

Abstract
Degradable cationic polymers are desirable for in vivo nucleic acid delivery because they offer significantly decreased toxicity over non-degradable counterparts. Peptide linkers provide chemical stability and high specificity for particular endopeptidases but have not been extensively studied for nucleic acid delivery applications. In this work, enzymatically degradable peptide-HPMA copolymers were synthesized by RAFT polymerization of HPMA with methacrylated peptide macromonomers, resulting in polymers with low polydispersity and near quantitative incorporation of peptides. Three peptide-HPMA copolymers were evaluated: (i) pHCathK10, containing peptides composed of the linker phe-lys-phe-leu (FKFL), a substrate of the endosomal/lysosomal endopeptidase cathepsin B, connected to oligo-(l)-lysine for nucleic acid binding, (ii) pHCath(d)K10, containing the FKFL linker with oligo-(d)-lysine, and (iii) pH(d)Cath(d)K10, containing all (d) amino acids. Cathepsin B degraded copolymers pHCathK10 and pHCath(d)K10 within one hour while no degradation of pH(d)Cath(d)K10 was observed. Polyplexes formed with pHCathK10 copolymers show DNA release by 4 hrs of treatment with cathepsin B; comparatively, polyplexes formed with pHCath(d)K10 and pH(d)Cath(d)K10 show no DNA release within 8 hrs. Transfection efficiency in HeLa and NIH/3T3 cells were comparable between the copolymers but pHCathK10 was less toxic. This work demonstrates the successful application of peptide linkers for degradable cationic polymers and DNA release.
Keywords: non-viral gene delivery, cathepsin B, RAFT polymerization
1. Introduction
Polycations are frequently used as nucleic acid delivery materials because of their ability to package nucleic acids and promote cellular uptake [1]. However, polycation toxicity is frequently a limiting factor for in vivo application, thus impeding clinical translation [2–4]. Both nucleic acid delivery efficiency and cytotoxicity have been correlated with increasing molecular weight for several polycations [5, 6]. Higher molecular weight polycations provide stronger binding to nucleic acids, resulting in improved stability in serum and circulation time after systemic delivery [7–9]. Lower molecular weight polycations are more readily displaced from their nucleic acid cargo; this release is necessary for biological activity [10]. Lower MW materials are also less toxic to cells when compared to their higher molecular weight counterparts [11, 12]. Biodegradable polycations with controllable degradation have been of great interest to the nucleic acid delivery field because these materials can provide combine advantages of both high and low molecular weight polycations.
The most extensively studied degradable cationic polymers have utilized either reducible or hydrolysable bonds [13–16], but these approaches provide limited control over the in vivo site of degradation. For example, poly(beta-amino esters) are degraded more quickly at neutral pH than acidic pH so that delivery vehicles would be susceptible to degradation once reconstituted in aqueous solution and administered in vivo [16]. Reducible linkers aim to take advantage of the highly reducing cytosolic environment for controlled intracellular degradation [17], but extracellular reduction has been implicated as the primary site of degradation for some polymeric systems [18]. We recently synthesized reducible HPMA-co-oligolysine copolymers by including disulfide linkers between the HPMA backbone and oligolysine pendant peptides [19]. Although reduced cytotoxicity was observed with these materials, transfection efficiency was also significantly decreased. This was partially attributed to the instability of the disulfide bond by sulfhydryl exchange and metal-catalyzed oxidation of the free sulfhydryl groups leading to crosslinking between polymer strands.
A controlled release material with polymer degradation triggered by specific intracellular enzymatic activity is highly attractive for gene delivery applications. Enzyme-catalyzed degradation combines chemical stability and high specificity. Peptide-based linkers act as selective substrates for specific extra- or intracellular peptidases, with enzymatic specificity determined by the amino acid sequence. In addition, linkers have varying rates of enzymatic cleavage, adjustable by changing the recognition sequence or neighboring residues [20, 21], further enabling the control of degradation. Peptide linkers have been used successfully for other biological applications, such as antibody-drug conjugates for cancer therapy [22] and degradable cross-linked scaffolds for tissue engineering [23], but have not been extensively studied for nucleic acid delivery applications. Two groups recently reported enzymatically-cleavable linkers for release of targeting ligands [24, 25], but, to our knowledge, no reports using peptide linkers in main or side-chain architectures in gene delivery materials as an approach for specific polymer degradation are available.
In nucleic acid delivery applications, endosomal degradation of carriers has several advantages. Materials need to remain intact in the extracellular environment to protect nucleic acids from nucleases present in serum. However, polycations such as polyethylenimine (PEI) and poly-(l)-lysine (PLL) have been shown to trigger mitochondrial-mediated apoptosis after polymer interaction with the outer mitochondrial membrane [26]. Polymers that degrade prior to cytosolic release are therefore hypothesized to be less cytotoxic than non-degraded polymers. Cathepsin B is a papain-like cysteine protease with both endopeptidase and exopeptidase activity that is involved in protein degradation and turnover in cells [10]. Cathepsin B functions primarily in the endo/lysosomal compartments [27–29] and has therefore been the target for enzyme-triggered, intracellular drug delivery [30–32]. We therefore designed cationic, peptide-based polymers for nucleic acid delivery with cathepsin B linker substrates for specific endosomal degradation as a means to reduce cytotoxicity through enzyme-mediated degradation.
In this work, peptide-HPMA copolymers were synthesized via reverse addition-fragmentation chain transfer (RAFT) polymerization of HPMA with methacrylamido-peptide monomers. The peptide monomers contained an oligolysine motif that was capped with a short, four amino acid cathepsin B substrate, thereby enabling endosomal degradation of the cationic copolymers. In total, three peptide-HPMA copolymers were evaluated: (i) pHCathK10, containing peptides composed of the linker phe-lys-phe-leu (FKFL), a substrate of cathepsin B, connected to oligo-(l)-lysine for nucleic acid binding; (ii) pHCath(d)K10, containing the FKFL linker with oligo-(d)-lysine; and (iii) pH(d)Cath(d)K10, containing all (d) amino acids. These polymers were evaluated for sensitivity to cathepsin B degradation, polyplex destabilization and DNA release, cytotoxicity and transfection efficiency.
2. Materials and Methods
2.1 Materials
N-(2-hydroxypropyl)methacrylamide (HPMA) was purchased from Polysciences (Warrington, PA). The initiator VA-044 was purchased from Wako Chemicals (Richmond, VA). Fmoc-protected amino acids and HBTU were purchased from AAPPTec (Louisville, KY), N-succinimidyl methacrylate from TCI America (Portland, Oregon), and Rink Amide Resin from EMD Biosciences (Darmstad, Germany). Human liver cathepsin B was purchased from Enzo Life Sciences (Plymouth Meeting, PA). All other materials were reagent grade or better and were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Endotoxin-free plasmid pCMV-Luc2 was prepared using the Qiagen Plasmid Giga kit (Qiagen, Hilden, Germany) according to manufacturer's recommendations.
