 (IL-12), which up-regulates interferon-γ (IFN-γ) synthesis via signal transduction and activator of transcription 4 (STAT4) signalling, or Th2 cells in the presence of IL-4. This stimulates STAT1 activation and T-bet transcription factor expression, leading to a Th1 phenotype. Conversely, IL-4 activates STAT6 signalling, which induces GATA3 transcription factor expression and determines Th2 cell differentiation. The Th17 phenotype develops in response to IL-6, transforming growth factor-β (TGF-β) and IL-23 via STAT3 and Smads signalling and the up-regulation of the transcription factor retinoic acid-related orphan receptor RORγt (RORC2 in humans) expression. Interleukin-23 through its receptors induces tyrosine phosphorylation of Jak2 and Tyk2. Jak2 next phosphorylates all STATs. STAT3/STAT4 alone and STAT3 through RORγt enhance Th17 differentiation. TGF-β acting together with IL-6 up-regulates RORγt and induces Th17 differentiation. In addition, Th1 and Th2 cytokines potently inhibit Th17 differentiation. Conversely, TGF-β inhibits the Th1 and Th2 differentiation both by inhibiting the IFN-γ and IL-4 synthesis by effector Th1 and Th2 cells and by blocking the IFN-γ and IL-4 activity on naive T cells. APC, antigen-presenting cells; TLR, Toll-like receptor; TNF-α, tumour necrosis factor-α;
IL-23 in autoimmune inflammatory diseases
Interleukin-23 is a major cytokine bridging the innate and adaptive arms of the immune response.37 It is essential for driving early local immune responses.2 Interleukin-23 was also initially shown to induce the production of IFN-γ,5,28,50 which is important in Th1 responses and cell-mediated immunity against intracellular pathogens. Additionally, IL-23 plays a leading role in the activation of NK cells, enhancement of T-cell proliferation and regulation of antibody production.
Copious evidence supports the theory that increased amounts of IL-23 are associated with several autoimmune diseases including psoriasis, inflammatory bowel disease (IBD), rheumatoid arthritis (RA) and multiple sclerosis (MS). Meanwhile IL-23 was sufficiently indicated acting as a maturation factor for Th17 cells, which had just been discovered and identified as a main player in autoimmunity.28 Furthermore, there is ample evidence showing a protective role of IL-17 against bacterial infections,51,52 whereas IL-17 promotes angiogenesis leading to tumour progression.53
IL-23 in the pathogenesis of psoriasis
Psoriasis is a common immune-mediated chronic epithelial inflammatory type 1 cytokine-related skin disease, characterized by abnormal keratinocyte proliferation and differentiation and also characterized by infiltrates of activated memory T cells that have a high IFN-γ expression.41 There are five types of psoriasis: plaque, guttate, inverse, pustular and erythrodermic; the most common form is plaque psoriasis. It is remarkable that the effect of anti-human IL-23 monoclonal antibody (mAb) was as efficacious as anti-TNF mAb, which is currently the standard therapy for psoriasis.54 A recent study further expands on the importance of blocking the IL-23 pathway on both the epithelial as well as the immunological components of psoriasis.55 While regarding IFN-γ as one of the major cytokines in psoriasis, IL-12 and IL-23 have also been specifically implicated as having crucial roles in the pathogenesis of psoriasis secondary to their role in linking the innate and adaptive immune responses.56 Functionally, IL-12 induce and sustains Th1 immune responses, leading to the secretion of IFN-α and the homing of T cells to the skin. However, IL-23 functions to activate macrophages, and maintain chronic autoimmune inflammation via the regulation of T memory cells and the induction of IL-17.57–59
It has been suggested that the expression of IL-12 might play a pivotal role in certain inflammatory skin diseases. Transgenic mice that over-express IL-12p40 developed an eczematous skin disease, characterized by hyperkeratosis, focal epidermal spongiosis and an inflammatory infiltrate in keratinocytes.60 Because IL-12p40 is a shared subunit of IL-12 and IL-23, either or both cytokines could potentially be involved in the skin lesions that develop in these mice. Accumulating data, however, show the presence of IL-23 in psoriasis lesions. The p40 transgenic mice constitutively produced IL-23, but not IL-12, in basal keratinocytes; furthermore, injections of recombinant IL-23 in non-transgenic littermates contributed to an inflammatory skin disease similar to that of p40 transgenic mice.7 Overall, these studies on transgenic p40 mice clearly indicate that there may be a crucial role for IL-23 rather than IL-12 in psoriasis. Several human studies have reported increased levels of IFN-γ mRNA and IL12p40 in psoriatic lesions.61–63 No significant differences in levels were observed among different clinical types of psoriasis. On the other hand, over-expression of p19 induces inflammation in multiple organs and epithelial tissues, including the skin, and premature death.31,64 These data indicate that IL-23 is a dominant cytokine regulating inflammation in peripheral tissues and epithelial tissues.
With IL-23 as one vital cytokine for the activation of memory T cells to produce IFN-γ, its increased expression may promote the perpetuation of the inflammation process in this disease. Interestingly, neither IFN-γ nor IL-17 gene expression showed any variation after anti-IL-23 treatment in Tonel's mouse model, whereas the Th17 cytokine IL-22 was expressed at low levels but showed a downward trend with anti-IL-23 treatment,55 which is in agreement with previous studies showing that IL-22 is particularly important for IL-23-induced skin inflammation and acanthosis.65,66 Not the IL-12/Th1 axis but the IL-23/Th17 axis is active in several immune-mediated inflammatory autoimmune diseases.15 Therefore, recent interest in psoriasis research has focused on the functional, applied and potential therapeutic roles of the IL-23/Th17 axis.67
Serum level of IL-23 was negatively correlated with the disease duration. Interleukin-23 was higher in the earlier lesion. This might suggest that it could be an important early mediator in the induction of psoriatic lesions. It might also play a role in the stimulation of innate immunity.
IL-23 in the pathogenesis of IBD
Inflammatory bowel diseases, which are chronic inflammatory disorders of the intestinal tract of unknown aetiology, include Crohn's disease and ulcerative colitis. Environmental and genetic factors promote an autoimmune response leading to chronic inflammation. Crohn's disease is identified by its Th1 cytokine pattern, whereas ulcerative colitis has a unique Th2 pathway.
