

Abstract
Mutations in the gene encoding DJ-1 have been identified in patients with familial Parkinson's disease (PD) and are thought to inactivate a neuroprotective function. Oxidation of the sulfhydryl group to a sulfinic acid on cysteine residue C106 of DJ-1 yields the “2O ” form, a variant of the protein with enhanced neuroprotective function. We hypothesized that some familial mutations disrupt DJ-1 activity by interfering with conversion of the protein to the 2O form. To address this hypothesis, we developed a novel quantitative mass spectrometry approach to measure relative changes in oxidation at specific sites in mutant DJ-1 as compared with the wild-type protein. Treatment of recombinant wild-type DJ-1 with a 10-fold molar excess of H2O2 resulted in a robust oxidation of C106 to the sulfinic acid, whereas this modification was not detected in a sample of the familial PD mutant M26I exposed to identical conditions. Methionine oxidized isoforms of wild-type DJ-1 were depleted, presumably as a result of misfolding and aggregation, under conditions that normally promote conversion of the protein to the 2O form. These data suggest that the M26I familial substitution and methionine oxidation characteristic of sporadic PD may disrupt DJ-1 function by disfavoring a site-specific modification required for optimal neuroprotective activity. Our findings indicate that a single amino acid substitution can markedly alter a protein's ability to undergo oxidative modification, and they imply that stimulating the conversion of DJ-1 to the 2O form may be therapeutically beneficial in familial or sporadic PD.
Parkinson's disease (PD)1 is a neurodegenerative disorder characterized by muscular rigidity, slowness of voluntary movement, poor balance, and resting tremor (1). These symptoms are caused by the death of neurons in a region of the midbrain called the substantia nigra. The neurons that survive in this region exhibit a defect in complex I of the mitochondrial electron transport chain and show signs of oxidative damage (2–4). In addition, surviving neurons contain characteristic cytosolic inclusions named “Lewy bodies” that are enriched with aggregated forms of the presynaptic protein α-synuclein (aSyn) (5). It is hypothesized that reactive oxygen species (ROS) accumulate as a result of mitochondrial impairment and contribute to neurodegeneration by causing the oxidation of lipid, proteins, and DNA (2–4). A buildup of ROS may promote the formation of harmful aSyn aggregates in the brains of PD patients (6–8). ROS accumulation is thought to occur preferentially in nigral dopaminergic neurons because of the catabolism and auto-oxidation of dopamine, reactions that result in the generation of H2O2 (2, 9). In addition, an abundance of iron in the substantia nigra promotes the decomposition of H2O2 to OH· by Fenton chemistry (10).
A number of patients with familial, early-onset, recessive PD have been found to harbor loss-of-function mutations (e.g. M26I, E64D, A104T, D149A, E163K, and L166P) in the gene encoding DJ-1, a protein that is abundant throughout the central nervous system (11, 12). Dysfunction of wild-type DJ-1 as a result of destabilizing oxidative modifications is also thought to play a role in more common sporadic forms of PD (13–15). DJ-1 has been reported to suppress neurodegeneration in cellular and animal models by activating antioxidant responses (16–21), up-regulating or carrying out a molecular chaperone function (20–24), and/or inducing prosurvival signaling responses (12, 19, 25). DJ-1 is a homodimer (subunit molecular weight = 20 kDa) for which there is evidence that cysteine 106 (C106) in the subunit sequence is readily oxidized to the sulfinic acid under oxidizing conditions, yielding the “2O” form of the protein (14, 16, 17, 26). Structural studies indicate that C106 is located in a pocket at the interface between the DJ-1 subunits that is lined with polar residues from both subunits, and oxidation of C106 to the sulfinic acid is facilitated by structure(s) within the pocket (16, 27–29). The design of this pocket suggests that controlled oxidation of DJ-1 at position 106 is advantageous for optimal DJ-1 function. Consistent with this idea, the oxidation of C106 to the 2O form is apparently critical for the ability of DJ-1 to translocate to mitochondria (16), inhibit mitochondrial fragmentation (30–32), or suppress fibril formation by recombinant aSyn (24). In contrast, overoxidation of DJ-1 leading to the conversion of C106 to the sulfonic acid (“3O”) form results in thermodynamic instability (15) and a loss of chaperone function (24).
Data from various groups indicate that L166P has a pronounced protein folding defect, resulting in impaired homodimer formation, rapid protein turnover, and a high propensity to form large protein complexes (33–35). Clearly, pronounced structural defects lead to the compromised functionality of L166P. Substitutions at other locations destabilize the structure of DJ-1 to a lesser extent (15, 36, 37), raising questions about why they are functionally diminished. As one possibility, we hypothesized that some familial DJ-1 mutants may have altered abilities to undergo key oxidative modifications, namely, (1) a decreased propensity to undergo oxidation at position 106 to yield the 2O form, and/or (2) an increased susceptibility to undergo oxidative modifications with potentially deleterious effects on DJ-1 function at other sites in the polypeptide chain. To address this hypothesis, we examined the impact of one familial substitution (the M26I mutation) on the ability of DJ-1 to undergo oxidation at C106 and other residues in the amino acid sequence. Using a novel mass spectrometry approach, we quantified site-specific oxidative modifications of M26I as compared with wild-type DJ-1 after treatment with different amounts of H2O2. Our results indicate that the M26I substitution has a profound disruptive effect on the ability of DJ-1 to undergo oxidation at C106.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise specified, chemicals were obtained from Sigma Chemical Co. (St. Louis, MO). Isopropylthiogalactoside (IPTG) was purchased from Gold Biotechnologies (St. Louis, MO). PreScission™ Protease was obtained from GE Healthcare (Piscataway, NJ). The bicinchoninic acid (BCA) protein assay kit was purchased from Pierce Biotechnology (Rockford, IL). Immobilized trypsin and tris (2-carboxyethyl) phosphine (TCEP) were purchased as Pierce products from Thermofisher Scientific (Rockford, IL). Amicon Ultra-0.5 ml (Ultracel YM-10) and Amicon Ultra-4 (Ultracel-3) centrifugal filters and H2O2 were purchased from Millipore (Billerica, MA). Trifluoroacetic acid and high-performance liquid chromatography (HPLC) grade CH3CN were purchased from Mallinckrodt Chemicals (Phillipsburg, NJ). Materials for two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) (immobilized pH gradient (IPG) strips, sample rehydration buffer, Criterion XT 12% Bis-Tris precast gels, XT MOPS 2D PAGE running buffer, protein standards) were obtained from Bio-Rad (Hercules, CA). Urea was obtained from Mallinckrodt Laboratories (Phillipsburg, NJ), and agarose was purchased from Invitrogen (Carlsbad, CA).
Preparation of Bacterial Expression Constructs
Human wild-type DJ-1 and the familial mutant M26I were expressed as N-terminal glutathione-S-transferase (GST) fusions. A construct encoding GST-DJ-1M26I in the pGEX-6P1 vector (courtesy of Dr. Soumya Ray, Brigham and Women's Hospital (38)) was converted to a new construct encoding GST-DJ-1WT by PCR using the QuikChange method (Stratagene, La Jolla, CA). The sequence of the DJ-1-encoding insert in each construct was verified using an Applied Biosystems (Foster City, CA; ABI 3700) DNA sequencer.
Purification of Recombinant DJ-1
Wild-type and mutant DJ-1 were purified as described (38). Cells of the BL21(DE3) strain of Escherichia coli were transformed with each pGEX-6P-1 GST-DJ-1 construct by electroporation. The transformed cells were grown to an OD600 of 0.4–0.6 in LB plus ampicillin (100 μg/ml) at 37 °C, and IPTG was added to a final concentration of 1 mm. The cells were grown under inducing conditions for 18 h at 18 °C, harvested by centrifugation, and resuspended in buffer G (25 mm KPi, pH 7.0, 200 mm KCl). The cells were lysed by incubation on ice in the presence of lysozyme (1 mg/ml, 30 min) followed by passage through a French pressure cell (p.s.i. > 1000). After centrifugation (20,000 × g, 20 min), the supernatant was applied to a GSTPrep FF column (GE Healthcare), from which GST-DJ-1 was eluted in 250 mm Tris HCl, pH 8.0, 500 mm NaCl, and 0.3% [w/v] reduced glutathione. Fractions most highly enriched with GST-DJ-1 were identified by SDS-PAGE with Coomassie Blue staining and pooled. The pooled fractions were dialyzed against buffer G plus dithiothreitol (DTT) (0.25 mm) to remove excess reduced glutathione. The overall protein concentration was determined with a BCA protein assay kit, and the fusion protein was cleaved with PreScission™ Protease (16 h, 4 °C, 1 unit protease per 133 μg DJ-1). Untagged DJ-1 was separated from uncleaved GST-DJ-1, free GST, and residual protease (which contains an uncleavable GST tag) by elution from a GSTPrep FF column equilibrated with buffer G. Fractions most highly enriched with DJ-1 were identified by SDS-PAGE with Coomassie Blue staining and pooled. The final protein sample (estimated purity, 95%) was supplemented with glycerol (5%, [v/v]) and DTT (2–3 mm), and aliquots were frozen at −80 °C. For all of the analyses outlined below, the concentration of recombinant DJ-1 was estimated using the BCA assay and verified with quantitative amino acid analysis (Purdue University Proteomics Core).