2.2 Synthesis and Enzymatic Evaluation of Peptide Monomers
2.2.1 Synthesis of Peptide Monomers
Three peptides were synthesized using (d) and (l) amino acids and 6-aminohexanoic acid (Ahx): AhxFKFLAhxK10 (composed of only (l) amino acids); AhxFKFLAhx(d)K10 (composed of (l) amino acid linker with oligo-(d)-lysine); and Ahx(d)FKFLAhx(d)K10 (composed of only (d) amino acids). Peptides were synthesized on a solid support with Rink amide linker following standard Fmoc chemistry on an automated PS3 peptide synthesizer (Protein Technologies, Phoenix, AZ). Prior to peptide cleavage from the resin, the amino termini of the peptides were deprotected and coupled with N-succinimidyl methacrylate. These functionalized peptide monomers are respectively called MaAhxFKFLAhxK10, MaAhxFKFLAhx(d)K10, and MaAhx(d)FKFLAhx(d)K10. Synthesized peptides were cleaved from resin by treatment of solid support with a solution of TFA/triisopropylsilane (TIPS)/1,3-dimethoxybenzene (92.5:2.5:5, v/v/v) for 2.5 hours under gentle mixing. Cleaved peptide monomers were precipitated in cold ether, dissolved in methanol and re-precipitated twice in cold ether. Each peptide monomer was analyzed by RP-HPLC and MALDI-TOF MS and shown to have greater than 95% purity after cleavage. The expected molecular weight of the peptides was 2128.82 Da. Experimentally measured molecular weights determined by MALDI-TOF MS were 2129.137 Da, 2128.526 Da, and 2128.689 Da for MaAhxFKFLAhxK10, MaAhxFKFLAhx(d)K10, and MaAhx(d)FKFLAhx(d)K10, respectively.
2.2.2 Cathepsin B Cleavage of Peptide Monomers
Specific cleavage at the FKFL linker by cathepsin B was determined by adapting a method from Dubowchik et al [20]. For this and subsequent cathepsin B cleavage studies, human liver cathepsin B (0.351 mg/mL stock) was added to activation buffer (30 mM DTT, 15 mM EDTA) for a final concentration of 0.117 mg/mL cathepsin B, 20 mM DTT, 10 mM EDTA and incubated at 37 °C for 15 min. Reaction buffer (25 mM acetate, 1 mM EDTA, pH 5, pre-warmed at 37 °C) and peptide solution (10 mM stock) were added to the enzyme solution such that the final concentration of the reaction solution was 1.28 μg/mL cathepsin B, 24.6 mM acetate, 1.1 mM EDTA, 0.33 mM DTT, 65 μM peptide, and the reaction solution was incubated at 37 °C. Aliquots were removed at various time points and enzymatic activity stopped by addition of thioprotease inhibitor E-64 (Thermo Scientific, Waltham, MA) to a 26 μg/mL final concentration. Aliquots were analyzed qualitatively via MALDI-TOF MS.
2.2.3 Serum Stability of Peptide Macromonomers
Linker susceptibility to serum proteases was evaluated using freshly isolated mouse serum. For each peptide, peptide (5 μL of 10 mM stock) was added to serum (120 μL, pre-incubated at 37 °C). At various time points, 20 μl aliquots of the mixture were withdrawn and 5 μL of ice-cold 15% trichloroacetic acid (w/v) was added to stop enzymatic degradation via protein precipitation. Precipitated solutions were centrifuged at 13000 rpm for 15 min and supernatants were analyzed by MALDI-TOF MS.
2.3 RAFT Polymerization and Characterization of HPMA-peptide Copolymers
2.3.1. Polymer Synthesis
Three polymers were synthesized: HPMA-co-MaAhxFKFLAhxK10 (pHCathK10), HPMA-co-MaAhxFKFLAhx(d)K10 (pHCath(d)K10), and HPMA-co-MaAhx(d)FKFLAhx(d)K10 (pH(d)Cath(d)K10). Each polymer was synthesized with target degree of polymerization (DP) of 190 with 20% mole feed peptide to yield polymers with target molecular weight of approximately 103 kDa. Monomers were dissolved in acetate buffer (1 M, pH 5.1) such that the final monomer concentration of the solution was 0.7 M. The RAFT chain transfer agent (CTA) used was ethyl cyanovaleric trithiocarbonate (ECT, molecular weight 263.4 g/mol) and the initiator (I) used was VA-044. The molar ratios of total monomer:CTA:I at the start of polymerization were 190:1:0.1. The reaction solutions were transferred to round bottom flasks, capped with a rubber septum, purged with argon for 10 min, and the submerged in a 44 °C oil bath to initiate polymerization. The polymerization was allowed to proceed for 48 hrs. The flasks were removed from the oil bath and polymers dialyzed against distilled H2O to removed unreacted monomers and buffer salts. The dialyzed polymers were lyophilized dry.
2.3.2. Size Exclusion Chromatography
Molecular weight analysis was carried out by size exclusion chromatography. The copolymers were dissolved at 10 mg/mL in running buffer (150 mM acetate buffer, pH 4.4) for analysis by size exclusion chromatography-multiangle light scattering (SEC-MALS). Analysis was carried out on an OHpak SB-804 HQ column (Shodex) in line with a miniDAWN TREOS multiangle light scattering detector (Wyatt, Santa Barbara, CA) and an OptiLab rEX refractive index detector (Wyatt). Absolute molecular weight averages (Mn, Mw) were calculated using ASTRA software (Wyatt).
2.3.3 Amino Acid Analysis
The actual incorporated amount of peptide and HPMA in the final copolymers was determined through modified amino acid analysis following the method of Bidlingmeyer and coworkers [33]. Briefly, hydrolyzed lysine and HPMA (which results in 1-amino-2-propanol) were derivatized with o-phthalaldehyde/β-mercaptopropionic acid and run on a Poroshell 120 EC-C18 (Agilent Technologies, Santa Clara, CA) HPLC column with pre-column derivatization to label hydrolyzed lysine and 1-amino-2-propanol. Calibration curves were generated using serial dilutions of (l)-lysine and reagent grade 1-amino-2-propanol.
2.4 Cathepsin B-mediated Polymer Degradation
Linker recognition and subsequent release of oligolysine side-chains from the polymer were assessed by enzymatic treatment with cathepsin B. For each polymer, cathepsin B reactions were set up as described previously with a final polymer concentration of 44 μg/mL. At various time points, aliquots were removed and E-64 was added to stop the enzymatic reaction. Solutions were analyzed by SEC and the relative differential refractive index profiles were compared. MALDI-TOF MS was also performed.
2.5 Polyplex Characterization and Enzymatic Susceptibility
2.5.1 Polyplex Formation
pCMV-Luc2 plasmid was diluted in ddH2O to a concentration of 0.1 mg/mL and mixed with an equal volume of polymer (in ddH2O water) at the desired amine to phosphate (N:P) ratio. After mixing, polyplexes were allowed for form for 10 minutes at room temperature. The charge densities of the oligolysine-co-HPMA copolymers, PEI, and PLL are approximately 290 mg/mmol amine, 43 mg/mmol amine, and 208 mg/mmol amine, respectively.