Studies in mice and humans highlight IL-23 as a drug target for the development of novel therapies for IBD. Both IL-12 and IL-23 are increased in the intestine of patients with Crohn's disease. However, various animal models of colitis suggest that IL-23 is more pathogenic than IL-12 in the gut. For instance, administration of anti-IL-23p19 antibodies was able to cure established experimental colitis in mice.68 Moreover, activities that were previously caused by IL-12 may have been mediated via IL-23. Especially, the IL-12p40 mAb that neutralizes IL-12 and IL-23 has shown clinical efficacy in patients with moderate and severe Crohn's disease,69,70 but the therapeutic effect ascribed to the neutralization of IL-12 and/or IL-23 should still be fully defined. Maybe the best opinion in terms of controlling the inflammatory pathway is to block the activities of both IL-12 and IL-23. Additionally, IL-12-dependent responses such as Th1 and cytotoxic T-cell responses are crucial in host protective immunity, and their blockade may leave patients defenceless against infection and cancer. It was shown that anti-CD40-induced systemic inflammation and elevation of concentrations of pro-inflammatory cytokines in the serum were dependent on IL-12, but the local intestinal inflammation and production of IL-17 in the intestine were controlled by IL-23.71 The selective profile of IL-23 in the mucosal inflammatory response makes it a hopeful therapeutic target from the perspective of ablating intestinal immune pathology while sparing systemic immunity. Furthermore, the variants of the IL-23R gene are associated with IBD susceptibility.72 Genetic susceptibility linked to the IL-23R is for generalized gastrointestinal mucosal inflammation.73
The target population of IL-23 is a subset of CD4+ T cells. Studies of experimental models of IBD and clinical research indicate that CD4+ T cells play an important role in initiating and shaping the immunopathological process of IBD.74,75 CD4+ lymphocytes are activated and increased in intestinal lamina propria in various colitis models and in IBDs.76 Moreover, the function of CD4+ lymphocytes has been shown to lead a lethal inflammatory colitis in a severe combined immune deficient mouse model.77 The production of distinct characteristics of cytokines determines the ability of CD4+ lymphocytes to modify the consequence of the intestinal tissue-lesion inflammation.78 Externally, the stimulation of DCs with toll-like receptor ligands induces the synthesis of cytokines that promoted differentiation of IL-17-producing CD4+ T cells.79 The ability of cytokines derived by Th17 to enhance the recruitment and facilitate the activation of neutrophils and motivate the production of defensins by epithelial cell is considered to be significant in the protection of the host particularly at mucosal surfaces.80
In accordance with these findings, studies from several laboratories have led to the identification of more complicated networks of cytokine interactions in IBD tissue, shedding light on the role of Th17 cells in the pathogenesis of colitis.43,68,81 A great increase of Th17 cells and IL-17-secreting macrophages was observed in lesions of patients with Crohn's disease compared with controls.43,47 Interleukin-17-producing T cells are higher in patients with Crohn's disease than in normal gut mucosa and some of these cells also secret IFN-γ.82 The Th17-related cytokines can provoke several inflammatory responses ascribed to their ability to enhance the synthesis of inflammatory cytokines (e.g. IL-1, IL-6, TNF-α, granulocyte–macrophage colony-stimulating factor), chemokines (e.g. IL-8, CXCL1, CXCL8, monocyte chemoattractant protein-1, monocyte-inhibitor protein-3α), cyclo-oxygenase 2, and tissue-degrading matrix metalloproteinases.83,84 These cytokines are found at high levels in the inflamed mucosa of patients with IBD.
In a recent study, it was shown that IL-23 presented in large numbers in Crohn's disease tissue and was produced predominantly by a subset of cells expressing both macrophage and DCs,85 which induced lamina propria cells to make IFN-γ rather than IL-17 production through an IL-23 and TNF-α-dependent mechanism.86 The expression of IL-17 increased in the serum and intestinal mucosa of both Crohn's disease patients and ulcerative colitis patients.87 In patients with active ulcerative colitis, the IL-17-expressing cells are localized typically within the lamina propria, whereas, in active Crohn's disease, these cells are distributed throughout the submucosa and muscularis propria.
Even though the increased expression of these cytokines is augmented in the intestinal mucosa of IBD patients and animal colitis models, the role of IL-23 and IL-17 in IBD pathogenesis remains to be ascertained.
IL-23 in the pathogenesis of RA
Rheumatoid arthritis is a chronic, systemic inflammatory disease characterized by progressively destructive joint inflammation, destruction of articular cartilage and bone and synovial hyperplasia. It principally targets the synovial membrane, cartilage and bone, but can affect many other tissues and organs. The rheumatoid joints contain several cell types, such as macrophages, monocytes, T cells, fibroblast, DCs and plasma cells, which are involved in chronic joint inflammation as well as progressive cartilage and bone destruction. Cytokines secreted by these cells are not only implicated in the inflammatory and immune processes, but also are associated with the pathogenesis of RA. A number of cytokines are expressed and functionally active in synovial tissues. Interleukin-1β, TNF-α, IL-6, IL-10, IL-12 or IL-23 may serve as useful prognostic factors of RA. Importantly, TNF is now targeted in the standard treatment of patients with RA and other cytokines are being tested as targets in the clinic, with promising results.
Both IL-17 and IL-23 are present in the serum, synovial fluid and synovial tissue of patients with RA, whereas they are both absent in healthy joints and osteoarthritis.33,88 The expression of IL-23p19 mRNA and protein, which produces an inflammatory response in RA synovial tissue and erodes bone by increased production of IL-6 and IL-8, is up-regulated by IL-1β and TNF-α in synovial fibroblasts. A similar observation was made in p19 transgenic mice where the p40 subunits of IL-23 were not detectable in serum and which had a high level of IL-1β and TNF-α.33 The IL-17 correlates with the destruction of collagen, increases bone desorption by inducing osteoclastogenesis, destroys human cartilage, and also activates synovial fibroblasts to produce IL-6, IL-8 and vascular growth factors.89–91
Additionally, IL-17 could contribute to up-regulate IL-23p19 expression through the activation of phosphatidyl inositol 3-kinase/AKt (protein kinase B), NF-κB and p38 mitogen-activated protein kinase (MAPK) in RA synovial fibroblasts, which perpetuates synovial inflammation in RA.92 It has been further confirmed that NF-κB and p38 MAPK signalling pathways have dominant roles in the regulation of IL-23p19 production in human microglia, which indicates that IL-23p19 may have an important role in the pathogenesis of MS and experimental autoimmune encephalitis (EAE).93 However, high levels of IL-17 have been found correlated with disease severity in EAE and in collagen-induced arthritis in mice. These data are in line with findings concerning the IL-23p19/IL-17 immune axis in the pathogenesis of MS and RA. Importantly, IL-23 regulates the proliferation of Th17 cells and induces IL-17 production from Th17 cells. Then IL-17 stimulates IL-1, TNF-α and receptor activator of NF-κB ligand (RANKL) expression, leading to the aggravation of the synovial inflammation, and joint destruction.94–96 Interleukin-17 alone and synergistically in combination with other pro-inflammatory cytokines can induce cartilage proteoglycan degradation and collagen breakdown.97,98 Moreover, neutralizing or blocking IL-17 during reactivation of antigen-induced arthritis moderates joint swelling, joint inflammation and bone erosion.91,99 Serum and synovial fluid levels of IL-23 are correlated positively not only with the IL-17 concentration, but also with the IL-1β and TNF-α, implying that IL-23 is closely linked to the production of other pro-inflammatory and anti-inflammatory cytokines in the course of RA. Hence, the IL-23p19/IL-17 axis is an essential inflammatory mediator for the offensive and destructive phases of autoimmune arthritis.
Therefore, these findings highlight the importance of regulating the IL-23/IL-17 axis and emphasize the crucial role of IL-23 in the development and maintenance of chronic inflammatory autoimmune diseases.
