

Abstract
Many MHC class I molecules contain unpaired cysteine residues in their cytoplasmic tail domains, the function of which remains relatively uncharacterized. Recently, it has been shown that in the small secretory vesicles known as exosomes, fully folded MHC class I dimers can form through a disulphide bond between the cytoplasmic tail domain cysteines, induced by the low levels of glutathione in these extracellular vesicles. Here we address whether similar MHC class I dimers form in whole cells by alteration of the redox environment. Treatment of the HLA-B27-expressing Epstein–Barr virus-transformed B-cell line Jesthom, and the leukaemic T-cell line CEM transfected with HLA-B27 with the strong oxidant diamide, and the apoptosis-inducing and glutathione-depleting agents hydrogen peroxide and thimerosal, induced MHC class I dimers. Furthermore, induction of apoptosis by cross-linking FasR/CD95 on CEM cells with monoclonal antibody CH-11 also induced MHC class I dimers. As with exosomal MHC class I dimers, the formation of these structures on cells is controlled by the cysteine at position 325 in the cytoplasmic tail domain of HLA-B27. Therefore, the redox environment of cells intimately controls induction of MHC class I dimers, the formation of which may provide novel structures for recognition by the immune system.
Keywords: antigen presentation, apoptosis, MHC/HLA
Introduction
Major histocompatibility complex (MHC) class I molecules function by presenting short peptides, normally of eight or nine amino acids in length, to T cells of the immune system.1 In this manner they provide a sensitive mechanism for the detection and elimination of pathogen-infected cells. Extensive polymorphism in the residues lining the peptide-binding groove of MHC class I molecules ensures that many different pathogenic peptides can be recognized.2 MHC class I molecules are also ligands for the extensive family of killer cell immunoglobulin-like receptors (KIR) expressed on natural killer (NK) cells.3
MHC class I molecules are composed of three main domains, with the α1 and α2 domains forming the peptide-binding groove, supported underneath by the α3 domain and the non-covalently attached β2-microglobulin.4 A transmembrane-spanning domain is then followed by a cytoplasmic tail domain, the full function(s) of which remain somewhat unclear, though roles in recycling,5 targeting for degradation by ubiquitination6 and influencing recognition by NK receptors have been demonstrated.7
All classical MHC class I molecules contain structurally conserved cysteines involved in disulphide bonding to maintain overall stability, located in the α1/α2 domain linking positions 101–164, and in the α3 domain linking positions 203–259. However, MHC class I molecules often also contain a number of unpaired cysteine residues, most notably at position 67 in the peptide-groove, which in the case of HLA-B27 has been shown to be involved in the formation of partially unfolded heavy-chain homodimers,8–10 and at position 42 on the external face of the molecule, which in HLA-G allows the formation of fully folded dimers.11,12 Significantly, there are also unpaired cysteine residues in the transmembrane domain region of HLA-B molecules at position 308, and in the cytoplasmic tail domain of many HLA-B molecules at position 325, and at position 339 in HLA-A molecules. The precise role, if any, of these cysteine residues remains unclear, though modification by palmitylation,7 involvement in dimer formation,13 transient interactions in the MHC class I peptide-loading complex,14 and NK receptor recognition have all been demonstrated.7
We recently identified that the cytoplasmic tail domain cysteines were intimately involved in the formation of fully folded MHC class I dimers in exosomes.15 These 50–150 nm vesicles form in the endocytic pathway in multivesicular bodies, some of which are released into the extracellular environment.16 They are released by a wide range of both normal and tumour cells, and have been implicated in a number of biological processes. We established that the formation of MHC class I dimers in exosomes was a function of the low level of glutathione (GSH) detected in these vesicles when compared with whole cell lysates, and hypothesized that exosomes cannot maintain the reducing environment of the normal cytoplasm, hence allowing disulphide bonds to form between the cytoplasmic tails.15
To address whether there were also circumstances wherein MHC class I dimers could be induced to form by mimicking the low GSH levels seen in exosomes, we set up experimental systems to modify the cellular redox environment, both by using a strong oxidant treatment, and by inducing apoptosis with agents known to cause a depletion of intracellular GSH. Our data indicate that apoptosis-induced alterations to cellular redox do indeed lead to the induction of MHC class I dimers.