2.5.2 Sizing of Polyplexes by Dynamic Light Scattering (DLS)
Polyplexes (0.5 μg DNA, 10 μL) were formed with pHCathK10, pHCath(d)K10, pH(d)Cath(d)K10, branched polyethylenimine (PEI, 25 kDa, PDI ~2.5) or poly-(l)-lysine (PLL, 15–30 kDa, PDI 1.2) at 3, 4, and 5 N:P ratios and were mixed with either 90 μL of ddH2O or PBS such that the final salt concentration was 150 mM. Particle size was determined by dynamic light scattering (ZetaPLUS, Brookhaven Instruments Corp, Holtsville, NY).
2.5.3 Polyplex Destabilization by DLS
Polyplexes were formulated at 4 N:P with pHCathK10, pHCath(d)K10, or pH(d)Cath(d)K10. The DLS instrument and cuvette with cap were pre-equilibrated at 37 °C. For each polyplex solution, cathepsin B reactions were set up as described previously with a final concentration of DNA of 5.33 μg/mL. Reaction solutions were transferred to the cuvette and capped to prevent evaporation. Enzymatic reaction was allowed to proceed for 8 hrs at 37 °C with particle size measured every 15 min.
2.5.4 Polyplex Unpackaging by YOYO-1 Fluorescence Quenching Assay
pCMV-Luc2 plasmid was mixed with the bis-intercalating dye YOYO-1 iodide (Invitrogen, Carlsbad, CA) at a dye/base pair ratio of 1:50 and incubated at room temperature for 1 hour. Polyplexes were formed at 4 N:P by complexing YOYO-1-labeled DNA with pHCathK10, pHCath(d)K10, and pH(d)Cath(d)K10. For each polyplex solution, cathepsin B reactions were set up as described previously with a final concentration of DNA at 5.33 μg/mL. At various time points, aliquots were removed and reaction stopped with addition of E-64, diluted to 100 μL total volume with ddH2O, and transferred to a 96-well plate. After the final time point, the fluorescence from each well of all the plates was measured on a Tecan Safire2 plate reader (Männerdorf, Switzerland) with excitation at 491 nm and emission at 509 nm. The fluorescence signal for each time point was normalized against a plasmid-only signal.
2.6 In vitro Transfection Efficiency
HeLa and NIH/3T3 cells were transfected as previously described [34]. Briefly, polyplexes (1 μg DNA) were formed at 3, 4, and 5 N:P, diluted to 200 uL in MEM Reduced Serum Medium (Hyclone), and added to cells for 4 hrs. After an additional 44 hrs, luciferase expression was quantified using a luciferase assay kit (Promega, Fitchburg, WI) according to the manufacturer's instructions, except with an additional freeze-thaw cycle at −20 °C to ensure complete cell lysis. Luminescence intensity was measured on the plate reader with 1 sec integration; total protein content was measured using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions so luciferase activity could be normalized to the total protein content. Each sample was tested in quadruplicate.
2.7 Cytotoxicity of Polymers and Polyplexes
The cytotoxicity of the polymers and polyplexes was evaluated in vitro using the MTS assay as described previously [34]. Polymers of various concentrations or polyplexes at 3, 4, and 5 N:P (0.2 μg DNA) were prepared in ddH2O water and then diluted 10-fold in MEM Reduced Serum Medium. The cells were rinsed once with PBS and incubated with 40 μL of the polymer or polyplex solution for 4 hrs. At 48 hrs, 20 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (Promega) was added to each well. Cells were then incubated for 3 hrs and absorbance measured at 490 nm using a plate reader. For polymers, IC50 values were determined using a nonlinear fit (four-parameter variable slope) in GraphPad Prism v.5 (San Diego, CA). For polyplexes, cellular viability was normalized against untreated cells.
Results
3.1 Enzymatic Degradation of Peptides by Cathepsin B and Mouse Serum
Methacrylated oligolysine (K10) peptides containing the FKFL cathepsin B substrate sequence flanked on each side by a six-carbon spacer were synthesized by solid phase peptide synthesis. Three peptides were synthesized, differing only in the use of d- versus l-amino acids to impart peptidase resistance; thus, all full length peptides have MW 2128 Da. Cathepsin B-mediated peptide cleavage at the FKFL linker was monitored over 30 min enzyme incubation (Figure 1). Rapid recognition and specific cleavage of the MaAhxFKFLAhxK10 and MaAhxFKFLAhx(d)K10 peptides at the FKFL linker results in disappearance of the 2128 Da peak and simultaneous emergence of a 1673 Da peak corresponding to the FLAhxK10/ FLAhx(d)K10 fragment, respectively. Fragmentation of the original peptide is complete within 15 min of enzymatic exposure. The FLAhxK10 peptide fragment is susceptible to exopeptidase degradation as well, with the systematic removal of C-terminal lysines over time; however, this process is considerably slower than the endopeptidase cleavage at the linker. The FLAhx(d)K10 fragment shows resistance to exopeptidase activity. The MaAhx(d)FKFLAhx(d)K10 peptide has no observable degradation within the 30 min of cathepsin B treatment.
Figure 1.

MALDI-TOF MS time-point study of cathepsin B-mediated fragmentation of (a) MaAhxFKFLAhxK10, (b) MaAhxFKFLAhx(d)K)10, and (c) MaAhx(d)FKFLAhx(DK10
Extracellular enzymatic stability of the peptide and specificity of the FKFL linker for intracellular peptidases was tested by exposing peptides to freshly isolated mouse serum and monitoring degradation over 4 hrs. No endopeptidase cleavage of the FKFL linker was detected but degradation of the MaAhxFKFLAhxK10 peptide by non-specific exopeptidase hydrolysis was observed by 1 hr post-incubation (Figure 2a), yielding the shortened fragment MaAhxFKFLAhxK (976 Da). In contrast, intact MaAhxFKFLAhx(D)K10 peptide is observed even after serum incubation for 4 hours. Some endopeptidase degraded product (1672 Da = FLAhx(D)K10) is observed at the 2 and 4 hr time points (Figure 2b). No FLAhxK10 fragments are observed in 2a, likely due to rapid exopeptidase degradation by the first 30 min time point. Peaks at 656 and 1796 Da correspond to matrix and a peptide found in serum, respectively.
Figure 2.

MALDI-TOF MS time-point study of mouse serum enzymatic digest of (a) MaAhxFKFLAhxK10, (b) MaAhxFKFLAhx(d)K10
3.2 Enzymatic Degradation of HPMA-co-oligolysine Copolymers by Cathepsin B
Three HPMA-oligolysine copolymers were synthesized by RAFT polymerization of peptide monomers with HPMA as described previously [34, 35]. The synthesized polymers displayed properties close to targeted values as summarized by Table 1. In general, the copolymers had Mn ~ 5–15% lower than expected. Polydispersity of all the copolymers was below 1.2. Amino acid analysis of the copolymers revealed peptide mole incorporation around 25–27%.
Table 1.