IL-23 receptor antagonists in development
The level of interest in this target can be seen from the fact that 15 IL-23 receptor antagonists are now reported to be in clinical or pre-clinical development, from 12 different companies. Some of the antagonists identified by the company compound code number are shown in Table 1.
Table 1.
Identified interleukin-23 receptor (IL-23R) antagonists and their reported development status as of 2010
| Drug | Target | Company | Status | Therapy area |
|---|---|---|---|---|
| Ustekinumab | IL-12R; IL-23R | Centocor Ortho Biotech | Launched | Plaque psoriasis |
| Phase III | Psoriatic arthritis | |||
| Phase II/III | Crohn's disease | |||
| Discontinued | Multiple sclerosis | |||
| Briakinumab | IL-12R; IL-23R | Abbott | Pre-registration | Psoriasis |
| Discontinued | Crohn's disease | |||
| Discontinued | Multiple sclerosis | |||
| Anti-IL-23 therapeutic treatment | IL-23R | Schering-Plough | Phase I | Chronic inflammatory conditions |
| MP-196 | IL-23R | TcL Pharma | Clinical | Autoimmune disease |
| IL-12/IL-23 inhibitors | IL-12R; IL-23R | Synta Pharmaceuticals | Discovery | Crohn's disease; rheumatoid arthritis; multiple sclerosis |
| FM-202 | IL-12R; IL-23R | Femta Pharmaceuticals | Discovery | Psoriasis |
| FM-303 | IL-23R | Femta Pharmaceuticals | Discovery | Inflammatory bowel disease |
| IL-23 Adnectin | IL-23R | Bristol-Myers Squibb | Discovery | Immune disorder |
| IL-23 receptor antagonist | IL-23R | Oscotec | Discovery | Arthritis |
| Anti-IL-23 immunotherapy | IL-23R | Peptinov SAS | Discovery | Inflammatory disease |
| ADC-1012 | IL-23R | Alligator Bioscience AB | Discontinued | Inflammatory disease; cancer |
| Anti-IL-12p40/IL-23p40 HumAbs | IL-12R; IL-23R | Theraclone Sciences | Discontinued | Autoimmune disease |
| Anti-IL-23 HumAbs | IL-23R | Theraclone Sciences | Discontinued | Autoimmune disease |
| Apilimod | IL-12R; IL-23R | Synta Pharmaceuticals | Discontinued | Psoriasis; rheumatoid arthritis; common immunodeficiency |
| LY-2525623 | IL-23R | Eli Lilly | Discontinued | Multiple sclerosis; psoriasis |
All of the antagonists shown in Table 1 are being developed for the treatment of psoriasis, MS, IBD and RA. Ustekinumab, developed by Centocor Ortho Biotech (Horsham, PA), has reached the market; briakinumab, developed by Abbott (Chicago, IL), has pre-registration status. Schering-Plough (Merck & Co., Kenilworth, NJ) has recently reported obtaining positive clinical data. Other IL-23 inhibitors have progressed to pre-clinical development, although for most of them there is no information on the format nor is it indicated whether a development candidate has been selected. The following will discuss the development of compounds shown in Table 1.
Ustekinumab
Centocor Ortho Biotech [a Johnson & Johnson (J&J) subsidiary] has developed and launched ustekinumab (CNTO-1275, Stelara™), an injectable (subcutaneous) mAb against the IL-12/IL-23p40 subunit. The mAb is a high-affinity IgG1κ HumAb, which was originally isolated using Medarex Inc.'s HumAb transgenic mouse technology (Medarex Inc., Princeton, NJ). No details of this process are available.
Ustekinumab prevents the interaction of IL-12/IL-23 with their receptor, thereby blocking subsequent signalling, differentiation and cytokine production central to inflammatory diseases. This candidate has been studied for four indications: psoriasis, psoriatic arthritis, Crohn's disease and MS, although it is not effective in reducing the number of MS lesions and it is assumed to have been discontinued for this indication. In 2009, the drug had been launched for chronic plaque psoriasis in Europe and USA. In Japan, this product was additionally indicated for the treatment of psoriatic arthritis in March 2011.
Psoriasis clinical studies
Integrated analysis and assessment that included data from phase III ustekinumab trials including the pivotal PHOENIX 1, PHOENIX 2 and ACCEPT studies were published.100–103 The proportion of patients with at least 75% improvement from baseline in the Psoriasis Area and Severity Index score (PASI-75) was 74·2% for 90 mg and 54·6% for 45 mg in heavier patients (> 100 kg), but the proportion with a response of at least 75% improvement from baseline in PASI score was similar between doses (80·8% versus 76·9%) in lighter patients (≤ 100 kg), and was only 3–4% in placebo-treated patients at 12 weeks. Serum ustekinumab concentrations were also affected by weight, with lower serum concentrations observed in heavier patients at each dose.
In the PHOENIX 1 trial, 766 patients with moderate-to-severe psoriasis were randomized to receive ustekinumab 45 mg (n = 255) or 90 mg (n = 256) at weeks 0 and 4 and then every 12 weeks; or placebo (n = 255) at weeks 0 and 4, with subsequent cross-over to ustekinumab at week 12. The primary end-point was the proportion of subjects who achieve ≥ 75% improvement in their PASI from baseline at week 12. After two doses, 67 and 66% of patients in the 45-mg and 90-mg groups, respectively, achieved PASI-75 compared with 3% of patients in the placebo group. A total of 42 and 37% of patients in the 45-mg and the 90-mg arms achieved PASI-90 or nearly complete clearance of psoriasis, respectively, compared with 2% of placebo-treated patients. Of those patients that continued ustekinumab treatment after week 40, 87 and 90% of subjects in the 45-mg and 90-mg groups had a sustained PASI-75 response, respectively, compared with 64 and 62% of patients who switched to placebo. Furthermore, 66 and 73% of patients achieved PASI-90 after receiving 45 and 90 mg of ustekinumab, respectively, and response rates were maintained until week 52 with continued treatment.
In the PHOENIX 2 trial, 1230 patients with moderate-to-severe plaque psoriasis in the USA and Canada were randomly assigned to receive subcutaneous ustekinumab (45 or 90 mg) or placebo at weeks 0 and 4, then every 12 weeks until week 52. The primary end-point was the proportion of patients who achieved ≥ 75% improvement in PASI at week 12. At week 12, 67 and 76% of patients in the 45-mg and 90-mg groups achieved a PASI-75 response, respectively, compared with 4% of placebo-treated patients. At the same time-point, 42 and 51% of patients in the 45-mg and 90-mg groups achieved PASI-90, compared with 1% of placebo-treated patients. A number of patients who received an additional dose at week 16 maintained a response through to week 28. In addition, within 4 weeks of treatment, significant improvements in quality-of-life measures were seen in ustekinumab-treated patients compared with placebo patients. Dermatology Life Quality Index (DLQI) scores of 6·0 were observed in both of the ustekinumab groups, compared with a score of 1·0 in the placebo group. At week 12, 72 and 77% of 45-mg-treated and 90-mg-treated patients had a significant reduction in their DLQI score, compared with 21% of placebo-treated patients. A total of 49, 53 and 48% of placebo-treated, 45-mg-treated and 90-mg-treated patients, respectively, experienced adverse events. Discontinuation for adverse events occurred in 0·2 and 1% of patients in the 45-mg and 90-mg groups, compared with 2% of patients in the placebo group. In all, 2% and 1% of 45-mg-treated and 90-mg-treated patients experienced at least one serious adverse event, compared with 2% of placebo-treated patients.