Materials and methods
Cell lines
The human lymphoblastoid lines .221 (gifted by Salim Khakoo, Imperial College, London, UK) and CEM (gifted by Antony Antoniou, UCL, London, UK), the human Epstein–Barr virus-transformed B-cell line Jesthom (Health Protection Agency line no. 88052004), and the rat C58 thymoma line (gifted by Geoff Butcher; Babraham Institute, Cambridge, UK) were cultured in RPMI-1640 (Gibco, Paisley, UK) supplemented with 10% fetal bovine serum (Gibco). Transfectants of these lines expressing HLA-B27 with site-directed mutations C308A (position 308 cysteine to alanine), C325A (position 325 cysteine to alanine), S42C (position 42 serine to cysteine), and HLA-A2 devoid of its cytoplasmic tail domain were maintained in 1 mg/ml G418 (Melford, Ipswich, UK), and have been described previously.15
Antibodies
HC10 (anti HLA-B and C) and HCA2 (anti HLA-A) were kind gifts from J. Neefjes (Amsterdam, the Netherlands). Anti V5 tag (Pk) was a kind gift from R. Randall (St Andrews, UK). BB7.2 (anti HLA-A2) was a kind gift from T. Elliott (Southampton, UK). Horseradish peroxidase -coupled anti-mouse IgG was obtained from Sigma (Poole, UK). CH-11 (anti-FasR/CD95) was obtained from Beckman Coulter, High Wycombe, UK.
Exosome isolation
Approximately 30 × 106 cells were incubated overnight in serum-free RPMI-1640. Cells were removed by centrifugation at 1000 g. Supernatants were alkylated with 10 mmN-ethylmaleimide (Sigma), and then spun at 10 000 g for 30 min to remove debris, and 100 000 g for 2 hr to isolate exosomes. Pellets were resuspended directly in non-reducing sample buffer.
Cell treatments
Approximately 1 × 106 cells were treated with 1 mm diamide (Sigma) in RPMI-1640, 10% fetal bovine serum for 20 min at 37°. A similar number of cells were incubated with the indicated concentration of hydrogen peroxide up to 1 mm (Sigma), 5 μm thimerosal (Sigma) and 0·5 μg/ml anti-CD95 antibody for 16 hr in RPMI-1640, 10% fetal bovine serum. Cells were then isolated by centrifugation and lysed in 50 μl lysis buffer (1% nonidet P-40, 150 mm NaCl, 10 mm Tris–HCl pH 7·6, 1 mm PMSF, 10 mmN-ethylmaleimide). Lysates were centrifuged at 20 000 g for 5 min and the supernatant was heated with an equal volume of non-reducing sample buffer. For immunoprecipitation, 10 × 106 diamide-treated cells were lysed in 0·5 ml lysis buffer and immunoprecipitated with 100 μl BB7.2 antibody supernatant and 20 μl Protein G–Sepharose beads (Sigma). Washed beads were resuspended in 40 μl non-reducing sample buffer. For staining of apoptotic cells with propidium iodide (Sigma), cells were washed twice in PBS, fixed in 70% ethanol at 4° for at least 30 min, washed twice in PBS and then resuspended in PBS containing 8 μg/ml propidium iodide. Apoptosis was also measured by staining with Annexin V-FITC. Briefly, 1 × 105 cells were resuspended in 100 μl binding buffer (10 mm HEPES, pH 7·4, 140 mm NaCl, 2·5 mm CaCl2), and 5 μl FITC-Annexin V (Invitrogen, Paisley, UK) for 10 min at room temperature. Cells were then analysed on a FACScan (BD Biosciences, Oxford, UK) using Cellquest software.
Assessment of cellular redox activity
Incubation of 1 × 105 of the indicated cells in 100 μl medium with 10 μl of Dojindo cell counting kit-8 (CCK-8/WST-8) reagent (NBS Biologicals, Cambs, UK) for 3 hr at 37° was followed by reading of the resulting colour shift at 495 nm on a Dynex MRX plate reader. The same number of cells were incubated with 50 μm monochlorobimane (Sigma) for 20 min at 37°, the supernatant was then removed carefully, and cells were lysed in PBS containing 0·1% SDS. Samples were then read by excitation at 340 nm and fluorescence at 520 nm in a Fluostar Optima (BMG Labtech, Aylesbury, UK) using automatic gain adjustment.