Properties of HPMA-oligolysine copolymers
| HPMA-oligolysine Copolymer | Peptide Monomer | Degradation Type | Targeted Mn (kDa)a | Determined Mn (kDa)b | Mw/Mnb | Mole% Peptide Monomerc |
|---|---|---|---|---|---|---|
| pHCathK10 | MaAhxFKPLAhxK10 | Endo/exopeptidase | 102.63 | 96 64 | 1.17 | 27.4 |
| pHCath(d)K10 | MaAhxFKPLAhx(d)K10 | Exopeptidase only | 102.63 | 93.26 | 1.17 | 24.8 |
| pH(d)Cath(d)K10 | MaAhx(d)FKFLAhx(d)K10 | None | 102.63 | 88.57 | 1.18 | 26.0 |
Based on Mn=[Mo]/[CTAo] × conversion × FW with no counterions.
Determined by SEC-MALS.
Determined by amino acid analysis.
Cathepsin-B mediated degradation of HPMA-oligolysine copolymers was evaluated by treating polymers with cathepsin B and analyzing polymers at various time points by size exclusion chromatography (Figure 3). Un-degraded polymers elute from the column ~13 min. Degradation of pHCathK10 and pHCath(D)K10 occurs within 10 min and continues up to 1 hr, as evidenced by the increased elution time of lower molecular weight polymers (Figures 3a and 3b). A secondary peak simultaneously emerges, eluting around 20 min for both copolymers. This secondary peak is attributed to released peptide after endopeptidase cleavage of the FKFL linker (FLAhxK10/FLAhx(D)K10 and respective C-terminally shortened fragments). This is supported by the shifting profile of the secondary peak in the pHCathK10 due to exopeptidase activity on the FLAhxK10 fragment compared to a stable profile in exopeptidase-resistant pHCath(D)K10. This is also confirmed by MALDI-TOF MS on the reaction mixture (Supplementary Information, Figure 1). pH(D)Cath(D)K10 shows no degradation over the two hours of enzymatic treatment due to its resistance to peptidases (Figure 3c).
Figure 3.
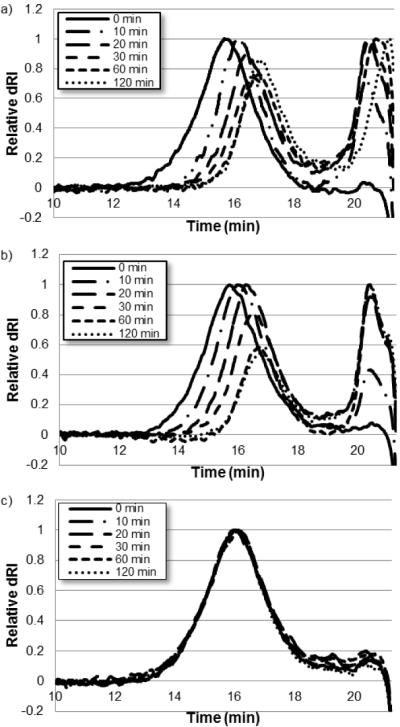
Overlaid size exclusion chromatography of cathepsin B degradation of (a) pHCathK10, (b) pHCath(d)K10, and (c) pH(d)Cath(d)K10.
3.3 Polyplex Sizing and Colloidal Stability
The HPMA-oligolysine copolymers were complexed with plasmid DNA at 3, 4, and 5 N:P ratios to form nanoparticles called “polyplexes.” The average hydrodynamic diameters of polyplexes in both water and 150 mM PBS were determined by dynamic light scattering. All the HPMA-oligolysine copolymers formed polyplexes with average hydrodynamic diameter of 120 nm water (Figure 4a). PEI and PLL also formed slightly smaller particles around 100 nm in diameter. In PBS, HPMA-oligolysine polyplexes remain relatively stable. The average diameter of polyplexes formed at 5 N:P increased only by 25% to 150 nm. In general, increases in size are seen with decreasing N:P ratios for all materials. In contrast, PEI and PLL particles aggregate in PBS, as indicated by the increased average diameter ranging from 500 nm to over 1 micron.
Figure 4.

DLS sizing of polyplexes in (a) ddH2O and (b) 150mM PBS
3.4 Cathepsin B-mediated Polyplex Destabilization and Unpackaging
The effects of cathepsin B exposure to pHCathK10, pHCath(D)K10, and pH(D)Cath(D)K10 polyplexes were determined by monitoring particle size by DLS and DNA condensation by dye quenching assay. Polyplexes were formulated at 4 N:P and particle size measured every 15 min for 8 hrs of cathepsin B treatment (Figure 5a). Polyplexes formed with pHCathK10 show particle size increases from ~150 nm to about 500 nm over 4hrs before plateauing. pHCath(D)K10 shows size increases over 8 hrs up to ~600 nm in diameter. In contrast, pH(D)Cath(D)K10 maintain particles ~150 nm in diameter with no observable size change over 8 hrs.
Figure 5.

Polyplex destabilization and unpackaging of pHCathK10/DNA, pHCath(d)K10/DNA, and pH(d)Cath(d)K10/DNA polyplexes as measured by (a) dynamic light scattering and (b) YOYO-1 fluorescence quenching assay.
To monitor DNA condensation, plasmid DNA was labeled with the DNA-intercalating fluorophore YOYO-1 before complexation with polymer at 4 N:P. Plasmid condensation results in self-quenching of the YOYO-1 fluorescence due to electronic interactions between nearby YOYO-1 molecules [13]. The YOYO-1 fluorescence, normalized to that of uncomplexed plasmid, is shown in Figure 5b as a function of time of cathepsin B exposure. For the first 3 hrs, all polyplex solutions have fluorescence around 10% of free plasmid indicating efficient condensation; starting around 4 hrs, the pHCathK10 polyplexes begin to show a trend of increasing fluorescence, up to 70% by 8 hrs. In contrast, pHCath(D)K10 and pH(D)Cath(D)K10 did not show any significant increase in fluorescence (unpackaging) over 8 hrs.
3.5 Plasmid DNA Delivery
Transfection efficiency of the polyplexes formed at 3, 4, and 5 N:P was tested using both HeLa and NIH/3T3 cells. The luciferase reporter transgene was used to assess gene delivery efficiency with cytotoxicity evaluated by BCA protein assay (Figure 6). In general, the HPMA-oligolysine copolymers transfected with higher efficiency than PLL over all N:P ratios. Efficiencies and trends were cell-line dependent. HeLa cells showed no significant difference in normalized transfection between the 3, 4, and 5 N:P ratios for the copolymers while PEI shows trends of increasing efficiency with increasing N:P ratio; however, copolymer materials are still within an order of magnitude less effective than PEI in transfection efficiency for N:P ratios of 4 and 5. For NIH/3T3 cells, transfection efficiencies of HPMA-oligolysine copolymers were higher with increasing N:P ratios. pHCath(D)K10 showed the best transfection of the three copolymers, with normalized luciferase expression comparable to PEI.
Figure 6.

Normalized luminescence per mg protein as a measure for transfection efficiency in (a) HeLa and (b) NIH/3T3 cells. Protein expression normalized to untreated cells for (c) HeLa and (d) NIH/3T3 cells.
