

Abstract
The nucleus accumbens shell (NAc) is a key brain region mediating emotional and motivational learning. In rodent models, dynamic alterations have been observed in synaptic NMDA receptors (NMDARs) within the NAc following incentive stimuli, and some of these alterations are critical for acquiring new emotional/motivational states. NMDARs are prominent molecular devices for controlling neural plasticity and memory formation. Although synaptic NMDARs are predominately located postsynaptically, recent evidence suggests that they may also exist at presynaptic terminals and reshape excitatory synaptic transmission by regulating presynaptic glutamate release. However, it remains unknown whether presynaptic NMDARs exist in the NAc and contribute to emotional and motivational learning. In an attempt to identify presynaptically located NMDARs in the NAc, the present study uses slice electrophysiology combined with pharmacological and genetic tools to examine the physiological role of the putative presynaptic NMDARs in rats. Our results show that application of glycine, the glycine-site agonist of NMDARs, potentiated presynaptic release of glutamate at excitatory synapses on NAc neurons, whereas application of 5,7-dichlorokynurenic acid or 7-chlorokynurenic acid, the glycine-site antagonists of NMDARs, produced the opposite effect. However, these seemingly presynaptic NMDAR-mediated effects could not be prevented by application of d-APV, the glutamate-site NMDAR antagonist, and were still present in the mice in which NMDAR NR1 or NR3 subunits were genetically deleted. Thus, rather than suggesting the existence of presynaptic NMDARs, our results support the idea that an unidentified type of glycine-activated substrate may account for the presynaptic effects appearing to be mediated by NMDARs.
Introduction
The forebrain region nucleus accumbens shell (NAc) is an essential brain site for emotion- and motivation-related learning and memory (Cardinal and Everitt, 2004; Kelley, 2004). Serving as plasticity triggers, synaptic NMDA receptors (NMDARs) within the NAc critically contribute to the acquisition of new physiological and pathophysiological emotional and motivational responses (Kelley et al., 1997; Brown et al., 2011). Because of the predominant postsynaptic distribution, most NAc NMDAR-mediated behavioral alterations have been attributed to postsynaptic NMDARs. More recently, evidence from cortical and subcortical regions suggests that NMDARs may also be present at the presynaptic axonal terminals to influence presynaptic release of neurotransmitters (Berretta and Jones, 1996; Sjöström et al., 2003; Bardoni et al., 2004; Yang et al., 2006; Corlew et al., 2007, 2008; Brasier and Feldman, 2008; Larsen et al., 2011). Given the potentially distinct cellular behaviors of presynaptic and postsynaptic NMDARs (Engelman and MacDermott, 2004; Rodríguez-Moreno et al., 2010), it is important to understand whether NMDARs are also expressed presynaptically within the NAc, and if so, what roles they play in NAc-based emotional and motivational learning.
In an attempt to detect presynaptic NMDARs within the NAc, we focused on the NMDAR glycine-binding site, which has been commonly targeted pharmacologically to delineate the behavioral roles of NAc NMDARs (Goldstein et al., 1994; Tricklebank et al., 1994; Wlaź et al., 1994; Kretschmer and Schmidt, 1996; Di Ciano and Everitt, 2001; Huang et al., 2008). NMDARs are thought to exist as tetramers, usually containing obligatory glycine-binding NR1 subunits and glutamate-binding NR2 subunits (Cull-Candy and Leszkiewicz, 2004; Lau and Zukin, 2007). Whereas activation of NR1/NR2 NMDARs requires the binding of both glycine and glutamate, the glycine-site is tonically occupied by endogenous agonists in an unsaturated manner under physiological conditions (Wilcox et al., 1996; Li et al., 2009). In some NMDARs, NR2 subunits are partially or completely substituted by NR3 subunits, which, similar to NR1 subunits, have glycine-, but not glutamate-, binding sites (Chatterton et al., 2002; Low and Wee, 2010). Thus, the glycine site of NMDARs can serve as a regulatory site and a druggable target; NMDAR-mediated primary and secondary responses can be regulated bidirectionally by pharmacological manipulation of NR1- or NR3-located glycine-binding site (Jansen and Dannhardt, 2003; Javitt, 2006; Awobuluyi et al., 2007; Madry et al., 2007).