Immunoblotting
Samples were analysed on 8% SDS–PAGE gels, transferred to nitrocellulose (BA85, Whatman), and probed with antibodies in PBS with 0·1% Tween-20 (PBST). Detection was performed by chemiluminescence with Femto Western reagents (Perbio, Cramlington, UK) and imaged on a Fuji LAS-3000 analyser. Densitometric analysis was performed using ImageJ (http://rsbweb.nih.gov/ij/).
Results
Diamide induces MHC class I dimers on whole cells
MHC class I molecules can be detected in a dimeric form on exosomes secreted from a number of different cell lines and in human plasma.15 The formation of these dimeric (molecular weights approximately 80 000–85 000) MHC class I structures, in the case of HLA-B27, is strictly dependent on the cysteine located at position 325 in the cytoplasmic tail domain, as demonstrated by immunoblotting of exosomes secreted from the HLA-B27 transfected .221 human B-cell line expressing single amino acid substitutions of position 308 (C308A, cysteine to alanine) and position 325 (C325A, cysteine to alanine) in the HLA-B27 heavy chain, as shown in Fig. 1 (left panel). Removal of the cytoplasmic tail domain from the HLA-A2 molecule, which includes the unpaired cysteine at position 339, also prevents dimers forming in exosomes released from transfected rat C58 cells (Fig. 1, right panel). Hence cytoplasmic tail domain cysteine residues are crucial to the formation of exosomal MHC class I dimers.
Figure 1.
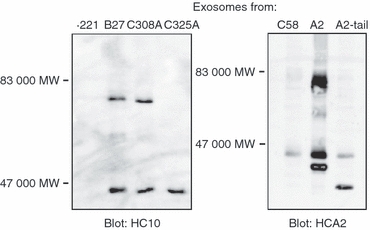
MHC class I dimers are detected on exosomes. Exosomes, purified by ultracentrifugation from supernatants of the indicated .221 and C58 transfected and control cells were immunoblotted for HLA-B (HC10) or HLA-A (HCA2). In .221.B27 cells HLA-B27.C325A fails to form dimers (left panel), as does the cytoplasmic tail domain deleted HLA-A2 molecule (right panel), the monomeric heavy chain of which migrates faster in SDS–PAGE. In .221.B27 cells 44·0% of the heavy chain is in dimeric form, in .221.B27.C308A cells 45·0% is in dimeric form, and in C58.A2 cells 62·1% is in dimeric form.
We identified a low level of glutathione in exosomes compared with whole cell lysates, which we proposed allowed the formation of these exosomal MHC class I dimers by disulphide linkages between unpaired cysteines in the tail domains. We also reported that treatment of cells with the strong oxidant diamide, which rapidly depletes intracellular glutathione, induced similar MHC class I dimers in the HLA-B27-expressing Jesthom B-cell line.15
To determine if the MHC class I dimers induced on whole cells by diamide were also controlled by the same tail domain cysteine, we treated HLA-B27-transfected CEM cells with diamide (Fig. 2a). Immunoblotting revealed the formation of HLA-B27 dimers in wild-type B27, and mutant C308A (cysteine 308 mutated to alanine). No dimers were induced in mutant C325A, demonstrating that cellular, oxidizing-induced MHC class I dimers are controlled by the same cysteines as in exosomes. Similar results were obtained with .221 cells transfected with the same B27 mutants (data not shown). Jesthom cells also displayed diamide-induced dimers, as previously reported (Fig. 2a). We also studied an HLA-B27 mutant (S42C) mutated to mimic the non-classical MHC class I molecule HLA-G, which forms extracellular dimers though cysteine at position 42. The HLA-B27.S42C mutant formed an enhanced level of dimer formation even in the absence of diamide, suggesting that it forms a similar structure to HLA-G. Diamide treatment failed to induce further dimer formation.
Figure 2.