3.6 Polymer Toxicity
Cytotoxicity of polymers and polyplexes was determined by MTS and BCA assay, respectively. MTS was used to determine the mitochondrial activity in HeLa and NIH/3T3 cells that had been treated with the different polymers. A range of polymer concentrations was evaluated in order to determine the IC50 value (concentration of polymer for 50% cell survival) for each polymer in amine equivalents. The three synthesized polymers had essentially indistinguishable IC50 values with overlapping confidence intervals. Determined IC50 values were higher in general for NIH/3T3 cells than HeLa cells and HPMA-oligolysine copolymers were less toxic than both PEI and PLL (Table 2).
Table 2.
Polymer Toxicity
| HeLa | NIH/3T3 | |||
|---|---|---|---|---|
| Polymer | IC50 (μg/mL) | IC50 (μM 1° N) | IC50 (μg/mL) | IC50 (μM 1° N) |
| pHCathK10 | 16.2 | 53 | 29.7 | 97.3 |
| pHCath(d)K10 | 16.3 | 53.5 | 28.7 | 94 |
| pH(d)Cath(d)K10 | 14.3 | 46.7 | 30.3 | 99.3 |
| PEI | 4.5 | 26.4 | 8.3 | 48.3 |
| PLL | 7.2 | 34.4 | 17.9 | 86.1 |
The BCA assay was conducted to determine the total amount of cellular protein in lysates of transfected cells 48 hrs after transfection. Cell viability was estimated by comparing the protein levels of transfected cells with control untreated cells. Generally, a N:P of 3 was nontoxic in all polymers screened, including PLL and PEI. Toxicity increased at higher N:P ratios for the HPMA-oligolysine polymers and PLL. A trend of decreased toxicity in the pHCathK10 polyplexes compared to pHCath(D)K10 and pH(D)Cath(D)K10 polyplexes for all N:P ratios in both cell types was observed (p < 0.01). This trend is confirmed in HeLa cells treated with polyplexes via the MTS assay (Figure 7).
Figure 7.
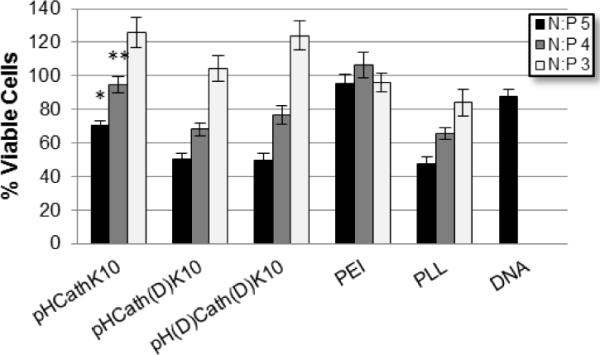
Cellular viability of HeLa cells treated with polyplexes measured via MTS assay. pHCathK10 are significantly less toxic than pHCath(d)K10, pH(d)Cath(d)K10, and PLL at *N:P 5 and **N:P 4 (p < 0.01).
4. Discussion
In previous work, we reported the synthesis of pendant peptide copolymers with HPMA synthesized by living polymerization and their application for nucleic acid delivery [34, 35]. RAFT polymerization of this class of materials resulted in better controlled peptide incorporation, lower polydispersity and decreased cytotoxicity compared to similar polymers prepared by free radical polymerization [19, 36]. In previous work, we found optimal polymers contained 20 mole percent K10 with transfection efficiency of these polymers in cultured cells similar to that of branched PEI [34]. The goal of this work is to develop peptide-HPMA materials that undergo triggered endosomal-degradation and cargo release in response to specific enzymatic activity. Our strategy is to include an enzymatically cleavable linker between the peptide and the polymer HPMA backbone. Because peptide content in these polymers is high, complete cleavage would result in a 60% reduction in polymer molecular weight and the release of short oligolysine peptides from an HPMA-based polymer backbone.
Cathepsin B was selected as the targeted enzyme for its endo-lysosomal activity. With a papain-like structure, cathepsin B has two domains with the active site cleft along the interface [37]. The peptidyl substrate amino acids are conventionally named … P2-P1-P1'-P2'…, where the peptide bond is cleaved between P1 and P1' [38]. In this study, the sequence FKFL, where F=P2, K=P1, F=P1' and L=P2', was used as the cathepsin B substrate linkage. Several studies have systematically evaluated various amino acids to optimize the substrate sequence for cathepsin B. These studies show that a phenylalanine in P2 gives the highest cathepsin B hydrolysis rate (kcat/Km) [39–41]. Arginine is the most preferred amino acid for the P1 position, although a lysine in this position is also accepted with about 50% relative activity compared to arginine [39, 40]. Dubowchik and Firestone utilized a lysine at P1 in their immunoconjugate BR96-Dox because of potential improved systemic stability compared to arginine [20, 42]. Based on this rationale, a lysine at P1 was also used for this work. Initially a peptide was designed with lysines in the P1' and P2' positions (peptide MaAhxFK10) but no specific endopeptidase cleavage was observed with this substrate (data not shown). Because of reports showing that hydrophobic amino acids are preferred in both the P1' and P2' positions, the amino acids F and L were used, respectively. Phenylalanine was used at the P1' position due to reports showing optimal cathepsin B cleavage in sequences with this amino acid at P1' [40, 43]. Finally, a six carbon linker (6-aminohexanoic acid) was included flanking both sides of the FKFL to provide spacers for the cathepsin substrate sequence both from the polymer backbone and the cationic K10 nucleic acid binding domain in attempt to provide better access to cathepsin B. The resulting peptide, MaAhxFKFLAhxK10, was shown to be recognized by cathepsin B and cleaved within 15 min (Figure 1).
Cathepsin B exhibits both specific endopeptidase and exopeptidase activity, with the former dominating at pH 6–7 and the latter optimal at pH < 5.0 [44]. Three peptides were therefore synthesized: MaAhxFKFLAhxK10, which is susceptible to both endopeptidase and exopeptidase activity; MaAhxFKFLAhx(D)K10, which is susceptible only to endopeptidase activity; and MaAhx(D)FKFLAhx(D)K10, which is protease resistant. Sensitivity of these peptides to cathepsin B-mediated proteolysis was evaluated at pH 5.0. This pH was selected because it is representative of the late endosome milieu where both endopeptidase and exopeptidase activity would be expected [45]. At this pH, the endopeptidase activity is about 15% of optimum activity at pH ~6.2 while exopeptidase activity is about 80% of optimal activity. Degradation of the three peptides by cathepsin B followed expected patt erns with faster kinetics for endopeptidase activity compared to exopeptidase activity (Figure 1).
Ideally, the peptide-based polymers would remain stable during circulation to protect nucleic acids from serum nucleases. In addition to nucleases, serum contains many peptidases, such as dipeptidyl peptidases and endopeptidases [46–49]. To assess serum stability, peptides were incubated with freshly isolated mouse serum (Figure 2). Degradation of MaAhxFKFLAhxK10 was complete by 1 hr, resulting in the shortened MaAhxFKFLAhxK fragment. In contrast, full length MaAhxFKFLAhx(D)K10 is detected even after a 4 hr serum incubation, although some endopeptidase degraded product (1672 Da = FLAhx(D)K10) is observed after 1 hr. It should be noted that in the presence of cathepsin B, endopeptidase degradation of this peptide is observed within 5 min. The analogous FLAhxK10 fragment from MaAhxFKFLAhxK10 likely is rapidly degraded by carboxypeptidases and thus is not detected on MALDI. The MaAhxFKFLAhx(D)K10 peptide is a promising building block for polymers used in in vivo applications requiring specific intracellular degradation. While stability in murine serum may not correlate to human serum, high homology in the degradomes (> 80% strict orthologues) between mice and humans suggests information regarding enzymatic substrate specificity may be gained from these serum studies [50].