Controlled Oxidation of DJ-1
Aliquots of purified DJ-1 (wild-type or M26I) were dialyzed against 10 mm Tris at pH 8.0 to remove reductant, and the protein was treated with a 10-fold or 500-fold molar excess of H2O2 for 1 h at 22 °C. These conditions were previously found to be suitable to convert DJ-1 to the 2O or 3O form, respectively (24). Excess H2O2 was removed from each sample by exchanging the protein into fresh buffer (10 mm Tris at pH 8.0) using Amicon Ultra-4 centrifugal filters (molecular weight cutoff, 3 kDa).
Two-dimensional Polyacrylamide Gel Electrophoresis
Changes in the isoelectric point (pI) of DJ-1 following oxidation in the presence of H2O2 as outlined above were monitored via 2D-PAGE (16). Protein aliquots were dialyzed overnight against PBS. An aliquot of the protein (15 μg) was mixed with sample rehydration buffer (8 m urea, 2% [w/v] 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), 50 mm DTT, 0.2% [w/v] BioLyte 3/10 ampholyte, 0.001% [w/v] bromphenol blue) and recombinant aSyn as an internal standard (predicted pI = 4.67) and Bio-Rad protein standards in a total volume of 185 μl. The solution was added to an 11 cm IPG strip with a pH range of 4 to 7. Mineral oil was added to the top of the IPG strip to reduce evaporation during electrophoresis. The IPG strip was actively rehydrated at 20 °C for 12 h and subjected to isoelectric focusing in a Protean isoelectric focusing (IEF) Cell (Bio-Rad) with the following voltage parameters: step one, 250 V for 15 min; step two, 8000 V for 2.5 h; step 3, 8000 V for 4.4 h. The IPG strip was reduced with 2% [w/v] DTT in equilibration buffer (6 m urea, 0.375 m Tris HCl, pH 8.8, 2% [w/v] SDS, 20% [v/v] glycerol) for 15 min, followed by alkylation with 2.5% [w/v] iodoacetamide for 15 min. The IPG strip was loaded onto the top of a Criterion XT 12% Bis-Tris pre-cast gel (Bio-Rad) and sealed with 0.5% [w/v] agarose in 1 × XT MOPS buffer to ensure an even transfer of protein. The second-dimension gel was stained with Coomassie Blue and analyzed with a Typhoon Imaging System. Approximate pI values were determined by calibration with the aSyn internal standard and the ends of the IPG strip.
Intensities of spots on 2D-PAGE gels were quantitated using Image J software (National Institutes of Health). Each spot was selected using a rectangular selection tool. The dimensions of the selected rectangular area were kept constant through the analysis of all spots on multiple 2D-PAGE gels. Each selected spot was displayed as a peak, to which a baseline was manually fitted, and the area under the peak was calculated. After correcting for background (determined from a rectangular zone in an “empty” region of the 2D-PAGE gel), peak areas were used to calculate the relative amount of 2O isoform in each sample.
Proteolytic 18O Labeling with Immobilized Trypsin
Proteolysis and peptide labeling were carried out using a modified version of a previously described method (39). Each protein (∼10 μg) was dried, denatured in guanidine HCl (6 m, 15 min, 22 °C), reduced with TCEP hydrochloride (1 mm, 60 min, 60 °C), alkylated with iodoacetamide (10 mm, 10 min, 22 °C), and mixed overnight with 1 μg of immobilized trypsin in 0.1 m ammonium bicarbonate (pH 8.0). Prior to this step the immobilized trypsin was prewashed four times with acetate buffer (pH 5), 20 volumes per wash, to eliminate any noncovalently bound protease on the surface of the agarose beads. After trypsin digestion, the samples were completely dried by vacuum centrifugation and resuspended in H216O or H218O in CH3CN (25%, v/v). The samples were then incubated in the presence of a fresh aliquot of immobilized trypsin (1 μg) in 25% (v/v) CH3CN (pH 6.0) for at least 7 h at 22 °C. Immobilized trypsin was used in both the digestion and labeling steps to prevent exchange of the 16O and 18O label that would occur if the labeled and unlabeled peptides were mixed in the presence of residual free protease. As previously described (40), the decoupling of the digestion and labeling procedures enabled us to carry out each step at its optimal pH (i.e. pH 8.0 and 6.0, respectively). The labeling reaction occurred at an acidic pH of 6.0 and in an aqueous solution containing 25% (v/v) CH3CN because previous studies showed that the addition of organic solvents accelerates trypsin-catalyzed oxygen exchange (39, 40). After adding phenylmethylsulfonylfluoride to inhibit the trypsin, the mixture was filtered using Amicon Ultra-0.5 ml centrifugal filters (molecular weight cutoff, 10 kDa), and the labeled and unlabeled peptides were recovered from the filtrate.
Mass Spectrometry
Aliquots of the labeled and unlabeled peptide pools were mixed and analyzed by online capillary chromatography mass spectrometry. Tryptic peptides were separated on an Agilent Zorbax C18 column (0.2 × 150 mm) using a nano-UPLC instrument (Waters instruments Inc., Milford, MA) at 0.2 μl/min. Solvent A was 0.01% trifluoroacetic acid in deionized H2O (di-H2O) and solvent B was 95% CH3CN/0.01% trifluoroacetic acid in di-H2O. The nano-UPLC instrument is coupled to a Q-STAR work station (Applied Biosystems, Framingham, MA) equipped with a nano-electrospray ionization source to ensure high sensitivity. An 85 min linear gradient (from 0% B to 60% B) was used in the separation of the peptides. Tandem MS (MS/MS) spectra were obtained in the positive ion mode via collision-induced dissociation using 2000 V of ionization voltage at a sampling rate of one spectrum per second. The top three peptides with charges ranging from 2 to 4 were monitored by MS/MS. The centroid value of MS/MS peaks was determined using the following parameters. The merge distance was set at 100 ppm with minimum and maximum widths of 10 ppm and 500 ppm, respectively. The percentage height was set at 50% and MASCOT was used for all searches.
Mascot Database Searching and Peptide Quantitation
The MS and MS/MS data files were sent to an in-house mascot distiller software program (Version 2.3.0) connected to a MASCOT server (Version 2.2, Matrix Science (41)). The program's quantitation tool box is designed to automatically recognize light and heavy peaks, calculate the clustered peak areas, and connect these to the MS/MS profile corresponding to the fragmentation of the peptides. The DJ-1 protein sequence in the NCBI database (release date March 15, 2006) was searched with the following parameters: (1) the selected protease was trypsin, (2) the variable modifications accounted for by the search were deamidation (N and Q), methionine to isoleucine, carbamidomethyl cysteine, dioxidation of cysteine, oxidation of methionine, trioxidation of cysteine, and histidine to asparagine conversion, and (3) the number of missed cleavages permitted was four. The precursor mass tolerance was set to 100 ppm, and the fragment mass tolerance was set to 0.6 Da. In each case the 18O labeling quantitation method was specified. The software calculated the areas of the 16O and 18O monoisotopic peaks corresponding to the unlabeled and labeled peptides, respectively. These peak area values were then used to determine peak ratios. The software algorithm corrected for the incomplete incorporation of 18O label to calculate the relative abundance ratio. Strict criteria were used to reduce the risk of false positives. The individual MS/MS spectra had a maximum expectation value of 0.05, which corresponded to a score of at least four. The MS/MS spectra of modifications detected were manually examined in order to be considered a correct match.
Circular Dichroism
Secondary structure analyses of recombinant DJ-1 were carried out via far-ultraviolet circular dichroism (far-UV CD) as described (15). Protein solutions were dialyzed against 10 mm KPi, pH 7.0, 150 mm NaCl. Aliquots of the protein (0.4 ml, 0.06 mg/ml) were introduced into a 0.2 cm quartz cuvette and analyzed using a J810 spectropolarimeter (JASCO, Easton, MD). Full spectra were collected at 20 °C with a bandwidth of 2 nm, a response time of 1 s, and a data pitch of 1 nm. Molar ellipticity was calculated using the equation:
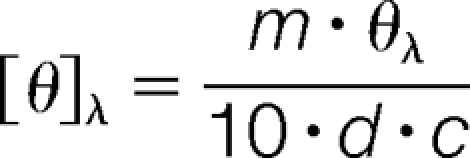 |
where [θ]λ is the molar ellipticity, m is the mean residual weight (g/mol), θλ is the ellipticity, d is the pathlength (cm), and c is the protein concentration (g/L).
Sedimentation-Equilibrium
Sedimentation-equilibrium runs were conducted at 22 °C in a Beckman XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) with absorbance optics, as described (15), using the methods of Laue and Stafford (42). Prior to ultracentrifugation, protein samples were dialyzed against 50 mm Tris HCl, pH 7.0, 200 mm NaCl (4 °C, 48 h, with a change in buffer after 24 h). Aliquots (110 μl) of the protein solution (0.8–2.8 mg/ml) were loaded into six-sector charcoal filled epon (CFE) sample cells, allowing three concentrations to be run simultaneously. Runs were performed at a minimum of three different speeds (2.0 × 104–2.4 × 104 rpm), and each speed was maintained until there was no significant difference in scans of r2/2 versus absorbance taken 2 h apart to ensure that equilibrium was achieved. Sedimentation-equilibrium data were evaluated using the program NONLIN, which employs a nonlinear least squares curve-fitting algorithm described by Johnson et al. (43). The program allows for analysis of both single and multiple data files and can be fit to models containing up to four associating species, depending upon which parameters are permitted to vary during the fitting routine. The protein's partial specific volume and the solvent density were estimated using the Sednterp program (44).