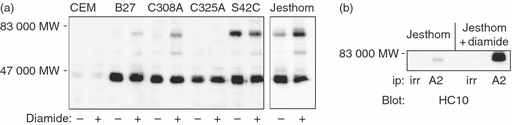
Diamide treatment induces MHC class I dimers on whole cells, and induces mixed HLA-A and HLA-B dimers. (a) The indicated cells were treated with or without diamide, lysed and analysed by immunoblotting for HLA-B (HC10) molecules. The HLA-B27.C325A mutant fails to form dimers. The HLA-B27.S42C mutant has a significantly higher pre-treatment level of dimers compared with wild-type HLA-B27. Jesthom cells are included as a positive control. In the treated B27 cells 17·2% of the heavy chain is in dimeric form, in C308A cells 37·3% is in dimeric form, and in S42C cells 36·9% is in dimeric form in untreated cells whereas 33·5% is in dimeric form in treated cells. In the Jesthom cells 23·7% is in dimeric form in untreated cells, and 45·1% is in dimeric form in treated cells. (b) Jesthom cells were treated with or without diamide, lysed and HLA-A2 molecules were isolated with BB7.2 antibody. After non-reducing SDS–PAGE, the presence of HLA-B molecules in the dimer region was revealed with anti HLA-B (HC10) antibody.
We also previously noted in the high-level expressing MHC class I B-cell line Jesthom, the formation of MHC class I dimers between HLA-B and HLA-A molecules, though at lower efficiency compared with B-B and A-A dimers.15 We confirm here that formation of A-B dimers in the Jesthom line can be further enhanced by diamide treatment. Cells were treated with or without diamide, alkylated and lysed, and immunoprecipitated with an irrelevant antibody (v5 tag), or with BB7.2 (anti-folded HLA-A2). The immunoprecipitates were then probed for the presence of HLA-B molecules with HC10, and as shown in Fig. 2(b), A-B dimers were clearly enhanced in diamide-treated cells.
Hydrogen peroxide and thimerosal-induced cell death induces MHC class I dimers
The use of the strong oxidant diamide clearly demonstrates the ability of dramatic alterations in the redox environment of cells to induce MHC class I dimer formation, but is highly non-physiological. However, we hypothesized that other perturbations of the cellular redox environment might also lead to dimer induction. We envisaged that one such redox alteration may be the induction of cell death by apoptosis.17,18 To test this idea we used both thimerosal19 and hydrogen peroxide20 as pro-apoptotic treatments to induce cell death, and monitored induction of MHC class I dimers by immunoblotting of cell lysates with HC10. Jesthom cells incubated with a range of thimerosal (1–5 μm) and hydrogen peroxide (0·125–1 mm) concentrations showed significant MHC class I dimer formation (Fig. 3a,c). Blotting for HLA-A molecules with HCA2 also showed similar dimer induction (data not shown). Annexin V staining of the Jesthom cells increased from 21·5% to 53·6% after hydrogen peroxide treatment (data not shown). Similarly, hydrogen peroxide (1 mm) and thimerosal (5 μm) treatment of CEM.B27.C308A and C325A cells demonstrated dimer induction in B27 and C308A cells, but not in C325A cells, indicating that the cysteine at position 325 was again responsible for disulphide-linked dimer formation (Fig. 3b,d). Thimerosal induction of MHC class I dimers was also detected in as little as 4 hr post-treatment (data not shown), suggesting that MHC class I dimers can appear rapidly upon the induction of cell death. Hence, thimerosal-induced and hydrogen peroxide-induced apoptotic cell death increase MHC class I dimer formation.
Figure 3.

Thimerosal and hydrogen peroxide-induced apoptosis induces MHC class I dimers. (a) Jesthom cells and (b) the indicated CEM cells were incubated overnight with thimerosal before immunoblotting with HC10. The amounts of the heavy chain in dimeric form in (a) are 14·6, 21·7, 22·2 and 42·0%. In (b) treated CEM.B27 and B27.C308A cells have 39·0% and 53·0% of the heavy chain induced in dimeric form, respectively. (c) Jesthom cells and (d) the indicated CEM cells were incubated with the indicated concentrations of hydrogen peroxide, and 1 mm in (d). Resulting cell lysates were probed for HLA-B molecules (HC10). In (c) the amounts of heavy chain in dimeric form from left to right are 27·0, 66·8, 69·9, 71·7 and 49·7%. In panel (d) the untreated and treated C308A cells have 22·4% and 34·1% of the heavy chain in dimeric form, respectively.