Copolymerization of the methacrylated peptides with HPMA by RAFT polymerization yielded polymers with narrow polydispersity, expected composition, and near-target molecular weights (Table 1). Cathepsin B-mediated degradation of polymers was evaluated at pH 5.0 by size exclusion chromatography (Figure 3). Accessibility of cathepsin B to linker sites incorporated in dendrimers has been shown to be restricted due to steric hindrance for some systems [51]. However, synthesized pHCathK10 and pHCath(D)K10 polymers susceptible to cathepsin B cleavage show rapid reduction in size within 10 min and complete cleavage of oligolysine side chains from the polymer backbone within one hour, leading to a theoretical reduction of ~60% in molecular weight. The kinetics of proteolysis for the peptide and polymers are similar as seen by comparing peptide monomer (Figure 1) and copolymer (Supplementary Figure 1) fragmentation. In comparison, pH(D)Cath(D)K10 shows complete enzymatic resistance. HPMA-co-MaAhxK10 copolymers were also treated with cathepsin B and the observed MW shift over 4hrs is comparable to 10 min with pHCathK10 (data not shown). In addition to rapid copolymer size reduction, labile side-chains can potentially increase accessibility of peptide side-chains for other peptidases; this is observed in the peptide peak (~20 min) released by degradation of pHCathK10 but not pHCath(D)K10 (Figures 3a and 3b).
All polymers efficiently complexed DNA and formed polyplexes with diameters ~100–200 nm that are stable in physiological salt concentrations (Figure 4). While poly-(l)-lysine-based polyplexes are not stable in 150 mM PBS, inclusion of HPMA in the backbone of the cationic polymer prevents salt-induced aggregation [34, 36, 52]. Controlled polyplex unpackaging and DNA release after cellular internalization is integral to efficient gene delivery and expression [10]. Effective delivery requires extracellular and endosomal stability with cytosolic unpackaging for eventual expression. Cathepsin B-mediated unpackaging of polyplexes formed from pHCathK10, pHCath(D)K10 and pH(D)Cath(D)K10 was studied by dynamic light scattering and YOYO-1 fluorescence quenching assay. pHCathK10 and pHCath(D)K10 polyplexes both show increasing particle size over time while pH(D)Cath(D)K10 particles remain unchanged, suggesting enzymatically-driven polyplex destabilization. DNA packaging was also assessed through YOYO-1 fluorescence quenching assay, which monitors DNA decondensation as YOYO-1 fluorescence is restored when these molecules are not in close proximity to each other. Decondensation of DNA from polyplexes is only observed with pHCathK10 and not pHCath(D)K10 or pHK10 (data not shown). Oligolysines as short as 8-mers are able to condense DNA [24, 53], so despite cleavage of oligolysine side chains from the HPMA backbone, oligolysine fragments may remain able to form relatively large and unstable, but intact, polyplexes. Since oligo-(D)-lysine is resistant to further degradation, it will remain bound to DNA thereby keeping it packaged; in contrast, oligo-(l)-lysine, subject to exopeptidase degradation, will eventually release DNA upon further degradation. This is supported by the observation that YOYO-1 fluorescence from polyplexes formed by oligo-(l)-lysine increases with time when incubated with cathepsin B but polyplexes formed by oligo-(D)-lysine do not (data not shown).
While reducible HPMA-oligolysine copolymers have significantly lower transfection efficiency compared to their non-degradable counterparts [19], the enzymatically-cleavable polymers described here (pHCathK10 and pHCath(D)K10) have similar transfection efficiencies to non-degradable pH(D)Cath(D)K10 in cultured cells (Figures 6a and 6b). Endosomal/lysosomal unpackaging may not increase transfection without compartmental escape for cytosolic release. Transfection was also evaluated in serum conditions (data not shown). Reduced transfection efficiency compared to serum-free transfections was observed, as reported previously for similar polymers [19]. No significant difference in transfection efficiency was noted between the three polymers even in serum conditions (data not shown), possibly because of altered protease activities in heat-inactivated serum. Cationic polymers have been implicated in toxicity through both necrotic and apoptotic routes [12, 26, 54, 55]. Electrostatic interactions with the plasma membrane cause permeabilization and pore formation, triggering cellular efflux of cytosolic contents and necrosis [54]. Compromised mitochondrial and lysosomal membranes lead to release of pro-apoptotic factors cytochrome c and cathepsins, respectively, initiating apoptotic cascades [56, 57]. Degradation of cationic polymers prior to cytosolic release or endo-lysosomal fusion can potentially decrease induction of apoptosis and reduce cytotoxicity. The relative toxicity pHCathK10, pHCath(D)K10 and pH(D)Cath(D)K10 was evaluated in HeLa and NIH/3T3 cells using the MTS assay (Table 2). Results suggest similar toxicity between the three synthesized polymers regardless of enzymatic susceptibility. One possible explanation is that for free polymer the primary route of toxicity might be through plasma membrane disruption and permeabilization as implicated for PEI [12]. Therefore, intracellularly-controlled degradation will not significantly affect toxicity. This hypothesis is supported by the observation that polyplexes formed with pHCathK10 are less toxic than pHCath(D)K10 and pH(D)Cath(D)K10 polyplexes at all tested charge ratios and in both HeLa and NIH/3T3 cells (Figures 6c and 6d, Figure 7). This result was confirmed both by protein content analysis and MTS assay of cells exposed to polyplexes. In the polyplex form, a reduced amount of free polymer is available to disrupt the cellular membrane and the effect of intracellular degradation on cytotoxicity may be more pronounced. pHCathK10 can be degraded additionally by exopeptidases, allowing for accelerated degradation and reduced toxicity compared to pHCath(D)K10. Still, substantial toxicity is still observed with the pHCathK10 polyplexes formed at higher N:P ratios. One potential approach that may further reduce toxicity of future generations of these polymers is to decrease the cationic charge density by including non-charged amino acids in the nucleic acid binding sequence. High charge density has been correlated with cytotoxicity for several cationic polymers [12, 58, 59].
Conclusions
In summary, peptide-based polycations that are susceptible to cathepsin B-catalyzed degradation were synthesized. The polymers were shown to undergo rapid degradation in the presence of cathepsin B. When packaged into nanoparticles by complexation with nucleic acids, linkers remain sufficiently available to fit into catalytic pockets of endopeptidases and be specifically degraded. Thus, we have demonstrated triggered nanoparticle degradation by a selective enzymatic mechanism.
Supplementary Material
Scheme 1.