Pymol Analysis
Electrostatic and hydrogen bonding interactions involving H115 were identified with PyMol software using the “find polar contacts” command. Distance measurements were carried out in the “Editing Mode” using the commands >dist (pk1), (pk2) in which pk1 = atom 1, and pk2 = atom 2. To identify structural changes we compared the following pair of proteins: 1SOA.pdb versus 1P5F.pdb and 2RK4.pdb versus 1P5F.pdb. Each pair was uploaded on the Pymol Viewer and superimposed using the align molecule command. 1P5F.pdb, wild type DJ-1 unoxidized (28); 1SOA.pdb, oxidized wild type DJ-1 (16); 2RK4.pdb, M26I DJ-1 (unoxidized) (36).
RESULTS
Strategy Overview
The primary objectives of this study were (1) to develop a mass spectrometry method for quantitative measurement of fold changes in DJ-1 oxidation at specific sites under different oxidizing conditions, and (2) to use this method to determine the impact of the M26I substitution on DJ-1 oxidation at C106 and other residues. To accomplish these goals, we implemented a strategy that combines oxidation of recombinant wild-type DJ-1 or M26I (via treatment with a 10- or 500-fold molar excess of H2O2), 18O labeling, and liquid chromatography (LC)-MS analysis (Fig. 1). Sequence-specific oxidative modifications were then quantitated by measuring the ratio of 18O- to 16O-labeled peptides. Next, the mass spectrometry data were validated by characterizing wild-type DJ-1 and M26I in terms of their propensities to undergo C106 oxidation via 2D-PAGE. Finally, to better understand why wild-type DJ-1 and M26I exhibit different fold changes in oxidation, the two proteins were compared in terms of their secondary and quaternary structures in both the untreated and 2O forms.
Fig. 1.

Schematic overview of the strategy used to quantitate oxidative modifications of DJ-1. Wild-type DJ-1 and M26I were untreated or exposed to a 10- or 500-fold molar excess of H2O2. The proteins were digested with trypsin, and the tryptic peptides were labeled by trypsin-catalyzed 16O/18O labeling. The 16O- and 18O-labeled peptides were mixed and analyzed by nanoscale LC/MS/MS.
Oxidative Modifications of Wild-type DJ-1 and M26I (Mass Spectrometry)
Approximately 6400 spectra were obtained for each analysis. The following post-translational modifications were identified and quantified: (1) oxidation of two cysteine residues to the sulfinic acid (C106) or the sulfonic acid (C53, C106); (2) oxidation of four methionine residues (M17, M26, M133, and M134) to the sulfoxide; and (3) oxidation of a histidine residue (H115) to asparagine. Representative mass spectrometry data that enabled us to identify these modifications are described in detail below (Supplemental Table S1).
Identification of Oxidized Cysteine Residues
Treatment of DJ-1 with H2O2 resulted in a dose-dependent oxidation of the protein's cysteine residues to the sulfinic or sulfonic acid form. In Fig. 2 we show the MS/MS spectrum of peptide 99KGLIAAICAGPTALLAHEIGFGSK122 derived from wild-type DJ-1 following oxidation with a 10-fold molar excess of H2O2. The “y” and “b” ion peaks in the spectrum were assigned labels corresponding to their m/z values. The mass difference between the b(8) ion (m/z = 802.464 Da) and the b(7) ion (m/z = 667.47 Da) was equal to the mass of a cysteine sulfinic acid (134.99 Da), suggesting that residue C106 had undergone oxidation to the 2O form under these conditions. In supplemental Fig. S1, we show the MS/MS spectrum of peptide 99KGLIAAICAGPTALLAHEIGFGSK122 derived from wild-type DJ-1 after oxidation with a 500-fold molar excess of H2O2. In this case the mass difference between b(8) (m/z = 818.44 Da) and b(7) (m/z = 667.45 Da) was equal to the mass of cysteine sulfonic acid (150.99 Da), suggesting that C106 was converted to the 3O form under the more stringent oxidizing conditions. Similarly, the MS/MS spectrum of peptide 49DVVICPDASLEDAKK63 derived from M26I following treatment with a 500-fold molar excess of H2O2 clearly revealed a difference between b(5) (m/z = 578.249 Da) and b(4) (m/z = 427.255 Da) equal to the mass of cysteine sulfonic acid (supplemental Fig. S2), suggesting that C53 was oxidized to the 3O form upon exposure to the high dose of peroxide.
Fig. 2.

MS/MS analysis of peptide 99KGLIAAICAGPTALLAHEIGFGSK122 in wild-type DJ-1 oxidized with a 10-fold molar excess of H2O2. (Top) MS/MS spectrum with all of the y and b ions assigned. The y and b ion map is superimposed on the peptide sequence. In this figure and in Fig. 3 and Supplemental Figs. S1–S4, the peptide sequence is shown in forward and reverse at the top of the spectrum. Solid lines denote peaks that have been identified, whereas dashed lines denote peaks that are missing. The black dots represent peaks that have been chosen and for which a mass has been calculated. (Bottom) C106 is oxidized to cysteine sulfinic acid (2O form) according to the Mascot identification. The difference between the masses of the b8 and b7 ions equals the mass of cysteine sulfinic acid.
Identification of Oxidized Methionine Residues
The identification of oxidized methionine residues is more complex because methionine sulfoxide (MetSO) undergoes a characteristic neutral loss of methanesulfenic acid (CH3SOH, 63.998 Da) during collision-induced dissociation (45, 46). The Mascot search engine accounts for this neutral loss when assigning peaks in the MS/MS spectrum. In Fig. 3 we show the MS/MS spectrum of peptide 13GAEEMETVIPVDVMR27 derived from wild-type DJ-1 following oxidation with a 500-fold molar excess of H2O2. The mass difference between y(11) (m/z = 1193.697 Da after the neutral loss of two CH3SOH groups) and y(10) (m/z = 1110.631 Da after the neutral loss of one CH3SOH group) was equal to the mass of methionine sulfoxide minus the neutral loss mass (83 Da = 147 Da - 64 Da). Additionally, the mass difference between y(2) (m/z = 258.105 Da after the neutral loss of CH3SOH) and y(1) (m/z = 175.106 Da) was equal to the mass of methionine sulfoxide minus the neutral loss mass (83 Da). These results indicate that both methionine residues in the parent peptide (M17 and M26) were oxidized to methionine sulfoxide under these H2O2 treatment conditions. Additional MS/MS data revealed the presence of methionine sulfoxide at positions 133 (supplemental Fig. S3) and 134 (supplemental Fig. S4) of peptide 131DKMMNGGMYTYSENRVEK148 derived from wild-type DJ-1 oxidized with a 500-fold molar excess of H2O2.
Fig. 3.

MS/MS analysis of peptide 13GAEEMETVIPVDVMR27 in wild-type DJ-1 oxidized with a 500-fold molar excess of H2O2. (Top) MS/MS spectrum with all of the y and b ions assigned. The y and b ion map is superimposed on the peptide sequence. (Bottom) Both methionine residues (M17 and M26) are oxidized to methionine sulfoxide according to the Mascot identification. The difference between the masses of y2 (after the neutral loss of one CH3SOH group, mass = 63.998 Da) and y1 equals 83 Da. The difference between the masses of y11 (after the neutral loss of two CH3SOH groups, total mass = 127.996 Da) and y10 (after the neutral loss of one CH3SOH group, mass = 63.998 Da) equals 83.06 Da. Addition of the neutral loss mass to the 83 Da value obtained for y2-y1 and y11-y10 yields the mass of methionine sulfoxide.
Methionine residues of DJ-1 are readily oxidized when the protein is incubated with a 500-fold molar excess of H2O2, whereas methionine oxidation occurs much less efficiently upon exposure of the protein to a 10-fold molar excess of the peroxide (24). Accordingly, methionine sulfoxides observed in DJ-1 after exposure to a 500-fold molar excess are the result of oxidation during both sample preparation and peroxide treatment, whereas methionine sulfoxides observed after treatment with a 10-fold molar excess of H2O2 are presumed to have originated during sample preparation.
Identification of the Oxidation of Histidine to Asparagine
Histidine residues can be oxidized to asparagine as a major product (47), resulting in a mass difference of –23.01. The mascot search engine detected the presence of asparagine in place of H115 based on the decrease in the mass of peptide 99KGLIAAICAGPTALLAHEIGFGSK122 derived from wild-type DJ-1 (but not M26I) oxidized with a 10- or 500-fold molar excess of H2O2.