Prior results suggest that activation of potential presynaptic NMDARs enhances synaptic transmission by facilitating presynaptic release of neurotransmitters (Berretta and Jones, 1996; Corlew et al., 2007; Brasier and Feldman, 2008; Larsen et al., 2011; but see Bardoni et al., 2004). Our current results show that application of glycine in the presence of glycine receptor antagonist strychnine increased presynaptic glutamate release at excitatory synapses on NAc neurons, whereas application of 5,7-dichlorokynurenic acid (DCKA) or 7-chlorokynurenic acid (7-Cl), the glycine-site antagonists of NMDARs, produced the opposite effect. However, these effects remain intact either in the presence of d-APV, the glutamate-site NMDAR antagonist, or in mice in which NR1 or NR3 subunits were genetically deleted. Thus, the seemingly presynaptic NMDAR-mediated effects of glycine and DCKA were likely mediated by an NMDAR-independent mechanism, possibly through an unidentified type of glycine-activated substrate. These results provide experimental evidence that may help reinterpret some of the prior results related to presynaptic NMDARs.
Materials and Methods
Animals.
Male Sprague Dawley rats (Simonsen Labs) were used for most experiments. The NR3A−/− mice and WT controls were generated by the Nakanishi laboratory as previously described (Das et al., 1998). The NR1−/− mice were generated by crossing NR1-floxed mice (generated by the Tonegawa laboratory) (Tsien et al., 1996) with Nex-Cre mice (generated by the Nave laboratory) (Goebbels et al., 2006) as described previously (Ultanir et al., 2007); both types of mice were generously provided by the Ghosh laboratory. The genotypes of all experimental animals were determined by PCR. Mice that were heterozygous for NR1-floxed allele or lack of Cre gene were used as controls and referred to as WT. All protocols were approved by the Washington State University Animal Care and Use Committee.
Preparation of acute brain slices.
Four- to 5-week-old rats or 3- to 4-week-old mice were decapitated following isoflurane anesthesia. Coronal slices (300 μm) containing the NAc were prepared on a vibratome (VT1200S; Leica) in 4°C cutting solution containing (in mm): 135 N-methyl-d-glucamine, 1 KCl, 1.2 KH2PO4, 0.5 CaCl2, 1.5 MgCl2, 20 choline-HCO3, and 11 glucose, saturated with 95% O2/5% CO2, pH adjusted to 7.4 with HCl. Slices were incubated in artificial CSF (aCSF) containing (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, saturated with 95% O2/5% CO2 at 37°C for 30 min and then allowed to recover for at least 30 min at room temperature before experimentation.
Electrophysiological recordings.
During recordings, slices were superfused with aCSF that was heated to 31–33°C by passing the solution through a feedback-controlled in-line heater (Warner Instruments) before entering the chamber. Recordings were made under visual guidance (40×, differential interference contrast optics) with electrodes (3–5 MΩ) filled with (in mm): 125 CsCH3O3S (or CsCl for recording mIPSCs), 15 CsCl, 5 TEA-Cl, 0.4 EGTA, 20 HEPES, 2.5 Mg-ATP, 0.25 Na-GTP, 1 QX-314, pH 7.3. Picrotoxin (100 μm) (or NBQX, 10–15 μm) was included in the external perfusion aCSF to block GABAA (or AMPA) receptors. Drugs were either bath-applied (see Figs. 1, 3) or locally superfused onto the slice through a wide-bore pipette (∼150 μm) placed near the recording area. In case of local perfusion, a control line containing only bath solution was included.
Figure 1.

Miniature EPSCs are bidirectionally regulated by glycine-binding site NMDAR agonists and antagonists. A, B, Example traces (A) and summarized data in cumulative distribution plots (B) show that bath application of d-APV (50 μm) did not affect mEPSCs in NAc MSNs; subsequent application of DCKA in the presence of d-APV reversibly and in a dose-dependent manner decreased the frequency and amplitude of mEPSCs. DCKA was applied at three concentrations (20, 50, and 100 μm) sequentially. Left insets in A show averaged mEPSCs recorded during control (black) and perfusion of DCKA (gray), and after being scaled to the same peak amplitude. Right inset in A shows a portion of mEPSCs in an extended time scale. C, Grand summary of the effects of NMDAR glycine-binding site agonists and antagonists on the frequency and amplitude of miniature events. mEPSCs and mIPSCs were recorded at the holding potential −70 mV in aCSF containing 1 μm TTX and 100 μm picrotoxin (for mEPSCs) or 10 μm NBQX (mIPSCs). Controls were obtained from ∼5–15 min following stabilization. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 3.