Synthetic scheme of HPMA-co-oligolysine copolymers
Acknowledgments
This work was supported by NIH/NINDS 1R01 NS064404, NSF DMR 0706647, and the Center for Intracellular Delivery of Biologics (Life Sciences Discovery Fund Grant 2496490). We thank Profs. Anthony Convertine and Patrick Stayton for generous donation of the ECT chain transfer agent. We also thank Drs. Peter Senter and Svetlana Dororina of Seattle Genetics for their advice in linker development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- [2].Chollet P, Favrot MC, Hurbin A, Coll JL. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J. Gene Med. 2002;4:84–91. doi: 10.1002/jgm.237. [DOI] [PubMed] [Google Scholar]
- [3].Ward CM, Read ML, Seymour LW. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 2001;97:2221–2229. doi: 10.1182/blood.v97.8.2221. [DOI] [PubMed] [Google Scholar]
- [4].Burke RS, Pun SH. Extracellular barriers to in vivo PEI and PEGylated PEI polyplex-mediated gene delivery to the liver. Bioconjug. Chem. 2008;19:693–704. doi: 10.1021/bc700388u. [DOI] [PubMed] [Google Scholar]
- [5].Grigsby CL, Leong KW. Balancing protection and release of DNA: tools to address a bottleneck of nonviral gene delivery. J. R. Soc. Interface. 2010;7(Suppl 1):S67–82. doi: 10.1098/rsif.2009.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Luten J, van Nostrum CF, De Smedt SC, Hennink WE. Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J. Control. Release. 2008;126:97–110. doi: 10.1016/j.jconrel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- [7].Fischer D, Osburg B, Petersen H, Kissel T, Bickel U. Effect of poly(ethylene imine) molecular weight and pegylation on organ distribution and pharmacokinetics of polyplexes with oligodeoxynucleotides in mice. Drug Metab. Dispos. 2004;32:983–992. [PubMed] [Google Scholar]
- [8].Mullen PM, Lollo CP, Phan QC, Amini A, Banaszczyk MG, Fabrycki JM, Wu D, Carlo AT, Pezzoli P, Coffin CC, Carlo DJ. Strength of conjugate binding to plasmid DNA affects degradation rate and expression level in vivo. Biochim. Biophys. Acta. 2000;1523:103–110. doi: 10.1016/s0304-4165(00)00104-5. [DOI] [PubMed] [Google Scholar]
- [9].Nishikawa M, Takemura S, Takakura Y, Hashida M. Targeted delivery of plasmid DNA to hepatocytes in vivo: optimization of the pharmacokinetics of plasmid DNA/galactosylated poly(L-lysine) complexes by controlling their physicochemical properties. J. Pharmacol. Exp. Ther. 1998;287:408–415. [PubMed] [Google Scholar]
- [10].Schaffer DV, Fidelman NA, Dan N, Lauffenburger DA. Vector unpacking as a potential barrier for receptor-mediated polyplex gene delivery. Biotechnol. Bioeng. 2000;67:598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- [11].de Wolf HK, de Raad M, Snel C, van Steenbergen MJ, Fens MHAM, Storm G, Hennink WE. Biodegradable poly(2-dimethylamino ethylamino)phosphazene for in vivo gene delivery to tumor cells. Effect of polymer molecular weight. Pharm. Res. 2007;24:1572–1580. doi: 10.1007/s11095-007-9299-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24:1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- [13].Breunig M, Lungwitz U, Liebl R, Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. P. Natl. Acad. Sci. USA. 2007;104:14454–14459. doi: 10.1073/pnas.0703882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mintzer MA, Simanek EE. Nonviral Vectors for Gene Delivery. Chem. Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- [15].Meng FH, Hennink WE, Zhong Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials. 2009;30:2180–2198. doi: 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]
- [16].Lynn DM, Langer R. Degradable poly(beta-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. J. Am. Chem. Soc. 2000;122:10761–10768. [Google Scholar]
- [17].Meister A, Anderson ME. Glutathione. Annu. Rev. Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- [18].Sun WC, Davis PB. Reducible DNA nanoparticles enhance in vitro gene transfer via an extracellular mechanism. J. Control. Release. 2010;146:118–127. doi: 10.1016/j.jconrel.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shi J, Johnson RN, Schellinger JG, Carlson PM, Pun SH. Reducible HPMA-co-oligolysine copolymers for nucleic acid delivery. Int. J. Pharm. 2011 doi: 10.1016/j.ijpharm.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002;13:855–869. doi: 10.1021/bc025536j. [DOI] [PubMed] [Google Scholar]
- [21].Hortin GL, Warshawsky I, Laude-Sharp M. Macromolecular chromogenic substrates for measuring proteinase activity. Clin. Chem. 2001;47:215–222. [PubMed] [Google Scholar]
- [22].Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003;21:778–784. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- [23].Levesque SG, Shoichet MS. Synthesis of enzyme-degradable, peptide-cross-linked dextran hydrogels. Bioconjug. Chem. 2007;18:874–885. doi: 10.1021/bc0602127. [DOI] [PubMed] [Google Scholar]
- [24].Wang Y, Mangipudi SS, Canine BF, Hatefi A. A designer biomimetic vector with a chimeric architecture for targeted gene transfer. J. Control. Release. 2009;137:46–53. doi: 10.1016/j.jconrel.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Grosse SM, Tagalakis AD, Mustapa MF, Elbs M, Meng QH, Mohammadi A, Tabor AB, Hailes HC, Hart SL. Tumor-specific gene transfer with receptor-mediated nanocomplexes modified by polyethylene glycol shielding and endosomally cleavable lipid and peptide linkers. FASEB J. 2010;24:2301–2313. doi: 10.1096/fj.09-144220. [DOI] [PubMed] [Google Scholar]
- [26].Hunter AC, Moghimi SM. Cationic carriers of genetic material and cell death: a mitochondrial tale. Biochim. Biophys. Acta. 2010;1797:1203–1209. doi: 10.1016/j.bbabio.2010.03.026. [DOI] [PubMed] [Google Scholar]
- [27].Authier F, Metioui M, Bell AW, Mort JS. Negative regulation of epidermal growth factor signaling by selective proteolytic mechanisms in the endosome mediated by cathepsin B. J. Biol. Chem. 1999;274:33723–33731. doi: 10.1074/jbc.274.47.33723. [DOI] [PubMed] [Google Scholar]
- [28].Blum JS, Fiani ML, Stahl PD. Proteolytic cleavage of ricin A chain in endosomal vesicles. Evidence for the action of endosomal proteases at both neutral and acidic pH. J. Biol. Chem. 1991;266:22091–22095. [PubMed] [Google Scholar]
- [29].Lautwein A, Kraus M, Reich M, Burster T, Brandenburg J, Overkleeft HS, Schwarz G, Kammer W, Weber E, Kalbacher H, Nordheim A, Driessen C. Human B lymphoblastoid cells contain distinct patterns of cathepsin activity in endocytic compartments and regulate MHC class II transport in a cathepsin S-independent manner. J. Leukoc. Biol. 2004;75:844–855. doi: 10.1189/jlb.0803367. [DOI] [PubMed] [Google Scholar]