The ACCEPT study is a randomized 12-week phase III clinical trial comparing ustekinumab with etanercept.103 The trial randomized 903 subjects into one of three groups: ustekinumab 45 mg dosed at weeks 0 and 4, ustekinumab 90 mg at weeks 0 and 4, or etanercept 50 mg twice weekly. The primary end-point, PASI-75 at week 12, was achieved by 74% of the ustekinumab 90-mg group, 68% of the ustekinumab 45-mg group, and 57% of the etanercept group. Treatment for the 90-mg ustekinumab group was significantly more efficacious compared with that for the etanercept group (P < 0·001). In general, patients given ustekinumab had better physician global assessment compared with the etanercept group.
Psoriatic arthritis clinical studies
A multi-centre, randomized, double-blind, placebo-controlled phase II trial was initiated in 146 patients with psoriatic arthritis.104 Patients received subcutaneous ustekinumab (90/63 mg) at weeks 0, 1, 2 and 3 and placebo at weeks 12 and 16, or placebo at weeks 0, 1, 2 and 3 and ustekinumab at weeks 12 and 16. The primary end-point was a ≥ 20% improvement from baseline in the American College of Rheumatology arthritis scores (ACR20) at week 12. The proportion of patients reaching the primary end-point (ACR20) was significantly higher in the ustekinumab group (42·1%) compared with the placebo group (14·3%). ACR50 and ACR70 were also achieved by a higher proportion of patients in the ustekinumab group (25 and 10·5%, respectively), compared with the placebo group (7·1 and 0%). A significantly greater decrease from baseline to week 12 in the Health Assessment Questionnaire Disability Index was also observed in ustekinumab-treated patients (mean change: −0·31) compared with placebo recipients (mean change: −0·04). Furthermore, 52·4% of patients in the ustekinumab group achieved a PASI-75 compared with 5·5% of placebo recipients. The proportion of patients who experienced an adverse event was similar in both groups. However, 3·9% of patients in the placebo group experienced a serious adverse event compared with 0% of patients in the ustekinumab group.
Crohn's disease clinical studies
A phase II randomized, double-blind, placebo-controlled, cross-over trial of the clinical effects of ustekinumab was performed in 104 patients with moderate to severe Crohn's disease of at least 6 weeks duration.105 Patients were randomized to one of four dose groups: subcutaneous placebo at weeks 0–3, then ustekinumab at weeks 8–11; subcutaneous ustekinumab at weeks 0–3, then placebo at weeks 8–11; intravenous placebo at week 0, then ustekinumab at week 8; or intravenous ustekinumab at week 0, then placebo at week 8. The primary end-point was clinical response [defined as a reduction in Crohn’ disease activity index (CDAI) score of 25% and 70 points]. At the week 8 end-point, 49% of the ustekinumab patients had achieved a complete response, compared with 39·6% of the placebo population. A second population composed entirely of non-responders to infliximab were given ustekinumab, either subcutaneously (90 mg; n = 14) at weeks 0, 1, 2 and 3, or intravenously (4·5 mg/kg; n = 13) at week 0. The rates for complete response for these patients at 8 weeks were 42·9 and 53·8%, respectively. No serious infections occurred in the first population but two were reported in the second population (disseminated histoplasmosis and food poisoning). Three patients developed injection-site reactions in both the placebo and ustekinumab groups. Ustekinumab demonstrated efficacy with the greatest response occurring in patients with a history of infliximab use.
Multiple sclerosis clinical studies
A phase I double-blind, placebo-controlled, sequential dose escalation trial enrolled 20 patients with remitting–relapsing MS.106 Four cohorts of five patients each were randomized to receive a single subcutaneous injection of ustekinumab or placebo at doses of 0·3, 0·75, 1·5 and 3·0 mg/kg. One subject was diagnosed with breast cancer 21 days after IL-12/23 antibody administration, whereas, another developed a rash 3 days after administration of the drug, which resolved within 3 days. In the phase II study in MS, adverse events occurred in 78% of the placebo-treated patients compared with 85% of those treated with ustekinumab, one (2%) of which was serious in the placebo arm compared with six (3%) in those receiving treatment.107 There was a dose-dependent increase in the number of patients who had injection-site reactions.
Briakinumab
Abbott, under license from Cambridge Antibody Technology (Cambridge, UK), is developing briakinumab (ABT-874, J-695), a recombinant, fully human, IgG1 mAb designed to target the shared p40 subunit of IL-12 and IL-23, for the potential subcutaneous treatment of psoriasis.57
Regulatory applications were submitted in the USA and Europe for the treatment of psoriasis in the third quarter of 2010. Briakinumab was previously developed as the agent for RA; however, no further development in this indication had been reported. The drug was also under development for Crohn's disease and MS, but these two indications had been terminated separately.
Briakinumab structure is similar to that of normal human IgGλ antibodies, only differing at the IL-12 p40-specific antigen binding region. Briakinumab binds to the soluble forms of IL-12 and IL-23, preventing binding of these interleukins with T cells and NK cells.108 Briakinumab was developed with a completely human protein sequence for the purpose of avoiding human reactions to foreign proteins.109 It was initially isolated by screening three separate single-chain variable fragment phage display libraries generated from human lymphoid cells for the purpose of selecting therapeutic antibodies to IL-12. Target affinity was improved by mutagenesis of the complementarity-determining regions (CDRs) simultaneously in both the heavy (H) and light (L) chains. The resulting V regions were classified and further modified by individually mutating specific contact positions.
Pre-clinical and clinical studies demonstrated that briakinumab significantly inhibited the production of pro-inflammatory cytokines, including IL-12, IFN-γ, TNF-α, IL-6, IL-23 and IL-17, but not IL-10 and IL-18. The magnitude of change in cytokine secretion did not correlate with the dose or regimen of briakinumab, but briakinumab dose-dependently inhibited IFN-γ production. Briakinumab has been studied in murine and primate animal models. Intravenous administration of briakinumab to monkeys resulted in minimal complement fixation and activation. No antibody-dependent cytotoxicity has been demonstrated, and in all experimental models briakinumab has been able to reduce the inflammatory reaction. In clinical trials, briakinumab was generally effective and well tolerated, with the most common adverse events being injection-site reactions and nasopharyngitis. Patients with psoriasis treated with briakinumab had higher clinical response rates compared with patients receiving placebo.