Presynaptic mechanisms dominate in DCKA-induced inhibition of EPSCs. A, Example traces (A1) and summarized data (A2) showing that glutamate (500 μm) puffing-induced AMPAR-mediated currents were not altered by application of DCKA (50 μm). Recordings were made in the presence of d-APV (50 μm), followed by sequential applications of DCKA and NBQX (15 μm). B, Example traces (from a control cell, B1) and summarized data (B2) showing that the peak amplitude of evoked EPSCs gradually decayed in the presence of Baf (2 μM), and the decay rate was significantly slower in the presence of DCKA.
Excitatory afferents were stimulated by a constant-current isolated stimulator (DS3; Digitimer), using a monopolar electrode (glass pipette filled with aCSF). Stimulus strength was adjusted so that the first EPSC was between 100 and 500 pA. Trains of pulses were generated using Clampex software (Molecular Devices). In some experiments (see Fig. 3), l-glutamate (500 μm dissolved in aCSF) was applied locally through a small-tip pipette (∼1 μm diameter) using a Picospritzer (Pressure System IIe, Toohey Co.); a ∼5 ms pulse of 20 psi was used to eject the solution. d-APV (50 μm) was included in the puffing pipette solution as well as in the bath to block NMDARs.
For all recordings, series resistance was 8–14 MΩ and was left uncompensated. Series resistance was monitored continuously during all recordings. Cells with a change in series resistance beyond 15% were not accepted for data analysis. Synaptic currents were recorded with a MultiClamp 700A amplifier (Molecular Devices), filtered at 2.6–3 kHz, amplified 5 or 10 times, and then digitized at 20 kHz with a Digidata 1322A analog-to-digital converter (Molecular Devices).
Viral vectors.
Recombinant adeno-associated vectors (rAAV) expressing venus-tagged channelrhodopsin-2 (ChR2) H134R (Makinodan et al., 1991) were pseudotyped with AAV1/2 capsid proteins. HEK293T cells were cotransfected with the plasmidspFΔ6 (adenoviral helper plasmid), pRVI (cap and rep genes for AAV serotype 2), pH21 (cap gene for AAV serotype 1 and rep gene for serotype 2) and the rAAV plasmid, using linear polyethyleneimine-assisted transfection (Kuroda et al., 2009). The helper plasmids were kindly provided by Dr. M. Schwarz (Max-Planck Institute for Medical Research, Heidelberg, Germany) (Pilpel et al., 2009). Cultures grown in DMEM (Biochrom) with 10% substituted FBS (Biochrom, #S0115) were harvested from 15 by 15 cm dishes after 48 h. rAAV were isolated and purified as described previously (Pilpel et al., 2009). Briefly, HEK293T cells were lysed with sodium deoxycholate and repeated freeze thawing cycles in the presence of Benzonase-Nuclease HC (Novagen). From the supernatant, rAAVs were isolated by iodixanol gradient centrifugation from the 40% and 54% interphase. rAAVs were then desalted by ultrafiltration and filtered through 0.2 μm Millex-GV filter units (Millipore) and stored at 4°C in 500 μl PBS with 10 mm MgCl2 and 25 mm KCl.
Stereotaxic injections.
Rats were anesthetized with a mixture of ketamine, xylazine, and acepromazine (0.1 ml/100 mg) and placed in a stereotaxic apparatus (Stoelting). A 26 gauge injection needle was used to bilaterally inject 1 μl (0.2 μl/min) of the AAV-ChR2-YFP solution via a Hamilton syringe into the infralimbic prefrontal cortex [anteroposterior (AP) +3.00, ML ±0.75, DV −4.00], the ventral tegmental area (AP −5.00, ML ±0.90, DV −7.65), or the basolateral amygdala (AP −2.50, ML ±4.80, DV −8.50) using a Thermo Orion M365 pump (Thermo Scientific). Injection needles were left in place for 5 min following injections. Rats were then sutured and allowed for viral expression for ∼3 weeks before experimentation.