Quantitation of Oxidative Modifications
In the next phase of our study we determined relative levels of post-translationally modified DJ-1 peptides generated in the presence of a low or high concentration of H2O2 via quantitative analysis of the MS peak intensities. The method involved dividing the peak intensity of each 18O-labeled peptide from H2O2-treated wild-type DJ-1 or M26I by the peak intensity of the identical 16O-labeled peptide derived from the untreated protein. Reproducible data from three separate labeling experiments revealed remarkable differences in relative levels of cysteine-modified peptides when comparing wild-type DJ-1 and M26I exposed to a 10- or 500-fold molar excess of H2O2 (Table I). These differences are summarized as follows: (1) wild-type DJ-1 exhibited a marked increase in the level of C106 sulfinic acid following incubation with a 10-fold molar excess of H2O2, whereas the 2O form of C106 was not detected in a sample of M26I exposed to identical oxidizing conditions; (2) the wild-type protein (but not M26I) exhibited a higher relative level of C106 sulfinic acid after exposure to a 10-fold molar excess of H2O2 compared with a 500-fold molar excess of the peroxide; (3) wild-type DJ-1 exhibited a dramatically higher relative level of C106 sulfonic acid compared with M26I after incubation with a 500-fold molar excess of H2O2 whereas the 3O form of C106 was essentially undetectable in samples of wild-type DJ-1 or M26I exposed to a 10-fold molar excess of the peroxide); (4) M26I (but not wild-type DJ-1) exhibited an increase in the level of C53 sulfonic acid following exposure to a 500-fold molar excess of H2O2 (whereas this modification didn't appear in any other forms of the protein); and (5) wild-type DJ-1 (but not M26I) exhibited an increase in the level of asparagine (an oxidation product of histidine) at position 115 following exposure to 10-fold or 500-fold molar excess of H2O2.
Table I. Quantitation of oxidative modifications of C53, C106, and H115 in wild-type DJ-1 and M26I exposed to a 10- or 500-fold molar excess of H2O2.
| Oxidative modification | MS ratioa |
|||||||
|---|---|---|---|---|---|---|---|---|
| 10-fold molar excess H2O2 |
500-fold molar excess H2O2 |
|||||||
| wild-type DJ-1 |
M26I |
wild-type DJ-1 |
M26I |
|||||
| average | S.E. | average | S.E. or range | average | S.E. or range | average | S.E. or range | |
| C53 sulfonic acid | NDb | NAb | NDb | NAb | NDb | NAb | 4.0 | 0.4 |
| C106 sulfinic acid | 12 | 3 | NDb | NAb | 5.1 | 4.9–6.5 | 4.0 | 3.4–4.7 |
| C106 sulfonic acid | NDb | NAb | NDb | NAb | 30.3c | 0.9 | 4.1c | 0.4 |
| H115 asparagine | 1.95 | 0.03 | NDb | NAb | 2.1 | 0.7 | NDb | NAb |
a MS ratios were determined by dividing the peak intensity of each 18O-labeled peptide derived from H2O2-treated DJ-1 by the peak intensity of the identical 16O-labeled peptide derived from untreated (control) protein. n = 2 or 3.
b ND, not detected; NA, not applicable.
c p value <0.01, Student's t test. Detailed results for the MS ratios for the three experiments can be found in supplemental Table S2.
In general, quantitative analysis of methionine oxidized peptides revealed greater variability across replicate runs and less pronounced differences between wild-type DJ-1 and M26I. Specifically, the following trends were noted (Table II): (1) wild-type DJ-1 exhibited a decrease in the level of a peptide containing methionine sulfoxide at positions 17 and 26 after incubation with a 10-fold molar excess of H2O2; (2) wild-type DJ-1 and M26I exhibited a decrease in the level of a peptide with methionine sulfoxide at position 133 after incubation with a 10-fold molar excess of H2O2; and (3) wild-type DJ-1 and M26I exhibited a decrease in the level of a peptide with methionine sulfoxide at position 134 after incubation with a 10-fold molar excess of H2O2.
Table II. Quantitation of methionine sulfoxide in wild-type DJ-1 and M26I exposed to a 10-fold molar excess of H2O2.
| Methionine residue(s) oxidized to methionine sulfoxide | MS ratioa |
|||
|---|---|---|---|---|
| wild-type DJ-1 |
M26I |
|||
| average | range | average | S.E. | |
| M17 and M26b | 0.41 | 0.39–0.44 | NAc | NA |
| M133 | 0.38 | 0.34–0.41 | 0.2 | 0.1 |
| M134 | 0.37 | 0.32–0.41 | 0.2 | 0.1 |
a MS ratios were determined by dividing the peak intensity of each 18O-labeled peptide derived from H2O2-treated DJ-1 by the peak intensity of the identical 16O-labeled peptide derived from untreated (control) protein. n = 2 or 3.
b Methionine sulfoxide was present at positions 17 and 26 of the same peptide derived from wild-type DJ-1 (see Fig. 3).
c NA, not applicable (M26 is substituted with isoleucine). Detailed results for the MS ratios for the three experiments can be found in supplemental Table S3.
Oxidative Modifications of Wild-type DJ-1 and M26I (2D-PAGE)
The results outlined above suggested that M26I has a reduced propensity to undergo oxidation to the 2O form. In the next phase of our study, we validated the mass spectrometry data by assessing the relative propensities of wild-type DJ-1 and M26I to undergo C106 oxidation to the 2O form using 2D-PAGE as an independent method. Each DJ-1 variant was either untreated or treated with a 10-fold molar excess of H2O2. The untreated forms of both variants consisted of two major species with estimated pI values of 5.9 and 6.2, similar to pI values previously assigned to the 2O and unoxidized DJ-1 isoforms, respectively (Fig. 4A, panels i and ii) (16). Data reported by several groups indicate that DJ-1 exists as multiple (> 2) species spanning a range of pI values in cell culture and in brain tissue (16, 48, 49), presumably because the protein undergoes various modifications (both oxidative and nonoxidative) in addition to the conversion to the 2O form observed here. We confirmed that the DJ-1 isoforms with pI values of 5.9 and 6.2 most likely correspond to the 2O and unoxidized species (respectively) by showing that the C106A mutant, which cannot undergo oxidation to the 2O form, only appears as the more basic species on a 2D-PAGE gel (data not shown). After exposure to H2O2 the wild-type protein consisted almost entirely of the lower pI species, whereas M26I was only partially converted to this acidic isoform (Fig. 4A, panels iii and iv). Quantitative analysis of spot intensities from replicate 2D-PAGE runs revealed a significant increase in the relative level of the 2O form in a sample of wild-type DJ-1, but not M26I, after peroxide treatment (Fig. 4B). From these data, we inferred that the mutant protein has a lower propensity to undergo oxidation at C106 upon treatment with a 10-fold molar excess of H2O2.
Fig. 4.

M26I has a lower propensity to undergo oxidation to the 2O form than WT DJ-1 in a cell free system. A, Representative 2D-PAGE gels showing the difference in the propensities of wild-type DJ-1 and M26I to convert to the 2O form. The wild-type protein (i, iii) and M26I (ii, iv) were incubated in the absence (i, ii) or presence (iii, iv) of a 10-fold molar excess of H2O2. The protein samples were then analyzed via 2D-PAGE. The numbers at the bottom of the gel images correspond to the approximate pI values of unoxidized DJ-1 (pI ≈ 6.2) and the 2O isoform (pI ≈ 5.9). The data are representative of three independent experiments. Because the gels in panels i-iv were stained separately, one cannot obtain meaningful quantitative information by comparing the intensities of spots on different gels, although it is possible to determine the relative abundance of species appearing on the same gel. B, Quantitative analysis of 2D-PAGE data. The 2O DJ-1 spot intensity is expressed as a percentage of the total intensity of all spots. “Unox” refers to DJ-1 samples incubated in the absence of H2O2. Mean ± S.E., n = 5 (WT DJ-1) or n = 4 (M26I); ** p < 0.01, one-way ANOVA with Neuman Keuls post-test; ns, not significant.
Secondary and Quaternary Structure of Wild-type DJ-1 and M26I
Next, we examined whether wild-type DJ-1 and M26I differed in their ability to undergo oxidation to the 2O form as a result of differences in their protein folds. We considered that M26I might have a decreased oxidation propensity at C106 because of (1) a local perturbation of structural elements in proximity to the cysteine residue, or (2) a more global perturbation (unfolding) of the mutant protein. Both types of perturbations would be expected to interfere with conversion to the 2O form by destabilizing the “C106 pocket,” which has polar residues in a configuration that favors C106 oxidation in the wild-type protein (16, 27–29). To address whether the overall fold of M26I was markedly different from that of wild-type DJ-1, we analyzed both proteins by far-UV CD and ultracentrifugation, two approaches that report on the protein's secondary and quaternary structures, respectively (15).
CD spectra recorded for unoxidized wild-type DJ-1 and M26I (Fig. 5) displayed broad negative ellipticity from 208 to 222 nm, consistent with a mixed α/β structure. The relative magnitude of the signals recorded for the two variants showed some variability, presumably because of differences in the extent to which the proteins were oxidized during purification (24). Although the spectrum of M26I occasionally overlapped closely with that of the wild-type protein, in most cases the mutant exhibited an overall reduction in negative ellipticity from 208 to 222 nm (Figs. 5A, 5B). Both variants exhibited a slightly reduced signal after treatment with a 10-fold molar excess of H2O2, suggesting that oxidation led to a small decrease in overall secondary structure (Figs. 5C, 5D). This observation was somewhat unexpected because x-ray crystallography data suggest that unoxidized wild-type DJ-1 and the 2O form have nearly identical three-dimensional structures (16, 22). A potential explanation for this discrepancy is that protein isoforms with modest structural differences detectable by CD analysis may adopt the same, energetically favorable conformation in a crystal lattice. Thus, slight differences in the solution structures of unoxidized and 2O DJ-1 may not be apparent in the 3D structures of these isoforms determined by x-ray crystallography.