- [30].Kopecek J, Kopeckova P, Minko T, Lu Z. HPMA copolymer-anticancer drug conjugates: design, activity, and mechanism of action. Eur. J. Pharm. Biopharm. 2000;50:61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- [31].Sutherland MS, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, Lewis TS, Meyer DL, Zabinski RF, Doronina SO, Senter PD, Law CL, Wahl AF. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem. 2006;281:10540–10547. doi: 10.1074/jbc.M510026200. [DOI] [PubMed] [Google Scholar]
- [32].Ulbrich K, Subr V, Strohalm J, Plocova D, Jelinkova M, Rihova B. Polymeric drugs based on conjugates of synthetic and natural macromolecules. I. Synthesis and physico-chemical characterisation. J. Control. Release. 2000;64:63–79. doi: 10.1016/s0168-3659(99)00141-8. [DOI] [PubMed] [Google Scholar]
- [33].Bidlingmeyer BA, Cohen SA, Tarvin TL. Rapid analysis of amino acids using pre-column derivatization. J. Chromatogr. 1984;336:93–104. doi: 10.1016/s0378-4347(00)85133-6. [DOI] [PubMed] [Google Scholar]
- [34].Johnson RN, Chu DS, Shi J, Schellinger JG, Carlson PM, Pun SH. HPMA-oligolysine copolymers for gene delivery: Optimization of peptide length and polymer molecular weight. J. Control. Release. 2011 doi: 10.1016/j.jconrel.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Johnson RN, Burke RS, Convertine AJ, Hoffman AS, Stayton PS, Pun SH. Synthesis of Statistical Copolymers Containing Multiple Functional Peptides for Nucleic Acid Delivery. Biomacromolecules. 2010 doi: 10.1021/bm100806h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Burke RS, Pun SH. Synthesis and characterization of biodegradable HPMA-oligolysine copolymers for improved gene delivery. Bioconjug. Chem. 2010;21:140–150. doi: 10.1021/bc9003662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guha S, Padh H. Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J. Biochem. Biophys. 2008;45:75–90. [PubMed] [Google Scholar]
- [38].Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- [39].Cezari MH, Puzer L, Juliano MA, Carmona AK, Juliano L. Cathepsin B carboxydipeptidase specificity analysis using internally quenched fluorescent peptides. Biochem. J. 2002;368:365–369. doi: 10.1042/BJ20020840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cotrin SS, Puzer L, de Souza Judice WA, Juliano L, Carmona AK, Juliano MA. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: assays with human cathepsin B. Anal. Biochem. 2004;335:244–252. doi: 10.1016/j.ab.2004.09.012. [DOI] [PubMed] [Google Scholar]
- [41].Hasnain S, Hirama T, Tam A, Mort JS. Characterization of recombinant rat cathepsin B and nonglycosylated mutants expressed in yeast. New insights into the pH dependence of cathepsin B-catalyzed hydrolyses. J. Biol. Chem. 1992;267:4713–4721. [PubMed] [Google Scholar]
- [42].Dubowchik GM, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg. Med. Chem. Lett. 1998;8:3341–3346. doi: 10.1016/s0960-894x(98)00609-x. [DOI] [PubMed] [Google Scholar]
- [43].Stachowiak K, Tokmina M, Karpinska A, Sosnowska R, Wiczk W. Fluorogenic peptide substrates for carboxydipeptidase activity of cathepsin B. Acta Biochim. Pol. 2004;51:81–92. [PubMed] [Google Scholar]
- [44].Polgar L, Csoma C. Dissociation of ionizing groups in the binding cleft inversely controls the endo- and exopeptidase activities of cathepsin B. J. Biol. Chem. 1987;262:14448–14453. [PubMed] [Google Scholar]
- [45].Killisch I, Steinlein P, Romisch K, Hollinshead R, Beug H, Griffiths G. Characterization of early and late endocytic compartments of the transferrin cycle. Transferrin receptor antibody blocks erythroid differentiation by trapping the receptor in the early endosome. J. Cell Sci. 1992;103(Pt 1):211–232. doi: 10.1242/jcs.103.1.211. [DOI] [PubMed] [Google Scholar]
- [46].Durinx C, Lambeir AM, Bosmans E, Falmagne JB, Berghmans R, Haemers A, Scharpe S, De Meester I. Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur. J. Biochem. 2000;267:5608–5613. doi: 10.1046/j.1432-1327.2000.01634.x. [DOI] [PubMed] [Google Scholar]
- [47].Erdos EG, Skidgel RA. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J. 1989;3:145–151. [PubMed] [Google Scholar]
- [48].Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 1993;214:829–835. doi: 10.1111/j.1432-1033.1993.tb17986.x. [DOI] [PubMed] [Google Scholar]
- [49].Wang LH, Ahmad S, Benter IF, Chow A, Mizutani S, Ward PE. Differential processing of substance P and neurokinin A by plasma dipeptidyl(amino)peptidase IV, aminopeptidase M and angiotensin converting enzyme. Peptides. 1991;12:1357–1364. doi: 10.1016/0196-9781(91)90220-j. [DOI] [PubMed] [Google Scholar]
- [50].Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat. Rev. Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- [51].Etrych T, Strohalm J, Chytil P, Cernoch P, Starovoytova L, Pechar M, Ulbrich K. Biodegradable star HPMA polymer conjugates of doxorubicin for passive tumor targeting. Eur. J. Pharm. Sci. 2011;42:527–539. doi: 10.1016/j.ejps.2011.03.001. [DOI] [PubMed] [Google Scholar]
- [52].Kasuya Y, Lu ZR, Kopeckova P, Minko T, Tabibi SE, Kopecek J. Synthesis and characterization of HPMA copolymer-aminopropylgeldanamycin conjugates. J. Control. Release. 2001;74:203–211. doi: 10.1016/s0168-3659(01)00318-2. [DOI] [PubMed] [Google Scholar]
- [53].Mann A, Thakur G, Shukla V, Ganguli M. Peptides in DNA delivery: current insights and future directions. Drug Discov. Today. 2008;13:152–160. doi: 10.1016/j.drudis.2007.11.008. [DOI] [PubMed] [Google Scholar]
- [54].Hong S, Leroueil PR, Janus EK, Peters JL, Kober MM, Islam MT, Orr BG, Baker JR, Jr., Banaszak Holl MM. Interaction of polycationic polymers with supported lipid bilayers and cells: nanoscale hole formation and enhanced membrane permeability. Bioconjug. Chem. 2006;17:728–734. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- [55].Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Molecular Therapy. 2005;11:990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- [56].Klemm AR, Young D, Lloyd JB. Effects of polyethyleneimine on endocytosis and lysosome stability. Biochem. Pharmacol. 1998;56:41–46. doi: 10.1016/s0006-2952(98)00098-7. [DOI] [PubMed] [Google Scholar]
- [57].Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U, Salvesen GS, Stoka V, Turk V, Turk B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008;283:19140–19150. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- [58].Allen MH, Green MD, Getaneh HK, Miller KM, Long TE. Tailoring charge density and hydrogen bonding of imidazolium copolymers for efficient gene delivery. Biomacromolecules. 2011;12:2243–2250. doi: 10.1021/bm2003303. [DOI] [PubMed] [Google Scholar]
- [59].Hwang SJ, Bellocq NC, Davis ME. Effects of structure of beta-cyclodextrin-containing polymers on gene delivery. Bioconjug. Chem. 2001;12:280–290. doi: 10.1021/bc0001084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.