Induction of apoptosis through FasR/CD95 induces MHC class I dimers
Cross-linking of FasR/CD95 using antibody CH-11 induces apoptotic cell death and the depletion of intracellular GSH.21 We determined whether this route of apoptosis also induced MHC class I dimers. CEM.B27, CEM.B27.C308A and CEM.B27.C325A cells were incubated overnight with 0·5 μg/ml anti-Fas/CD95 antibody CH-11, then fixed and stained with propidium iodide before analysis by flow cytometry. Eighty-two per cent of the treated cells showed evidence of propidium iodide incorporation staining of DNA in a sub-G1 region, suggesting DNA-fragmentation associated with apoptosis after anti-FasR/CD95 treatment (Fig. 4b).21 Immunoblotting revealed that MHC class I dimer induction occurred in CEM.B27 and CEM.B27.C308A cells, but not CEM.B27.C325A cells. Hence, the cytoplasmic tail domain cysteine at position 325 also controls MHC class I dimer formation after FasR/CD95-induced cell death.
Figure 4.

FasR/CD95 induced apoptosis causes MHC class I dimer formation. (a) The indicated CEM cells were incubated overnight with 0·5 μg/ml anti-FasR/CD95 antibody. Cell lysates were probed for HLA-B molecules (HC10). The treated B27 and C308A cells display 27·0% and 30·2% of the heavy chain in dimeric form, respectively. (b) CEM.B27 cells treated with (shaded grey) or without (unshaded) anti-FasR/CD95 were fixed and labelled with propidium iodide (PI) and analysed by flow cytometry. The peak indicating cells in G1 and G2/M phases are indicated. 82% of the treated cells fall into a sub-G1 region suggesting fragmented DNA indicative of apoptosis. In (c) and (d) CEM and CEM.B27 cells were treated overnight with 1 mm hydrogen peroxide, 5 μm thimerosal, or 0·5 μg/ml anti-FasR/CD95 antibody, before incubation with reagent WST-8 (c) or incubation with 50 μm monochlorobimane. The whole culture was analysed at 495 nm in (a) and cell lysates at excitation/emission of 340/520 nm in (b). Samples were performed in triplicate and the data are displayed as the mean plus standard deviation.
Finally, to associate the appearance of the MHC class I dimers described herein with alterations in the redox potential of cells undergoing hydrogen peroxide, thimerosal and anti-CD95 treatments, we directly measured redox activities using two methods. First we used the water-soluble tetrazolium salt (WST-8) to determine general dehydrogenase activity in the cells, and second we used monochlorobimane, which gives a direct fluorescent readout of intracellular GSH content.22 With both assay systems treatment of cells with hydrogen peroxide and thimerosal resulted in a profound reduction in signal (Fig. 4c,d). Treatment with anti-CD95 resulted in less significant loss of signal, which is a broad agreement with the immunoblotting results of Figs 2–4, where anti-CD95 induces fewer MHC class I dimers.
Discussion
In our previous work, we established that fully folded (i.e. recognized by conformation-specific monoclonal antibodies) MHC class I dimers exist on secretory exosome vesicles, and that these form by disulphide linkage between available cysteine residues in the cytoplasmic tail of many HLA-A and HLA-B molecules.15 In this study we extend these observations and show that similar MHC class I dimers can be detected on cells in which the redox environment has been significantly altered, either by chemical oxidation with diamide, or chemically induced apoptosis with hydrogen peroxide and thimerosal, or by cross-linking of FasR/CD95. Control of dimer formation was likewise localized to the cytoplasmic tail domain cysteine located at residue 325, found in many HLA-B alleles. This is somewhat in contrast to previous observations wherein HLA-B27 dimer structures were observed even after removal of the cysteine at position 325,10,23 but this may potentially be accounted for by the use of different cell lines and overall expression levels of the HLA-B27 heavy chain in different systems. For example, it is notable that in our CEM transfectants there was very little HLA-B27 dimer present in cell lysates in the absence of oxidative stress, as shown in Fig. 2, whereas the Jesthom cell line, which expresses higher levels of cell surface HLA-B27 than the CEM lines, displays dimers under normal conditions. Similarly, we have previously noted that HLA-B27 dimers tend to form in dendritic cells only after activation and significant up-regulation of MHC class I expression.24 Therefore, MHC class I expression levels and the redox status of cells may both contribute to dimer formation.