Fig. 5.

Far-UV CD analysis of wild-type DJ-1 and M26I. The proteins were analyzed without peroxide treatment or after exposure to a 10-fold molar excess of H2O2. Panels A–D show pairwise comparisons of spectra recorded for the following variants: A, untreated wild-type DJ-1 versus untreated M26I; B, H2O2-treated wild-type DJ-1 versus H2O2-treated M26I; C, untreated wild-type DJ-1 versus H2O2-treated wild-type DJ-1; and D, untreated M26I versus H2O2-treated M26I.
In parallel with the CD measurements, we analyzed the quaternary structure of wild-type DJ-1 and M26I (unoxidized and 2O forms) via ultracentrifugation (Figs. 6 and 7). Sedimentation-equilibrium data obtained for unoxidized wild-type DJ-1 were best fit to a monomer-dimer model with estimated Kd values ranging from 3.6 ± 0.2 × 10−8 m to 2.8 ± 0.4 × 10−7 m (Table III). The data obtained for M26I were also best fit to a monomer-dimer model with an estimated Kd value of 6 ± 2 × 10−7 m, near the upper end of the range determined for the wild-type protein. Sedimentation data obtained for wild-type DJ-1 after treatment with a 10-fold molar excess of H2O2 were best fit to a monomer-dimer model with estimated Kd values ranging from 6 ± 1 × 10−8 m to 2.0 ± 0.7 × 10−7 m. In contrast, the data for M26I treated with a 10-fold molar excess of H2O2 were best fit to a monomer-tetramer model (Table III).
Fig. 6.

Sedimentation-equilibrium analysis of wild-type DJ-1 and M26I (without H2O2 treatment). The proteins were dissolved in 50 mm Tris, 200 mm sodium chloride, pH 7.0 buffer and centrifuged at 20,000, 22,000, and 24,000 rpm at 22 °C (only the data collected at 22,000 rpm are shown). The protein concentrations used were 2.2 (circles), 1.5 (squares), and 1.1 mg/ml (triangles) for wild-type DJ-1; and 2.8 (circles), 1.9 (squares), and 1.4 mg/ml (triangles) for M26I. Lower graphs illustrate r2/2 versus absorbance plots; symbols represent measured data points, and solid lines represent best-fit curves to a monomer-dimer model. Upper graphs illustrate the residuals from fitting the measured data points to a two-species model. The random, nonsystematic distribution of the residuals indicates a good fit of the data to the models.
Fig. 7.

Sedimentation-equilibrium analysis of wild-type DJ-1 and M26I after exposure to a 10-fold molar excess of H2O2. The proteins were dissolved in 50 mm Tris, 200 mm sodium chloride, pH 7.0 buffer, and centrifuged at 20,000, 22,000, and 24,000 rpm at 22 °C (only the data collected at 22,000 rpm are shown). The protein concentrations used were 1.6 (circles), 1.1 (squares), and 0.8 mg/ml (triangles) for wild-type DJ-1; and 2.8 (circles), 1.9 (squares), and 1.4 mg/ml (triangles) for M26I. Lower graphs illustrate r2/2 versus absorbance plots; symbols represent measured data points, and solid lines represent best-fit curves to a monomer-dimer model for wild-type DJ-1 and a monomer-tetramer model for M26I. Upper graphs illustrate the residuals from fitting the measured data points to a two-species model. The random, nonsystematic distribution of the residuals indicates a good fit of the data to the models.
Table III. Subunit association/dissociation parameters for wild-type DJ-1 and M26I analyzed by sedimentation-equilibrium.
| Variant | Best-fit model | Molecular weight (Da) | Kd |
|---|---|---|---|
| WT 2O | monomer-dimer | 37,980 | 6 ± 1 × 10−8 (M) |
| 34,485 | 2.0 ± 0.7 × 10−7 (M) | ||
| WT | monomer-dimer | 38,060 | 3.6 ± 0.2 × 10−8 (M) |
| 39,536 | 2.8 ± 0.4 × 10−7 (M) | ||
| M26I 2O | monomer-tetramer | 28,769 | 4.2 ± 0.6 × 10−17(M3) |
| M26I | monomer-dimer | 30,386 | 6 ± 2 × 10−7(M) |
Collectively, these results suggested that (1) unoxidized wild-type DJ-1 and M26I have similar secondary and quaternary structures, although M26I forms a less stable homodimer; (2) oxidation of wild-type DJ-1 in the presence of a 10-fold molar excess of H2O2, a treatment that results in selective oxidation of C106 to the sulfinic acid without affecting other oxidizable residues (24), does not alter the protein's quaternary structure or stability; and (3) oxidation of M26I in the presence of a 10-fold molar excess of H2O2 promotes higher order self-assembly.
DISCUSSION
Several loss-of-function mutations in the gene encoding DJ-1 have been identified in patients with familial PD (11, 12). One mutation (L166P) abrogates DJ-1 function by destabilizing the native homodimer, thus promoting aggregation or turnover of the protein (33–35). In contrast, other mutations have a less pronounced effect on DJ-1 stability, and it is unclear how they perturb DJ-1 function. In this study we addressed the hypothesis that the M26I familial mutant exhibits reduced protective activity as a result of (1) a decreased ability to undergo oxidation to the functionally important 2O form (16, 24, 30), and/or (2) an increased propensity to undergo oxidative modifications with potentially disruptive effects on DJ-1 function at other sites in the polypeptide chain.
Our experimental approach consisted of comparing recombinant wild-type DJ-1 and M26I in terms of fold changes in oxidation at C106 and other sites following treatment with a 10- or 500-fold molar excess of H2O2. This approach was reasonable because our focus was to compare the two DJ-1 variants in terms of their intrinsic propensities to undergo oxidation. Accordingly, we considered it important to avoid complicating factors that might mask or offset these intrinsic propensities, including effects of protein-protein interactions in a cellular or animal model. To quantitate fold changes in oxidation at each site, we developed a new method involving LC/MS/MS analysis of a mixture of 16O- and 18O-labeled peptides derived from untreated and H2O2-treated DJ-1, respectively. To our knowledge, 18O labeling technology has not been applied previously to quantitative analysis of oxidative protein modifications. An advantage of the 18O-labeling method is that it can be used to quantitate multiple oxidative modifications simultaneously, whereas other approaches enable quantitation of only a single type of modification (e.g. cysteine oxidation (50–55) and carbonylation (56)). While this manuscript was being written, Pimenova et.al (57) described a method to simultaneously measure multiple oxidative protein modifications using isobaric tags for relative and absolute quantitation labeling technology. A potential drawback of labels such as isobaric tags for relative and absolute quantitation is that these reagents can produce unpredicted fragmentation patterns which confound the interpretation of MS data (58). In contrast, the 18O-labeling approach described here avoids this pitfall because facile incorporation of the 18O isotope into the C termini of tryptic peptides does not result in anomalous fragmentation patterns.
Effects of the M26I Substitution on C106 Oxidation
Oxidative modifications of DJ-1 have been characterized by mass spectrometry in a number of earlier studies (13, 15, 24, 26). Here we describe fold changes in oxidation at specific sites deduced from quantitative MS data. We found that residue C106 of wild-type DJ-1 was preferentially oxidized to the sulfinic or sulfonic acid upon treatment of the protein with a 10- or 500-fold molar excess of H2O2 (respectively), consistent with previous findings (24). Wild-type DJ-1 (but not the M26I mutant) exhibited an increase in the level of C106 sulfinic acid after exposure to a 10-fold molar excess of H2O2. On the basis of semiquantitative 2D PAGE data we confirmed that wild-type DJ-1 had a greater propensity to convert to the 2O form under these oxidizing conditions.
From these observations, we infer that the M26I substitution alters the structure of DJ-1 in a way that decreases the reactivity of C106 with H2O2. A previous study showed that C106 is more readily oxidized than the single cysteine residue of bovine serum albumin (26), presumably because the “active site” pocket surrounding C106 contains a number of acidic and basic residues (including the protonated carboxylic acid sidechain of E18) that are suitably positioned to promote the transfer of two oxygen atoms to the cysteine sidechain (27–29). The M26I mutation may induce a conformational change that disrupts the configuration of residues in this pocket. Consistent with this idea, analysis of M26I by two-dimensional NMR revealed chemical shift changes in charged residues that may play a role in modulating C106 oxidation (37). In addition, the M26I substitution was shown by x-ray crystallography to induce a packing defect in the interior of the protein (36). Moreover, we found that C53 was oxidized to the sulfonic acid upon exposure of M26I (but not wild-type DJ-1) to a 500-fold molar excess of H2O2. C53 is located at the subunit interface (22, 29, 59, 60) and is less readily oxidized than C106 in the wild-type protein (16, 24). Therefore, increased oxidation of C53 in M26I may reflect a weakening of intersubunit contacts in the mutant homodimer. In support of this idea, our sedimentation data revealed that M26I has a slightly higher dimer dissociation constant than the wild-type protein. In turn, a structural perturbation at the dimer interface could contribute to the impaired ability of M26I to convert to the 2O form, given that the active site pocket surrounding C106 consists of residues from both subunits. Importantly, our far-UV CD and sedimentation data indicated that unoxidized wild-type DJ-1 and M26I have similar secondary and quaternary structures, consistent with previous reports that the M26I mutation has little impact on the global fold of the protein (36, 37). In an earlier report we presented evidence that M26I had substantially reduced secondary structure compared with wild-type DJ-1, even without exposure to H2O2 (15). However, in contrast to the experiments with untagged DJ-1 summarized here, the previous study was carried out using N-terminally histidine-tagged variants, and we have found that the presence of the histidine tag promotes oxidative modifications that induce M26I misfolding and aggregation (Hulleman and Rochet, unpublished observations).