Psoriasis clinical studies
A phase III randomized, double-blind, double-dummy, multi-centre 12-week study in patients with moderate to severe psoriasis (n = 347), compared the efficacy and safety of treatment with briakinumab to etanercept. Patients were randomized to receive briakinumab 200 mg at weeks 0 and 4, then 100 mg at week 8; etanercept 50 mg twice weekly; or placebo. The primary end-point was proportion of patients achieving a physician global assessment score of 0 or 1 and proportion of patients achieving PASI-75 at week 12. Briakinumab was statistically superior to both etanercept and placebo on both primary end-point measures, with 71% of briakinumab-treated patients achieving a physician global assessment score of 0 or 1 (39·7% with etanercept versus 2·9% with placebo) and 81·9% of briakinumab-treated patients achieved PASI-75 (56% with etanercept versus 7·4% with placebo).
A 12-week, randomized, double-blind, placebo-controlled, multi-centre, post-hoc phase III study involved a total of 180 adult psoriasis patients with at least 10% body surface area (BSA) involvement and a PASI score equal to or above 12. Patients were randomized to receive six cohorts: single dose of 200 mg briakinumab at week 0; 100 mg briakinumab once every 2 weeks for 12 weeks; 200 mg briakinumab every week for 4 weeks; 200 mg once every 2 weeks for 12 weeks; 200 mg briakinumab every week for 12 weeks; or placebo. The percentage of patients weighting ≤ 100 kg who achieved a PASI-75 response at week 12 was 71·7% (73% for patients over 100 kg). A PASI-75 response was likewise similar between patients with and without a history of psoriatic arthritis, at 83·7 and 86·9%, respectively. Similar response rates were also observed regardless of baseline PASI scores: 87·0% of patients with baseline PASI scores of ≤ 20 achieved PASI-75 at week 12 and 84·0% of patients with baseline PASI scores > 20 achieved a PASI-75 response at week 12. Previous psoriasis treatment (e.g. topical agents, phototherapy, systemic agents and other biological agents) had a minimal effect on the efficacy of briakinumab and did not appear to affect PASI-75 response rates.
A worldwide, randomized, double-blind phase III trial evaluated safety and efficacy, comparing briakinumab with methotrexate in subjects with moderate to severe plaque psoriasis. Patients (n = 317) were to receive briakinumab 200 mg at week 0, 4 and 100 mg at week 8 and every 4 weeks thereafter administered as a subcutaneous injection; or methotrexate 5·0–25 mg weekly. The primary end-point included the proportion of subjects achieving a PASI-75 response relative to baseline and a physician global assessment score of 0/1 at 24 and 52 weeks, respectively. At 24 weeks, 81·8% of briakinumab-treated patients achieved PASI-75 clearance, compared with 39·9% of those taking methotrexate. PASI-90 clearance was achieved by 63·6% of briakinumab-treated patients, versus 22·7% in patients treated with methotrexate. PASI-100 was achieved by 42·2% of briakinumab-treated patients, versus 8·6% of methotrexate-treated patients. At 52 weeks, 66·2% of briakinumab-treated patients achieved PASI-75 clearance compared with 23·9% with methotrexate. PASI-90 clearance was achieved by 59·7% of briakinumab-treated patients versus 17·8% of those taking methotrexate. PASI-100 was achieved by 45·5% of briakinumab-treated patients, versus 9·2% of methotrexate-treated patients.
Crohn's disease clinical studies
In a double-blind, placebo-controlled international trial, 79 patients (16 given placebo and 63 briakinumab-treated) with active Crohn's disease were given 1 or 3 mg/kg briakinumab subcutaneously weekly for 7 weeks. Cohort 1 was given a 4-week interval between the first and second injections and the second cohort received the seven weekly injections with no interruption. At the 18-week follow-up, briakinumab was found to be safe and well tolerated. Adverse events included nausea, abdominal pain, headache and fever, but were not significantly different from those of placebo (except for mild injection-site reactions). Clinical responses of patients treated in cohort 1 at 3 mg/kg were consistently higher than placebo rates, but this was not significant. Patients in cohort 2 receiving 3 mg/kg responded well to treatment. Clinical responses following the 3 mg/kg dose were 75% and 69% at the end of treatment and after 12 weeks follow-up, respectively, compared with placebo at 25% and 13%. After 12 weeks of follow-up, briakinumab produced remission in 38% and 50% of patients, respectively. The effects were durable throughout the 18 weeks of follow-up. In a subgroup study, briakinumab decreased lamina propria mononuclear cell Th1 cytokine production in patients achieving a clinical response. At the end of treatment, mean IL-12, IFN-γ and TNF-α levels were reduced from 153 to 2·8, 9835 to 1078, and 3486 to 1062 pg/ml, respectively. The 1 mg/kg dose did not exert a significant therapeutic effect.
In November 2007, a worldwide, multi-centre randomized, double-blind phase IIb study was initiated to evaluate safety, efficacy and pharmacokinetics of briakinumab compared with placebo in patients with moderate-to-severe active Crohn's disease. Patients (n = 246) were to receive briakinumab (400 or 700 mg) or placebo every 4 weeks. The primary end-point included the proportion of subjects achieving clinical remission, defined as a CDAI score of lower than 150. However, the study had been terminated by July 2010.
Multiple sclerosis clinical studies
A phase II trial in MS sponsored by Cambridge Antibody Technology was initiated in 2004. Patients were to be randomized in parallel groups to receive briakinumab dosed weekly or every other week or placebo for 24 weeks, which would be followed by a 24-week open-label extension phase. The primary outcome was to be the comparison over 24 weeks of the cumulative gadolinium-enhanced (T1-weighted) lesions. However, in 2007, Cambridge Antibody Technology reported that further development for this indication had ceased.
Anti-IL-23 (chronic inflammation) therapeutic treatment
Schering-Plough is presumed to be developing a mAb against IL-23 for the potential treatment of chronic inflammatory conditions. However, the company compound number or research code is unknown.
Pre-clinical data were published on testing of anti-IL-23p19-specific antibodies in the EAE animal model of human MS.110 Anti-IL-23p19 mAbs were compared with anti-p40 mAbs: anti-IL-23p19 treatment effectively blocked both acute disease and EAE relapse, and consistent with previous reports, anti-p40 treatment was also remarkably effective. Anti-IL-23p19 therapeutic treatment reduced the serum level of IL-17 as well as central nervous system expression of IFN-γ, IP-10, IL-17, IL-6, and TNF mRNA. The research has demonstrated that administration of anti-IL-19 mAbs blocked the invasion of inflammatory cells into the central nervous system and prevented EAE. In addition, therapeutic treatment with anti-IL-23p19 during active disease inhibited proteolipid protein epitope spreading and prevented subsequent disease relapse, which means that targeting IL-23 can suppress and reverse ongoing disease.
Pre-clinical observations on mice treated with an anti-mouse IL-23 antibody and then challenged with various pathogens showed that the anti-IL-23 mAbs did not increase the risk of infection as much as an anti-mouse p40-subunit mAb. In addition, cancer risk was decreased with the IL-23 antibody in different murine tumour models (unlike the anti-p40 antibody, which increased risk). The phase I clinical trial for chronic inflammatory conditions is ongoing, but the details are unavailable.
Other drugs in development status
TcL Pharma (now known as Effimune; Nantes, France) is developing MP-196, a mAb that selectively blocks the p19 subunit of IL-23, for the potential treatment of autoimmune diseases including rheumatoid arthritis and Crohn's disease. By September 2010, clinical proof-of-concept studies were ongoing.