In this current study we also generated a mutant form of HLA-B27 called S42C that mimics the dimer formed by the non-classical HLA-G molecule. None of the treatments applied in this current report significantly increased the dimer population over that already formed in the absence of treatment (Fig. 2a and data not shown), and indeed even the strong oxidant diamide failed to induce the formation of a 100% dimer population in all our studies. Hence, there may be a population of MHC class I monomers that cannot undergo dimerization, possibly because of steric hindrance or modifications to the crucial cytoplasmic tail domain cysteines.7,25
The recognition of cells by the immune system when undergoing redox stress is not well defined. Our data suggest that cells undergoing a redox stress that leads to a lowering in the levels of intracellular glutathione may begin to display MHC class I dimers on their cell surface. Such alterations in cellular glutathione levels have been reported to occur during T-cell activation26,27 and also during apoptosis.17,18,28 A more extensive study monitoring both MHC class I dimer formation and the level of glutathione in cells undergoing a variety of stimuli would, we consider, be of some worth. Furthermore, distinguishing whether both apoptotic and necrotic cell death pathways induce dimers would also be informative, alongside other pathological states such as viral infection, and many others that induce inflammatory responses and the production of reactive oxygen species. This would allow a pattern of conditions to be catalogued under which MHC class I dimer formation is induced.
Although various MHC class I dimers have been reported in the literature, their potential biological role remains enigmatic.8,13 Of significant note, however, are the observations that Ig-MHC class I dimers can act to tolerize T cells.29 In this study, Kourilsky and colleagues used a soluble H2-Kd molecule dimerized with a cross-linking antibody to demonstrate that an antigen-specific T-cell hybridoma was initially activated, followed by a state of unresponsiveness. It has also been demonstrated that antigen-specific occupancy of just one of the two peptide-binding grooves in an MHC class I dimer can have an effect on T cells,30 which may allow not only the HLA-B dimers we show here, but also the HLA-A-B dimers we describe in Fig. 2 to have some biological activity. Hence, the MHC class I dimers detected on apoptotic cells, and also on exosomes,15 may be capable of providing signal, including tolerogenic signals to immune cells. Similarly, tumour cells undergoing apoptosis, or releasing exosomes containing tumour-associated or tumour-specific antigens may influence T-cell behaviour. Of further interest is the possible recognition of MHC class I dimers by members of the NK cell receptor family. It has been shown that a disulphide-linked engineered version of the KIR2DL1 receptor has an increased affinity for HLA-C,31 and that KIR molecules can form an array of dimers and multimers in a zinc-dependent interaction.32 Hence interactions between dimers of both ligands and receptors may occur, potentially inducing extra stability for the generation of either inhibitory or activatory signals. As indicated above, defining the various conditions under which such dimers form would allow the design of experiments to directly study their potential influence on immune responses.
It is now possible to define three distinct types of dimeric MHC class I molecules that can exist on cells and exosomes, which are depicted in Fig. 5. Partially folded HLA-B27 molecules, linked by the relatively unique cysteine 67 residue in the peptide-binding groove have been detected both in vitro and in vivo,8,9,33,34 and may be a contributory factor to the development of the arthritic condition ankylosing spondylitis, either by altered NK receptor recognition at the cell surface,35 or by induction of intracellular unfolded protein cellular stress responses.36 HLA-G molecules form unique dimers by disulphide linkage at position 42 on an external loop of the peptide-binding groove.12 These dimers may be relevant in tolerizing signals in pregnancy and in regulatory T-cell subsets.11,37 Lastly, a population of folded MHC class I dimers can exist on exosomes and redox-altered normal cells, and apoptotic cells, induced by disulphide linkages between cysteines in the cytoplasmic tails.15
Figure 5.

Cartoon depiction of currently known MHC class I dimer structures. (a) The cysteine-67-linked HLA-B27 heavy chain dimer.38 (b) The cysteine-42-linked HLA-G dimer.39 (c) The cytoplasmic tail domain MHC class I dimer on redox-altered and apoptotic cells described herein, and exosomes.15
Acknowledgments
The work in this study was funded in part by the Chief Scientist's Office (CSO) of the Scottish Government.
Disclosures
No competing financial interests exist.