In addition to having conformational properties that potentially disfavor C106 oxidation, M26I may incur a loss of stability upon oxidation by H2O2. In turn, this loss of stability may have led to misfolding, aggregation, and thus a decreased recovery of M26I (2O form) prior to MS analysis. Consistent with this scenario, our ultracentrifugation data revealed that M26I obeyed a monomer-tetramer equilibrium after conversion to the 2O form, suggesting that the oxidized mutant protein has a relatively high propensity to form higher-order oligomers. Moreover, we and others have found that the M26I substitution induces a number of structural perturbations (see above) (15, 36, 37), and these could elicit misfolding and aggregation of the mutant protein when combined with C106 oxidation. Alternatively, our ultracentrifugation data are also consistent with a mechanism whereby oxidized DJ-1 forms higher-order oligomers in the absence of dimer dissociation.
Compared with wild-type DJ-1, M26I exhibited a 7.5-fold lower ratio of 3Otreated to 3Ountreated when the proteins were exposed to a 500-fold molar excess of H2O2. This difference in MS ratios may reflect the apparent decrease in C106 reactivity with H2O2 in the active site pocket of M26I (see above). A decrease in C106 reactivity is expected to interfere with the conversion of M26I to both the 2O and 3O forms, given that the oxidation of cysteine to the sulfonic acid presumably involves the formation of cysteine sulfinic acid as an obligatory intermediate. In addition, M26I is likely to undergo misfolding and aggregation upon oxidation to the 3O form (15, 36, 37), resulting in a diminished recovery of the mutant protein prior to MS analysis (as outlined above in the case of M26I oxidized to the 2O form).
Based on the results presented herein and data reported by Canet-Avilés et al. (16), we would predict that M26I should have a decreased ability to localize to mitochondria in oxidatively stressed cells. Surprisingly, however, the Cookson group reported data showing the opposite trend—namely, that M26I has a greater propensity to relocalize to mitochondria in oxidatively stressed cells than the wild-type protein (61). Therefore, we infer that oxidation to the 2O form may not always be a key regulatory event that determines whether wild-type or mutant forms of DJ-1 localize to mitochondria (i.e. because M26I, despite its reduced propensity to convert to the 2O form, has a strong ability to localize to mitochondria). Consistent with this inference, another group has reported that a significant level of DJ-1 is associated with mitochondria in cultured cells and animals even in the absence of oxidative stress (62).
The data presented here, together with observations that oxidation of DJ-1 to the 2O form is necessary for inhibition of aSyn fibril formation (24), suggest that M26I may have a reduced ability to suppress aSyn fibrillization compared with the wild-type protein. In support of this idea, a recent study revealed that M26I pretreated with H2O2 inhibited aSyn fibril formation less efficiently than the oxidized wild-type protein (38). Additional evidence suggests that the oxidation of DJ-1 at position 106 is necessary for protection against toxicity related to complex I inhibition (Liu and Rochet, unpublished data) (30). Accordingly, we infer that the decreased propensity of M26I to undergo conversion to the 2O form as reported here may result in various functional defects in familial PD patients, including a reduced ability of the protein to carry out a chaperone function in cytosolic and/or mitochondrial compartments.
Effects of the M26I Substitution on H115 Conversion to Asparagine
Treatment of wild-type DJ-1 (but not M26I) with a 10- or 500-fold molar excess of H2O2 led to the conversion of H115 to asparagine. We interpret this result to mean that H115 oxidation to asparagine is (1) disfavored by conformational rearrangements resulting from the M26I substitution, and/or (2) promoted by structural changes resulting from C106 oxidation in wild-type DJ-1. A comparison of the x-ray structures of the wild-type protein (unoxidized and 2O) and M26I (unoxidized) using PyMol software revealed similar residue configurations near H115 in all three structures (Fig. 8), although crystal lattice packing may have played a role in stabilizing the protein conformation in this region. NMR analysis revealed only very minor chemical shift changes between wild-type DJ-1 and M26I at position 115 (37). Thus, we favor the second interpretation outlined above, namely, that the wild-type protein more readily undergoes H-to-N conversion at position 115 because of its greater propensity to become oxidized at position 106. Because H115 is strictly conserved among eukaryotic DJ-1 variants (63), we predict that oxidation of this residue to asparagine could have functional consequences. PyMol analysis revealed that replacement of H115 with asparagine only subtly perturbed interactions with nearby residues in the structure of wild-type 2O DJ-1 (Fig. 8).
Fig. 8.

Pymol analysis of structural motifs surrounding residue 115 of wild-type DJ-1 and M26I. A, Comparison of the structures of wild-type DJ-1 and M26I (both unoxidized) focusing on H115. Wild-type DJ-1 (1P5F) and M26I (2RK4) are shown in purple and yellow, respectively. Polar contacts are shown as black and red dashed lines for wild-type DJ-1 and M26I, respectively. B, Comparison of the structures of unoxidized and oxidized DJ-1 focusing on H115. Unoxidized wild-type DJ-1 (1P5F) and 2O wild-type DJ-1 (1SOA) are shown in purple and yellow, respectively. Polar contacts are shown as black and red dashed lines for unoxidized and 2O DJ-1, respectively. C, D, The H115N substitution may alter interactions with nearby residues. C, View of 2O wild type DJ-1 (1SOA) showing electrostatic interactions and hydrogen bonds with H115. D, View of 2O wild-type DJ-1 (1SOA) showing electrostatic interactions and hydrogen bonds with asparagine modeled in place of histidine at position 115. Note the loss of an electrostatic interaction with the main-chain carbonyl group of L82.
Effects of Methionine Oxidation on DJ-1 Stability
The data in Table II revealed that peptides having undergone methionine oxidation at positions 17, 26, 133, or 134 were depleted in samples of wild-type DJ-1 exposed to a 10-fold molar excess of H2O2 compared with untreated samples. These results imply that DJ-1 isoforms with methionine sulfoxide at these sites have a high propensity to undergo misfolding and aggregation on further oxidation, and are thus lost from H2O2-treated samples prior to MS analysis. Consistent with this interpretation, all four methionine residues are highly conserved among vertebrate forms of DJ-1 (63), suggesting that the structural elements encompassing these residues are intolerant of chemical modifications. Accordingly, oxidation of these methionine residues may cause a partial loss of DJ-1 stability, thereby predisposing the protein to unfold and aggregate following additional modifications.
Because M26 is located near the subunit interface of DJ-1, oxidation at this site may destabilize the wild-type homodimer. This prediction is supported by evidence presented here and in other reports (15, 36, 37) that the M26I substitution elicits conformational changes that in turn may cause a decrease in dimer stability. Interestingly, MS analysis of DJ-1 from human postmortem brain samples reveals that methionine is oxidized to methionine sulfoxide at positions 17, 26, 133, and 134 in PD patients, whereas methionine sulfoxide is only detected at positions 17 and 26 in age-matched controls (13). These findings indicate that the oxidation of each methionine residue occurs under physiological conditions. Moreover, the abundance of DJ-1 isoforms with methionine sulfoxide at these sites may increase as the activity of MsrA, an enzyme that repairs methionine sulfoxide, decreases with age (64). An age-dependent decrease in MsrA activity coupled with increases in methionine oxidation of DJ-1 could contribute to the increased risk of PD with aging (14, 65). Importantly, DJ-1 isoforms with methionine sulfoxide at positions 133 and 134 are detected in the post-mortem brains of sporadic PD patients, but not age-matched controls (13). Our findings suggest that DJ-1 dysfunction, resulting from misfolding and aggregation of the methionine-oxidized protein under conditions that normally favor oxidation to the functionally relevant 2O form, may contribute to neurodegeneration in sporadic PD.
The more pronounced loss of methionine-oxidized M26I compared with methionine-oxidized wild-type DJ-1 in the presence of a 10-fold molar excess of H2O2 is consistent with the observation that M26I is less stable than the wild-type protein (15, 36, 37). Thus, the combined destabilizing effects of the M26I substitution and methionine oxidation may result in greater DJ-1 misfolding and aggregation (and a more pronounced loss of function) upon further oxidation than the milder destabilizing effect of methionine oxidation alone. In turn, the enhanced loss of function expected of methionine-oxidized M26I compared with methionine-oxidized wild-type DJ-1 may contribute to the earlier onset of disease in familial PD patients with the M26I mutation.