Synta (Lexington, MA) has also recently started to pursue the development of a series of small molecule, IL-12/IL-23 dual inhibitors, for the potential oral/topical treatment of inflammatory and autoimmune diseases. The IL-12/23 inhibitors had shown substantial efficacy in animal models of Crohn's disease, RA and MS but no candidate can currently be provided.
Femta Pharmaceuticals (San Diego, CA) is developing a series of humanized mAbs, including FM-202 and FM-303, created using ATLAb antibody engineering platform licensed from BioAtla (San Diego, CA). FM-202 targets IL-12/IL-23 for the potential treatment of psoriasis, whereas FM-303, targeting IL-23 selectively, is used to treat IBD. The ATLAb antibody engineering platform provides rapid humanization of non-human antibodies and significant improvement in antibody characteristics including affinity, specificity, effector function, stability and solubility.
Bristol-Myers Squibb (Uxbridge, UK) is investigating IL-23 Adnectin, a fusion protein antagonist of IL-23 isolated using Adnexus’ Adnectin technology (Adnexus, Waltham, MA), for the potential treatment of immune disorders. At December 2010, IL-23 Adnectin was still in the company's pipeline.
Alligator Bioscience (Lund, Sweden) is investigating ADC-1012, a recombinant protein IL-23 antagonist, created using Alligator's FIND (Fragment Induced Diversity) optimization technology, for the potential treatment of inflammation. Previously, the company was investigating the drug for use in cancer. However, by June 2010, this indication was no longer listed as under investigation on the company's pipeline. Proteins were created using Alligator's FIND optimization technology, a rapid in vitro evolution technology that mimics the natural process of creating protein diversity through recombination.
Oscotec Inc. (Chungnam, Korea) appears to be fairly active in seeking a series of small molecule IL-23 antagonists, which inhibit differentiation of Th17 cells, for the potential oral treatment of arthritis. In December 2009, the company planned to initiate pre-clinical studies in 2012 and a phase I trial in 2013; in April 2010, the programme was listed as being in hit identification.
Peptinov SAS (Paris, France) is investigating an immunotherapy, which selectively inhibits cytokine IL-23, for the potential treatment of inflammatory diseases including psoriasis and RA. Proof-of-concept studies had demonstrated that the therapy showed efficacy in animals by protecting mice against collagen-induced arthritis.
Conclusion
Interleukin-23 is a member of an intriguing family of cytokines, which has both pro-inflammatory and anti-inflammatory effects on the development of autoimmune pathology. Emerging evidence suggests that IL-23 is the dominant cytokine for many immunological functions. Expression of IL-23 exhibits great importance in the inflammatory reaction in psoriasis, RA and IBD, in terms of its biological activity, which elicits the activation of Th17 cells.
Therapeutically, much experience has been gained through pre-clinical and clinical studies of IL-12/IL-23 antibodies for the treatment of autoimmune inflammatory diseases. We have reviewed the IL-23 or IL-23R or IL-23/IL-17 axis novel target, which is important for chronic inflammation and autoimmunity, as well as a subset of the technological advances in the therapy for autoimmune inflammatory diseases. Interleukin-23 can be blocked by a human monoclonal recombinant antibody against IL-12/IL-23p40, such as ustekinumab and briakinumab. Similarly, numerous drugs, for instance, MP-196, FM-303, IL-23 Adnectin, etc. can block IL-23 specifically.
In conclusion, the inhibition of IL-23 might be a novel and promising therapeutic strategy, especially in the therapy of autoimmune inflammatory diseases like psoriasis, for which therapeutic effectiveness and safety data from ustekinumab are positive. Interleukin-23 can be targeted by using an antibody against IL-12/IL-23; nevertheless, it would be much more useful to design drugs that target the IL-23p19 or IL-23 receptor, so inhibiting IL-23 without modifying the effects of IL-12.
Acknowledgments
The work was supported by the Important National Science & Technology Specific Projects of China (No. 2009ZX09103-033), the National Natural Science Foundation of China (No. 30772647) and the Fundamental Research Funds for the Central Universities of China (No.2J10023).
Disclosures
The authors declare having no competing interests.
References
- 1.Kleinschek MA, Muller U, Brodie SJ, et al. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J Immunol. 2006;176:1098–106. doi: 10.4049/jimmunol.176.2.1098. [DOI] [PubMed] [Google Scholar]
- 2.McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23/IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Lupardus PJ, Garcia KC. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J Mol Biol. 2008;382:931–41. doi: 10.1016/j.jmb.2008.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerosa F, Baldani-Guerra B, Lyakh LA, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. 2008;205:1447–61. doi: 10.1084/jem.20071450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 6.Lankford CS, Frucht DM. A unique role for IL-23 in promoting cellular immunity. J Leukoc Biol. 2003;73:49–56. doi: 10.1189/jlb.0602326. [DOI] [PubMed] [Google Scholar]
- 7.Kopp T, Lenz P, Bello-Fernandez C, Kastelein RA, Kupper TS, Stingl G. IL-23 production by cosecretion of endogenous p19 and transgenic p40 in keratin 14/p40 transgenic mice: evidence for enhanced cutaneous immunity. J Immunol. 2003;170:5438–44. doi: 10.4049/jimmunol.170.11.5438. [DOI] [PubMed] [Google Scholar]
- 8.Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–31. doi: 10.1038/nri1648. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi F, Yanagawa Y, Onoe K, Iwabuchi K. Dendritic cell differentiation with prostaglandin E results in selective attenuation of the extracellular signal-related kinase pathway and decreased interleukin-23 production. Immunology. 2010;131:67–76. doi: 10.1111/j.1365-2567.2010.03275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux-Lombard P, Ferrari-Lacraz S, Dayer JM. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood. 2008;112:3696–703. doi: 10.1182/blood-2008-05-155408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trinchieri G. Proinflammatory and immunoregulatory functions of interleukin-12. Int Rev Immunol. 1998;16:365–96. doi: 10.3109/08830189809043002. [DOI] [PubMed] [Google Scholar]
- 12.Belladonna ML, Renauld JC, Bianchi R, et al. IL-23 and IL-12 have overlapping, but distinct, effects on murine dendritic cells. J Immunol. 2002;168:5448–54. doi: 10.4049/jimmunol.168.11.5448. [DOI] [PubMed] [Google Scholar]
- 13.Beadling C, Slifka MK. Regulation of innate and adaptive immune responses by the related cytokines IL-12, IL-23, and IL-27. Arch Immunol Ther Exp (Warsz) 2006;54:15–24. doi: 10.1007/s00005-006-0002-6. [DOI] [PubMed] [Google Scholar]
- 14.Bastos KR, de Deus Vieira de Moraes L, Zago CA, Marinho CR, Russo M, Alvarez JM, D'Imperio Lima MR. Analysis of the activation profile of dendritic cells derived from the bone marrow of interleukin-12/interleukin-23-deficient mice. Immunology. 2005;114:499–506. doi: 10.1111/j.1365-2567.2005.02118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–22. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol. 2008;38:2636–49. doi: 10.1002/eji.200838535. [DOI] [PubMed] [Google Scholar]