References
- 1.Antoniou AN, Powis SJ. Pathogen evasion strategies for the major histocompatibility complex class I assembly pathway. Immunology. 2008;124:1–12. doi: 10.1111/j.1365-2567.2008.02804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purcell AW, Elliott T. Molecular machinations of the MHC-I peptide loading complex. Curr Opin Immunol. 2008;20:75–81. doi: 10.1016/j.coi.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology. 2011;132:315–25. doi: 10.1111/j.1365-2567.2010.03398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–12. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 5.Santos SG, Antoniou AN, Sampaio P, Powis SJ, Arosa FA. Lack of tyrosine 320 impairs spontaneous endocytosis and enhances release of HLA-B27 molecules. J Immunol. 2006;176:2942–9. doi: 10.4049/jimmunol.176.5.2942. [DOI] [PubMed] [Google Scholar]
- 6.Hewitt EW, Duncan L, Mufti D, Baker J, Stevenson PG, Lehner PJ. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 2002;21:2418–29. doi: 10.1093/emboj/21.10.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gruda R, Achdout H, Stern-Ginossar N, et al. Intracellular cysteine residues in the tail of MHC class I proteins are crucial for extracellular recognition by leukocyte Ig-like receptor 1. J Immunol. 2007;179:3655–61. doi: 10.4049/jimmunol.179.6.3655. [DOI] [PubMed] [Google Scholar]
- 8.Allen RL, O'Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. J Immunol. 1999;162:5045–8. [PubMed] [Google Scholar]
- 9.Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem. 2004;279:8895–902. doi: 10.1074/jbc.M311757200. [DOI] [PubMed] [Google Scholar]
- 10.Bird LA, Peh CA, Kollnberger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol. 2003;33:748–59. doi: 10.1002/eji.200323678. [DOI] [PubMed] [Google Scholar]
- 11.Apps R, Gardner L, Sharkey AM, Holmes N, Moffett A. A homodimeric complex of HLA-G on normal trophoblast cells modulates antigen-presenting cells via LILRB1. Eur J Immunol. 2007;37:1924–37. doi: 10.1002/eji.200737089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyson JE, Erskine R, Whitman MC, et al. Disulfide bond-mediated dimerization of HLA-G on the cell surface. Proc Natl Acad Sci U S A. 2002;99:16180–5. doi: 10.1073/pnas.212643199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capps GG, Robinson BE, Lewis KD, Zuniga MC. In vivo dimeric association of class I MHC heavy chains. Possible relationship to class I MHC heavy chain-beta 2-microglobulin dissociation. J Immunol. 1993;151:159–69. [PubMed] [Google Scholar]
- 14.Chambers JE, Jessop CE, Bulleid NJ. Formation of a major histocompatibility complex class I tapasin disulfide indicates a change in spatial organization of the peptide-loading complex during assembly. J Biol Chem. 2008;283:1862–9. doi: 10.1074/jbc.M708196200. [DOI] [PubMed] [Google Scholar]
- 15.Lynch S, Santos SG, Campbell EC, Nimmo AM, Botting C, Prescott A, Antoniou AN, Powis SJ. Novel MHC class I structures on exosomes. J Immunol. 2009;183:1884–91. doi: 10.4049/jimmunol.0900798. [DOI] [PubMed] [Google Scholar]
- 16.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 17.Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–14. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 18.Macho A, Hirsch T, Marzo I, Marchetti P, Dallaporta B, Susin SA, Zamzami N, Kroemer G. Glutathione depletion is an early and calcium elevation is a late event of thymocyte apoptosis. J Immunol. 1997;158:4612–9. [PubMed] [Google Scholar]
- 19.Makani S, Gollapudi S, Yel L, Chiplunkar S, Gupta S. Biochemical and molecular basis of thimerosal-induced apoptosis in T cells: a major role of mitochondrial pathway. Genes Immunol. 2002;3:270–8. doi: 10.1038/sj.gene.6363854. [DOI] [PubMed] [Google Scholar]
- 20.Gupta S, Young T, Yel L, Su H, Gollapudi S. Differential sensitivity of naive and subsets of memory CD4+ and CD8+ T cells to hydrogen peroxide-induced apoptosis. Genes Immunol. 2007;8:560–9. doi: 10.1038/sj.gene.6364416. [DOI] [PubMed] [Google Scholar]