Conclusion
In summary, we have exploited the unique power of mass spectrometry and proteomic technology to determine the impact of a familial mutation on the propensity of DJ-1 to undergo oxidation at specific sites. We find that the M26I substitution interferes with the ability of the protein to undergo oxidation to the 2O form, a functionally important modification. This effect apparently involves subtle conformational perturbations that may alter the geometry of the active site pocket surrounding C106 rather than a global unfolding of the protein. These observations lay the groundwork for investigating whether other DJ-1 mutants without a global folding defect, similar to M26I but in contrast to L166P (36), also have a diminished ability to undergo conversion to the 2O form. Our data also suggest that the mild oxidation that normally results in functional enhancement of wild-type DJ-1 can cause destabilization and aggregation of DJ-1 isoforms having undergone initial stages of oxidative damage, including methionine oxidation characteristic of sporadic PD. Importantly, our approach of studying recombinant DJ-1 variants in an isolated system enabled us to characterize mutant and oxidized forms of the protein in terms of their intrinsic propensities to undergo a functionally relevant oxidative modification. We note that these intrinsic propensities are likely modulated by additional factors (e.g. protein-protein interactions) in the complex environment of the brain, and thus our findings set the stage for future research aimed at understanding the role of potential in vivo modulatory factors.
The results reported here represent new evidence that DJ-1 dysfunction resulting from a mutation or oxidative damage may contribute to neurodegeneration in familial or sporadic PD. Our findings also imply that the function of wild-type or mutant DJ-1 may be enhanced by small molecules which promote conversion to the 2O form by (1) selectively binding the C106 sulfinic acid (66), or (2) stabilizing structural motifs destabilized by methionine oxidation. Such compounds may be useful therapeutic agents to enhance DJ-1 function and alleviate neurodegeneration in PD patients.
Acknowledgments
We thank Dr. Soumya Ray (Brigham and Women's Hospital) for providing the pGEX-6P1-DJ-1M26I DNA.
Footnotes
* This work was supported by grants from the National Cancer Institute (grant number 1U24CA126480-01) (FER), National Institute of Aging (grant number 5R01AG025362-02) (FER), National Institute on Drug Abuse (grant number 1R03DA027111-01) (J-CR), American Parkinson Disease Association (J-CR), National Parkinson Foundation (J-CR, FER), American Foundation for Pharmaceutical Education (JDH), Protein Engineering Network of Centres of Excellence (CMK), and Alberta Cancer Board (CMK). A portion of the research described herein was conducted in a facility constructed with support from Research Facilities Improvement Program Grants Number C06-14499 and C06-15480 from the National Center for Research Resources of the National Institutes of Health (Department of MCMP, Purdue University).
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S3.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S3.
1 The abbreviations used are:
- PD
- Parkinson's disease
- aSyn
- α-synuclein
- ROS
- reactive oxygen species
- IPTG
- isopropylthiogalactoside
- TCEP
- tris(2-carboxyethyl) phosphine
- 2D-PAGE
- two-dimensional polyacrylamide gel electrophoresis
- BCA
- bicinchoninic acid
- far-UV CD
- far-ultraviolet circular dichroism
- IPG
- immobilized pH gradient
- GST
- glutathione-S-transferase.
REFERENCES
- 1. Dawson T. M., Dawson V. L. (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302, 819–822 [DOI] [PubMed] [Google Scholar]
- 2. Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., Greenamyre J. T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306 [DOI] [PubMed] [Google Scholar]
- 3. Banerjee R., Starkov A. A., Beal M. F., Thomas B. (2009) Mitochondrial dysfunction in the limelight of Parkinson's disease pathogenesis. Biochim. Biophys. Acta 1792, 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malkus K. A., Tsika E., Ischiropoulos H. (2009) Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson's disease: how neurons are lost in the Bermuda triangle. Mol. Neurodegener. 4, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spillantini M. G., Schmidt M. L., Lee V. M.-Y., Trojanowski J. Q., Jakes R., Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 6. Conway K. A., Rochet J.-C., Bieganski R. M., Lansbury P. T., Jr. (2001) Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science 294, 1346–1349 [DOI] [PubMed] [Google Scholar]
- 7. Rochet J. C. (2007) Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev. Mol. Med. 9, 1–34 [DOI] [PubMed] [Google Scholar]
- 8. Cookson M. R. (2009) alpha-Synuclein and neuronal cell death. Mol. Neurodegener. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Graham D. G. (1978) Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 14, 633–643 [PubMed] [Google Scholar]
- 10. Hirsch E. C., Faucheux B. A. (1998) Iron metabolism and Parkinson's disease. Mov. Disord. 13 Suppl 1, 39–45 [PubMed] [Google Scholar]
- 11. Bonifati V., Rizzu P., van Baren M. J., Schaap O., Breedveld G. J., Krieger E., Dekker M. C., Squitieri F., Ibanez P., Joosse M., van Dongen J. W., Vanacore N., van Swieten J. C., Brice A., Meco G., van Duijn C. M., Oostra B. A., Heutink P. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 [DOI] [PubMed] [Google Scholar]
- 12. Kahle P. J., Waak J., Gasser T. (2009) DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radic. Biol. Med. 47, 1354–1361 [DOI] [PubMed] [Google Scholar]
- 13. Choi J., Sullards M. C., Olzmann J. A., Rees H. D., Weintraub S. T., Bostwick D. E., Gearing M., Levey A. I., Chin L. S., Li L. (2006) Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 281, 10816–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meulener M. C., Xu K., Thomson L., Thompson L., Ischiropoulos H., Bonini N. M. (2006) Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. U.S.A. 103, 12517–12522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hulleman J. D., Mirzaei H., Guigard E., Taylor K. L., Ray S. S., Kay C. M., Regnier F. E., Rochet J. C. (2007) Destabilization of DJ-1 by familial substitution and oxidative modifications: implications for Parkinson's disease. Biochemistry 46, 5776–5789 [DOI] [PubMed] [Google Scholar]
- 16. Canet-Avilés R. M., Wilson M. A., Miller D. W., Ahmad R., McLendon C., Bandyopadhyay S., Baptista M. J., Ringe D., Petsko G. A., Cookson M. R. (2004) The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taira T., Saito Y., Niki T., Iguchi-Ariga S. M., Takahashi K., Ariga H. (2004) DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim R. H., Smith P. D., Aleyasin H., Hayley S., Mount M. P., Pownall S., Wakeham A., You-Ten A. J., Kalia S. K., Horne P., Westaway D., Lozano A. M., Anisman H., Park D. S., Mak T. W. (2005) Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Y., Gehrke S., Haque M. E., Imai Y., Kosek J., Yang L., Beal M. F., Nishimura I., Wakamatsu K., Ito S., Takahashi R., Lu B. (2005) Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 13670–13675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou W., Freed C. R. (2005) DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J. Biol. Chem. 280, 43150–43158 [DOI] [PubMed] [Google Scholar]
- 21. Liu F., Nguyen J. L., Hulleman J. D., Li L., Rochet J. C. (2008) Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson's disease. J. Neurochem. 105, 2435–2453 [DOI] [PubMed] [Google Scholar]
- 22. Lee S. J., Kim S. J., Kim I. K., Ko J., Jeong C. S., Kim G. H., Park C., Kang S. O., Suh P. G., Lee H. S., Cha S. S. (2003) Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J. Biol. Chem. 278, 44552–44559 [DOI] [PubMed] [Google Scholar]
- 23. Shendelman S., Jonason A., Martinat C., Leete T., Abeliovich A. (2004) DJ-1 Is a redox-dependent molecular chaperone that inhibits α-synuclein aggregate formation. PLoS Biol. 2, e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou W., Zhu M., Wilson M. A., Petsko G. A., Fink A. L. (2006) The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J. Mol. Biol. 356, 1036–1048 [DOI] [PubMed] [Google Scholar]
- 25. Aleyasin H., Rousseaux M. W., Marcogliese P. C., Hewitt S. J., Irrcher I., Joselin A. P., Parsanejad M., Kim R. H., Rizzu P., Callaghan S. M., Slack R. S., Mak T. W., Park D. S. (2010) DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 3186–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andres-Mateos E., Perier C., Zhang L., Blanchard-Fillion B., Greco T. M., Thomas B., Ko H. S., Sasaki M., Ischiropoulos H., Przedborski S., Dawson T. M., Dawson V. L. (2007) DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 14807–14812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tao X., Tong L. (2003) Crystal structure of human DJ-1, a protein associated with early onset Parkinson's disease. J. Biol. Chem. 278, 31372–31379 [DOI] [PubMed] [Google Scholar]