- 17.Liao JJ, Huang MC, Goetzl EJ. Cutting edge: alternative signaling of Th17 cell development by sphingosine 1-phosphate. J Immunol. 2007;178:5425–8. doi: 10.4049/jimmunol.178.9.5425. [DOI] [PubMed] [Google Scholar]
- 18.Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–55. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 20.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J Immunol. 1997;158:5507–13. [PubMed] [Google Scholar]
- 21.Manoury-Schwartz B, Chiocchia G, Bessis N, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J Immunol. 1997;158:5501–6. [PubMed] [Google Scholar]
- 22.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 23.Goriely S, Neurath MF, Goldman M. How microorganisms tip the balance between interleukin-12 family members. Nat Rev Immunol. 2008;8:81–6. doi: 10.1038/nri2225. [DOI] [PubMed] [Google Scholar]
- 24.Goriely S, Goldman M. The interleukin-12 family: new players in transplantation immunity? Am J Transplant. 2007;7:278–84. doi: 10.1111/j.1600-6143.2006.01651.x. [DOI] [PubMed] [Google Scholar]
- 25.Chang J, Voorhees TJ, Liu Y, Zhao Y, Chang CH. Interleukin-23 production in dendritic cells is negatively regulated by protein phosphatase 2A. Proc Natl Acad Sci U S A. 2010;107:8340–5. doi: 10.1073/pnas.0914703107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci U S A. 2007;104:12099–104. doi: 10.1073/pnas.0705268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Franks HA, Porte J, El Refaee M, Shah S, Crooks J, Patel PM, Jackson AM. Novel approach for interleukin-23 up-regulation in human dendritic cells and the impact on T helper type 17 generation. Immunology. 2011;134:60–72. doi: 10.1111/j.1365-2567.2011.03467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stojanovic I, Cvjeticanin T, Lazaroski S, Stosic-Grujicic S, Miljkovic D. Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology. 2009;126:74–83. doi: 10.1111/j.1365-2567.2008.02879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg M, Nadiv O, Luknar-Gabor N, Agar G, Beer Y, Katz Y. Synergism between tumor necrosis factor α and interleukin-17 to induce IL-23 p19 expression in fibroblast-like synoviocytes. Mol Immunol. 2009;46:1854–9. doi: 10.1016/j.molimm.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, Krueger JG. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–30. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brentano F, Ospelt C, Stanczyk J, Gay RE, Gay S, Kyburz D. Abundant expression of the interleukin (IL)23 subunit p19, but low levels of bioactive IL23 in the rheumatoid synovium: differential expression and Toll-like receptor-(TLR) dependent regulation of the IL23 subunits, p19 and p40, in rheumatoid arthritis. Ann Rheum Dis. 2009;68:143–50. doi: 10.1136/ard.2007.082081. [DOI] [PubMed] [Google Scholar]
- 34.Shimozato O, Ugai S, Chiyo M, et al. The secreted form of the p40 subunit of interleukin (IL)-12 inhibits IL-23 functions and abrogates IL-23-mediated antitumour effects. Immunology. 2006;117:22–8. doi: 10.1111/j.1365-2567.2005.02257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olleros ML, Vesin D, Martinez-Soria E, et al. Interleukin-12p40 overexpression promotes interleukin-12p70 and interleukin-23 formation but does not affect bacille Calmette–Guérin and Mycobacterium tuberculosis clearance. Immunology. 2007;122:350–61. doi: 10.1111/j.1365-2567.2007.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Vosse E, Lichtenauer-Kaligis EG, van Dissel JT, Ottenhoff TH. Genetic variations in the interleukin-12/interleukin-23 receptor (β1) chain, and implications for IL-12 and IL-23 receptor structure and function. Immunogenetics. 2003;54:817–29. doi: 10.1007/s00251-002-0534-9. [DOI] [PubMed] [Google Scholar]
- 37.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 38.Torti DC, Feldman SR. Interleukin-12, interleukin-23, and psoriasis: current prospects. J Am Acad Dermatol. 2007;57:1059–68. doi: 10.1016/j.jaad.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 39.Yoon C, Johnston SC, Tang J, Stahl M, Tobin JF, Somers WS. Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J. 2000;19:3530–41. doi: 10.1093/emboj/19.14.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beyer BM, Ingram R, Ramanathan L, Reichert P, Le HV, Madison V, Orth P. Crystal structures of the pro-inflammatory cytokine interleukin-23 and its complex with a high-affinity neutralizing antibody. J Mol Biol. 2008;382:942–55. doi: 10.1016/j.jmb.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 42.Pahan K, Jana M. Induction of lymphotoxin-α by interleukin-12 p40 homodimer, the so-called biologically inactive molecule, but not IL-12 p70. Immunology. 2009;127:312–25. doi: 10.1111/j.1365-2567.2008.02985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 2007;56:1333–6. doi: 10.1136/gut.2006.115402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tato CM, Cua DJ. Reconciling id, ego, and superego within interleukin-23. Immunol Rev. 2008;226:103–11. doi: 10.1111/j.1600-065X.2008.00715.x. [DOI] [PubMed] [Google Scholar]
- 45.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–42. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 46.Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney AL, De Sauvage FJ. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 2004;172:2827–33. doi: 10.4049/jimmunol.172.5.2827. [DOI] [PubMed] [Google Scholar]
- 47.Kikly K, Liu L, Na S, Sedgwick JD. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr Opin Immunol. 2006;18:670–5. doi: 10.1016/j.coi.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Mathur AN, Chang HC, Zisoulis DG, et al. STAT3 and STAT4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–7. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 49.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–17. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Happel KI, Zheng M, Young E, et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–6. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Happel KI, Dubin PJ, Zheng M, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–7. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 54.Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366:1367–74. doi: 10.1016/S0140-6736(05)67566-6. [DOI] [PubMed] [Google Scholar]
- 55.Tonel G, Conrad C, Laggner U, et al. Cutting edge: a critical functional role for IL-23 in psoriasis. J Immunol. 2010;185:5688–91. doi: 10.4049/jimmunol.1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reddy M, Davis C, Wong J, Marsters P, Pendley C, Prabhakar U. Modulation of CLA, IL-12R, CD40L, and IL-2Rα expression and inhibition of IL-12- and IL-23-induced cytokine secretion by CNTO 1275. Cell Immunol. 2007;247:1–11. doi: 10.1016/j.cellimm.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 57.Kimball AB, Gordon KB, Langley RG, Menter A, Chartash EK, Valdes J. Safety and efficacy of ABT-874, a fully human interleukin 12/23 monoclonal antibody, in the treatment of moderate to severe chronic plaque psoriasis: results of a randomized, placebo-controlled, phase 2 trial. Arch Dermatol. 2008;144:200–7. doi: 10.1001/archdermatol.2007.63. [DOI] [PubMed] [Google Scholar]
- 58.Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, Dooley LT, Lebwohl M. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007;356:580–92. doi: 10.1056/NEJMoa062382. [DOI] [PubMed] [Google Scholar]
- 59.Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. 2006;176:1908–15. doi: 10.4049/jimmunol.176.3.1908. [DOI] [PubMed] [Google Scholar]