- 21.Efferth T, Fabry U, Osieka R. Anti-Fas/Apo-1 monoclonal antibody CH-11 depletes glutathione and kills multidrug-resistant human leukemic cells. Blood Cells Mol Dis. 1996;22:2–9. doi: 10.1006/bcmd.1996.0002. discussion 10. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee S, Noack H, Possel H, Keilhoff G, Wolf G. Glutathione levels in primary glial cultures: monochlorobimane provides evidence of cell type-specific distribution. Glia. 1999;27:152–61. [PubMed] [Google Scholar]
- 23.Saleki K, Hartigan N, Lith M, Bulleid N, Benham AM. Differential oxidation of HLA-B2704 and HLA-B2705 in lymphoblastoid and transfected adherent cells. Antioxid Redox Signal. 2006;8:292–9. doi: 10.1089/ars.2006.8.292. [DOI] [PubMed] [Google Scholar]
- 24.Santos SG, Lynch S, Campbell EC, Antoniou AN, Powis SJ. Induction of HLA-B27 heavy chain homodimer formation after activation in dendritic cells. Arthritis Res Ther. 2008;10:R100. doi: 10.1186/ar2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufman JF, Krangel MS, Strominger JL. Cysteines in the transmembrane region of major histocompatibility complex antigens are fatty acylated via thioester bonds. J Biol Chem. 1984;259:7230–8. [PubMed] [Google Scholar]
- 26.Hehner SP, Breitkreutz R, Shubinsky G, Unsoeld H, Schulze-Osthoff K, Schmitz ML, Droge W. Enhancement of T cell receptor signaling by a mild oxidative shift in the intracellular thiol pool. J Immunol. 2000;165:4319–28. doi: 10.4049/jimmunol.165.8.4319. [DOI] [PubMed] [Google Scholar]
- 27.Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci U S A. 1998;95:3071–6. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slater AF, Nobel CS, Maellaro E, Bustamante J, Kimland M, Orrenius S. Nitrone spin traps and a nitroxide antioxidant inhibit a common pathway of thymocyte apoptosis. Biochem J. 1995;306(Pt 3):771–8. doi: 10.1042/bj3060771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abastado JP, Lone YC, Casrouge A, Boulot G, Kourilsky P. Dimerization of soluble major histocompatibility complex–peptide complexes is sufficient for activation of T cell hybridoma and induction of unresponsiveness. J Exp Med. 1995;182:439–47. doi: 10.1084/jem.182.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cebecauer M, Guillaume P, Mark S, et al. CD8+ cytotoxic T lymphocyte activation by soluble major histocompatibility complex–peptide dimers. J Biol Chem. 2005;280:23820–8. doi: 10.1074/jbc.M500654200. [DOI] [PubMed] [Google Scholar]
- 31.Fan QR, Long EO, Wiley DC. A disulfide-linked natural killer cell receptor dimer has higher affinity for HLA-C than wild-type monomer. Eur J Immunol. 2000;30:2692–7. doi: 10.1002/1521-4141(200009)30:9<2692::AID-IMMU2692>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 32.Vales-Gomez M, Erskine RA, Deacon MP, Strominger JL, Reyburn HT. The role of zinc in the binding of killer cell Ig-like receptors to class I MHC proteins. Proc Natl Acad Sci U S A. 2001;98:1734–9. doi: 10.1073/pnas.041618298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kollnberger S, Bird LA, Roddis M, et al. HLA-B27 heavy chain homodimers are expressed in HLA-B27 transgenic rodent models of spondyloarthritis and are ligands for paired Ig-like receptors. J Immunol. 2004;173:1699–710. doi: 10.4049/jimmunol.173.3.1699. [DOI] [PubMed] [Google Scholar]
- 34.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007;56:215–23. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 35.Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, Bowness P. Cell-surface expression and immune receptor recognition of HLA-B27 homodimers. Arthritis Rheum. 2002;46:2972–82. doi: 10.1002/art.10605. [DOI] [PubMed] [Google Scholar]
- 36.Turner MJ, Sowders DP, DeLay ML, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–48. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 37.Apps R, Gardner L, Moffett A. A critical look at HLA-G. Trends Immunol. 2008;29:313–21. doi: 10.1016/j.it.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Bowness P. HLA B27 in health and disease: a double-edged sword? Rheumatology (Oxford) 2002;41:857–68. doi: 10.1093/rheumatology/41.8.857. [DOI] [PubMed] [Google Scholar]
- 39.Shiroishi M, Kuroki K, Ose T, et al. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J Biol Chem. 2006;281:10439–47. doi: 10.1074/jbc.M512305200. [DOI] [PubMed] [Google Scholar]