- 28. Wilson M. A., Collins J. L., Hod Y., Ringe D., Petsko G. A. (2003) The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 9256–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Witt A. C., Lakshminarasimhan M., Remington B. C., Hasim S., Pozharski E., Wilson M. A. (2008) Cysteine pKa depression by a protonated glutamic acid in human DJ-1. Biochemistry 47, 7430–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blackinton J., Lakshminarasimhan M., Thomas K. J., Ahmad R., Greggio E., Raza A. S., Cookson M. R., Wilson M. A. (2009) Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J. Biol. Chem. 284, 6476–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Irrcher I., Aleyasin H., Seifert E. L., Hewitt S. J., Chhabra S., Phillips M., Lutz A. K., Rousseaux M. W., Bevilacqua L., Jahani-Asl A., Callaghan S., MacLaurin J. G., Winklhofer K. F., Rizzu P., Rippstein P., Kim R. H., Chen C. X., Fon E. A., Slack R. S., Harper M. E., McBride H. M., Mak T. W., Park D. S. (2010) Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 19, 3734–3746 [DOI] [PubMed] [Google Scholar]
- 32. Kamp F., Exner N., Lutz A. K., Wender N., Hegermann J., Brunner B., Nuscher B., Bartels T., Giese A., Beyer K., Eimer S., Winklhofer K. F., Haass C. (2010) Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macedo M. G., Anar B., Bronner I. F., Cannella M., Squitieri F., Bonifati V., Hoogeveen A., Heutink P., Rizzu P. (2003) The DJ-1 L166P mutant protein associated with early onset Parkinson's disease is unstable and forms higher-order protein complexes. Hum. Mol. Genet. 12, 2807–2816 [DOI] [PubMed] [Google Scholar]
- 34. Moore D. J., Zhang L., Dawson T. M., Dawson V. L. (2003) A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 87, 1558–1567 [DOI] [PubMed] [Google Scholar]
- 35. Olzmann J. A., Brown K., Wilkinson K. D., Rees H. D., Huai Q., Ke H., Levey A. I., Li L., Chin L. S. (2004) Familial Parkinson's disease-associated L166P mutation disrupts DJ-1 protein folding and function. J. Biol. Chem. 279, 8506–8515 [DOI] [PubMed] [Google Scholar]
- 36. Lakshminarasimhan M., Maldonado M. T., Zhou W., Fink A. L., Wilson M. A. (2008) Structural impact of three Parkinsonism-associated missense mutations on human DJ-1. Biochemistry 47, 1381–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malgieri G., Eliezer D. (2008) Structural effects of Parkinson's disease linked DJ-1 mutations. Protein Sci. 17, 855–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Logan T., Clark L., Ray S. S. (2010) Engineered disulfide bonds restore chaperone-like function of DJ-1 mutants linked to familial Parkinson's disease. Biochemistry 49, 5624–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown K. J., Fenselau C. (2004) Investigation of Doxorubicin Resistance in MCF-7 Breast Cancer Cells Using Shot-Gun Comparative Proteomics with Proteolytic 18O Labeling. J. Proteome Res. 3, 455–462 [DOI] [PubMed] [Google Scholar]
- 40. Miyagi M., Rao K. C. S. (2007) Proteolytic 18O-labeling strategies for quantitative proteomics. Mass Spectrom. Rev. 26, 121–136 [DOI] [PubMed] [Google Scholar]
- 41. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 42. Laue T. M., Stafford W. F., 3rd (1999) Modern applications of analytical ultracentrifugation. Annu. Rev. Biophys. Biomol. Struct. 28, 75–100 [DOI] [PubMed] [Google Scholar]
- 43. Johnson M. L., Correia J. J., Yphantis D. A., Halvorson H. R. (1981) Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys. J. 36, 575–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laue T. M., Shah B. D., Ridgeway T. M., Pelletier S. L. (1992) Analytical ultracentrifugation in biochemistry and polymer science. In: Harding S., Rowe A., eds., pp. 90–125, Royal Society of Chemistry [Google Scholar]
- 45. Guan Z., Yates N. A., Bakhtiar R. (2003) Detection and characterization of methionine oxidation in peptides by collision-induced dissociation and electron capture dissociation. J. Am. Soc. Mass Spectrom. 14, 605–613 [DOI] [PubMed] [Google Scholar]
- 46. Hoffman M. D., Sniatynski M. J., Rogalski J. C., Le Blanc J. C., Kast J. (2006) Multiple Neutral Loss Monitoring (MNM): A Multiplexed Method for Post-Translational Modification Screening. J. Am. Soc. Mass Spectrom. 17, 307–317 [DOI] [PubMed] [Google Scholar]
- 47. Berlett B. S., Stadtman E. R. (1997) Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316 [DOI] [PubMed] [Google Scholar]
- 48. Bandopadhyay R., Kingsbury A. E., Cookson M. R., Reid A. R., Evans I. M., Hope A. D., Pittman A. M., Lashley T., Canet-Aviles R., Miller D. W., McLendon C., Strand C., Leonard A. J., Abou-Sleiman P. M., Healy D. G., Ariga H., Wood N. W., de Silva R., Revesz T., Hardy J. A., Lees A. J. (2004) The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson's disease. Brain 127, 420–430 [DOI] [PubMed] [Google Scholar]
- 49. Betarbet R., Canet-Aviles R. M., Sherer T. B., Mastroberardino P. G., McLendon C., Kim J. H., Lund S., Na H. M., Taylor G., Bence N. F., Kopito R., Seo B. B., Yagi T., Yagi A., Klinefelter G., Cookson M. R., Greenamyre J. T. (2006) Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol. Dis. 22, 404–420 [DOI] [PubMed] [Google Scholar]
- 50. Sethuraman M., McComb M. E., Heibeck T., Costello C. E., Cohen R. A. (2004) Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol. Cell. Proteomics 3, 273–278 [DOI] [PubMed] [Google Scholar]
- 51. Sethuraman M., McComb M. E., Huang H., Huang S., Heibeck T., Costello C. E., Cohen R. A. (2004) Isotope-coded affinity tag (ICAT) approach to redox proteomics: Identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J. Proteome Res. 3, 1228–1233 [DOI] [PubMed] [Google Scholar]
- 52. Sethuraman M., Clavreul N., Huang H., McComb M. E., Costello C. E., Cohen R. A. (2007) Quantification of oxidative posttranslational modifications of cysteine thiols of p21ras associated with redox modulation of activity using isotope-coded affinity tags and mass spectrometry. Free Radic. Biol. Med. 42, 823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U. S. A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Winter J., Ilbert M., Graf P. C., Ozcelik D., Jakob U. (2008) Bleach Activates a Redox-Regulated Chaperone by Oxidative Protein Unfolding. Cell 135, 691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Held J. M., Danielson S. R., Behring J. B., Atsriku C., Britton D. J., Puckett R. L., Schilling B., Campisi J., Benz C. C., Gibson B. W. (2010) Targeted Quantitation of Site-Specific Cysteine Oxidation in Endogenous Proteins Using a Differential Alkylation and Multiple Reaction Monitoring Mass Spectrometry Approach. Mol. Cell. Proteomics 9, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Madian A. G., Regnier F. E. (2010) Proteomic identification of carbonylated proteins and their oxidation sites. J. Proteome Res. 9, 3766–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pimenova T., Pereira C. P., Gehrig P., Buehler P. W., Schaer D. J., Zenobi R. (2010) Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J. Proteome Res. 9, 4061–4070 [DOI] [PubMed] [Google Scholar]
- 58. Aggarwal K., Choe L. H., Lee K. H. (2006) Shotgun proteomics using the iTRAQ isobaric tags. Brief Funct Genomic Proteomic 5, 112–120 [DOI] [PubMed] [Google Scholar]
- 59. Honbou K., Suzuki N. N., Horiuchi M., Niki T., Taira T., Ariga H., Inagaki F. (2003) The crystal structure of DJ-1, a protein related to male fertility and Parkinson's disease. J. Biol. Chem. 278, 31380–31384 Epub 32003 Jun 31388 [DOI] [PubMed] [Google Scholar]
- 60. Huai Q., Sun Y., Wang H., Chin L. S., Li L., Robinson H., Ke H. (2003) Crystal structure of DJ-1/RS and implication on familial Parkinson's disease. FEBS Lett. 549, 171–175 [DOI] [PubMed] [Google Scholar]
- 61. Blackinton J., Ahmad R., Miller D. W., van der Brug M. P., Canet-Avilés R. M., Hague S. M., Kaleem M., Cookson M. R. (2005) Effects of DJ-1 mutations and polymorphisms on protein stability and subcellular localization. Brain Res. Mol. Brain Res. 134, 76–83 [DOI] [PubMed] [Google Scholar]
- 62. Zhang L., Shimoji M., Thomas B., Moore D. J., Yu S. W., Marupudi N. I., Torp R., Torgner I. A., Ottersen O. P., Dawson T. M., Dawson V. L. (2005) Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 14, 2063–2073 [DOI] [PubMed] [Google Scholar]
- 63. Bandyopadhyay S., Cookson M. R. (2004) Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol. Biol. 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petropoulos I., Mary J., Perichon M., Friguet B. (2001) Rat peptide methionine sulphoxide reductase: cloning of the cDNA, and down-regulation of gene expression and enzyme activity during aging. Biochem. J. 355, 819–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu F., Hindupur J., Nguyen J. L., Ruf K. J., Zhu J., Schieler J. L., Bonham C. C., Wood K. V., Davisson V. J., Rochet J. C. (2008) Methionine sulfoxide reductase A protects dopaminergic cells from Parkinson's disease-related insults. Free Radic. Biol. Med. 45, 242–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Miyazaki S., Yanagida T., Nunome K., Ishikawa S., Inden M., Kitamura Y., Nakagawa S., Taira T., Hirota K., Niwa M., Iguchi-Ariga S. M., Ariga H. (2008) DJ-1-binding compounds prevent oxidative stress-induced cell death and movement defect in Parkinson's disease model rats. J. Neurochem. 105, 2418–2434 [DOI] [PubMed] [Google Scholar]