- 60.Kopp T, Kieffer JD, Rot A, Strommer S, Stingl G, Kupper TS. Inflammatory skin disease in K14/p40 transgenic mice: evidence for interleukin-12-like activities of p40. J Invest Dermatol. 2001;117:618–26. doi: 10.1046/j.1523-1747.2001.01441.x. [DOI] [PubMed] [Google Scholar]
- 61.Cheng J, Tu Y, Li J, Huang C, Liu Z, Liu D. A study on the expression of interleukin (IL)-10 and IL-12 P35, P40 mRNA in the psoriatic lesions. J Tongji Med Univ. 2001;21:86–8. doi: 10.1007/BF02888047. [DOI] [PubMed] [Google Scholar]
- 62.Lew W, Lee E, Krueger JG. Psoriasis genomics: analysis of proinflammatory (type 1) gene expression in large plaque (Western) and small plaque (Asian) psoriasis vulgaris. Br J Dermatol. 2004;150:668–76. doi: 10.1111/j.0007-0963.2004.05891.x. [DOI] [PubMed] [Google Scholar]
- 63.Shaker OG, Moustafa W, Essmat S, Abdel-Halim M, El-Komy M. The role of interleukin-12 in the pathogenesis of psoriasis. Clin Biochem. 2006;39:119–25. doi: 10.1016/j.clinbiochem.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 64.Wiekowski MT, Leach MW, Evans EW, et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J Immunol. 2001;166:7563–70. doi: 10.4049/jimmunol.166.12.7563. [DOI] [PubMed] [Google Scholar]
- 65.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 66.Ma HL, Liang S, Li J, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–45. doi: 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- 68.Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–70. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 69.Mannon PJ, Fuss IJ, Mayer L, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–79. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 70.Fuss IJ, Becker C, Yang Z, et al. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15. doi: 10.1097/01.mib.0000194183.92671.b6. [DOI] [PubMed] [Google Scholar]
- 71.Uhlig HH, McKenzie BS, Hue S, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 72.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 74.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–21. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–57. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 76.Kappeler A, Mueller C. The role of activated cytotoxic T cells in inflammatory bowel disease. Histol Histopathol. 2000;15:167–72. doi: 10.14670/HH-15.167. [DOI] [PubMed] [Google Scholar]
- 77.Maerten P, Shen C, Colpaert S, et al. Involvement of interleukin 18 in Crohn's disease: evidence from in vitro analysis of human gut inflammatory cells and from experimental colitis models. Clin Exp Immunol. 2004;135:310–7. doi: 10.1111/j.1365-2249.2004.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheroutre H, Madakamutil L. Acquired and natural memory T cells join forces at the mucosal front line. Nat Rev Immunol. 2004;4:290–300. doi: 10.1038/nri1333. [DOI] [PubMed] [Google Scholar]
- 79.Fukata M, Breglio K, Chen A, et al. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–94. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–31. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 81.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bamba S, Andoh A, Yasui H, Araki Y, Bamba T, Fujiyama Y. Matrix metalloproteinase-3 secretion from human colonic subepithelial myofibroblasts: role of interleukin-17. J Gastroenterol. 2003;38:548–54. doi: 10.1007/s00535-002-1101-8. [DOI] [PubMed] [Google Scholar]
- 85.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, Meuer SC, Stallmach A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 86.Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Invest. 2008;118:2269–80. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu FL, Chen CH, Chu SJ, Chen JH, Lai JH, Sytwu HK, Chang DM. Interleukin (IL)-23 p19 expression induced by IL-1β in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-κB and AP-1 dependent pathway. Rheumatology (Oxford) 2007;46:1266–73. doi: 10.1093/rheumatology/kem055. [DOI] [PubMed] [Google Scholar]
- 89.Olsen NJ, Moore JH, Aune TM. Gene expression signatures for autoimmune disease in peripheral blood mononuclear cells. Arthritis Res Ther. 2004;6:120–8. doi: 10.1186/ar1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–9. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 92.Kim HR, Cho ML, Kim KW, et al. Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI3-kinase-, NF-κB- and p38 MAPK-dependent signalling pathways. Rheumatology (Oxford) 2007;46:57–64. doi: 10.1093/rheumatology/kel159. [DOI] [PubMed] [Google Scholar]
- 93.Li Y, Chu N, Hu A, Gran B, Rostami A, Zhang GX. Inducible IL-23p19 expression in human microglia via p38 MAPK and NF-κB signal pathways. Exp Mol Pathol. 2008;84:1–8. doi: 10.1016/j.yexmp.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HR, Kim HS, Park MK, Cho ML, Lee SH, Kim HY. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand J Rheumatol. 2007;36:259–64. doi: 10.1080/03009740701286813. [DOI] [PubMed] [Google Scholar]
- 96.Nistala K, Wedderburn LR. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (Oxford) 2009;48:602–6. doi: 10.1093/rheumatology/kep028. [DOI] [PubMed] [Google Scholar]
- 97.Chabaud M, Lubberts E, Joosten L, van Den Berg W, Miossec P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001;3:168–77. doi: 10.1186/ar294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD. Interleukin 17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Ann Rheum Dis. 2002;61:704–13. doi: 10.1136/ard.61.8.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167:141–9. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–74. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 101.Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008;371:1675–84. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 102.Lebwohl M, Yeilding N, Szapary P, et al. Impact of weight on the efficacy and safety of ustekinumab in patients with moderate to severe psoriasis: rationale for dosing recommendations. J Am Acad Dermatol. 2010;63:571–9. doi: 10.1016/j.jaad.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 103.Griffiths CE, Strober BE, van de Kerkhof P, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362:118–28. doi: 10.1056/NEJMoa0810652. [DOI] [PubMed] [Google Scholar]
- 104.Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009;373:633–40. doi: 10.1016/S0140-6736(09)60140-9. [DOI] [PubMed] [Google Scholar]
- 105.Sandborn WJ, Feagan BG, Fedorak RN, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–41. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 106.Kasper LH, Everitt D, Leist TP, Ryan KA, Mascelli MA, Johnson K, Raychaudhuri A, Vollmer T. A phase I trial of an interleukin-12/23 monoclonal antibody in relapsing multiple sclerosis. Curr Med Res Opin. 2006;22:1671–8. doi: 10.1185/030079906X120931. [DOI] [PubMed] [Google Scholar]
- 107.Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. [DOI] [PubMed] [Google Scholar]
- 108.Ding C, Xu J, Li J. ABT-874, a fully human monoclonal anti-IL-12/IL-23 antibody for the potential treatment of autoimmune diseases. Curr Opin Investig Drugs. 2008;9:515–22. [PubMed] [Google Scholar]
- 109.Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, Krusemeier M, Kay NE. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin Diagn Lab Immunol. 1994;1:365–72. doi: 10.1128/cdli.1.4.365-372.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen Y, Langrish CL, McKenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–26. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]