Drugs.
Tetrodotoxin (TTX), DCKA, 7-Cl, d-(−)-2-Amino-5-phosphonopentanoic acid (d-APV), NBQX, bafilomycin A1 (Baf) and cyclothiazide were purchased from Tocris Bioscience. One batch of DCKA and all other chemicals were obtained from Sigma-Aldrich.
Data acquisition and statistics.
All data were analyzed off-line. Miniature EPSCs (mEPSCs) were obtained at least 15 min after achieving whole-cell configuration. For analysis, a template was made by averaging ∼100 hand-picked miniature events using pClamp9 software (Molecular Devices). Approximately 250–2000 miniature events over a period of 1.5–5 min were analyzed under various conditions. mEPSC decay was fitted by a single exponential to obtain the decay time constant.
For variance-mean (V-M) analysis (see Fig. 4), the variance and mean amplitude were calculated for each of the five EPSCs in the train from ∼100 consecutive sweeps once stabilized. The parabola function is y = A*X − B*X2, where y is the variance and X is the mean amplitude; A and B can thus be derived through fitting (see Fig. 4, example curves). With mathematical assumptions and simplifications, the average quantal content (Q) = A, N = 1/B, and the release probability (Pr) at a particular stimulus in the train: Pi = Xi*B/A. Stimulus artifacts were truncated in evoked synaptic currents for clarity. Mean values are expressed ± SEM. Statistical significance was assessed using one- or two-factor ANOVA with Bonferroni post-tests, or t test as indicated.
Figure 4.

Variance-mean analysis reveals presynaptic effects of DCKA. A–C, Variance-mean plots of example EPSCs recorded from synapses within the VTA-to-NAc (A), PFC-to-NAc (B), and BLA-to-NAc (C) pathways. Insets showing individual (gray) and averaged (black) EPSCs evoked by a repeated train (5 pulses at 20 Hz) generated by channelrhodopsin-based light stimulation.
Results
In search of presynaptic NMDARs in the NAc, we focused on the NMDAR glycine-binding site, manipulation of which bidirectionally regulates the function of potential NMDARs and produces detectable presynaptically generated alterations during synaptic transmission.
Spontaneous glutamatergic synaptic currents are bidirectionally regulated by NMDAR glycine-site agonists and antagonists
In rat NAc medium spiny neurons (MSNs), mEPSCs were recorded under whole-cell voltage-clamp mode at −70 mV in the presence of picrotoxin (PTX, 100 μm) to block GABAA receptors, TTX (1 μm) to block action potential-dependent release events, and strychnine (1 μm) to block glycine receptors. Application of the NMDAR glycine-site competitive antagonist DCKA reversibly and in a dose-dependent manner reduced the frequency (F(2,19) = 18.34, p < 0.0001, one-way ANOVA; 20 μm DCKA, 77.4 ± 6.4% of baseline, p < 0.01, n = 5; 50 μm DCKA, 59.0 ± 7.9% of baseline, p < 0.001, n = 6, Bonferroni post-test; Fig. 1C) and peak amplitude of mEPSCs in NAc MSNs (F(2,19) = 25.73, p < 0.0001, one-way ANOVA; 20 μm DCKA, 84.5 ± 7.0% of baseline, p = 0.118, n = 5; 50 μm DCKA, 73.7 ± 3.6% of baseline, p < 0.001, n = 6, Bonferroni post-test; Fig. 1C). Alterations in both the frequency and amplitude of mEPSCs suggest that the effect of DCKA is mediated by both presynaptic and postsynaptic mechanisms (but see discussions below). To affirm the pharmacological effects of DCKA, DCKA from two different vendors (Tocris Bioscience and Sigma) was used and no qualitative difference was observed; nor was there difference observed whether DCKA was dissolved in water or DMSO (in the latter case DMSO was included in equal volume for control conditions). Data were thus combined. Moreover, the transmitter reuptake process or postsynaptic membrane properties do not appear to be involved as the effects of DCKA (50 μm) were not accompanied by detectable changes in either the decay kinetics of mEPSCs (decay τ: control, 5.3 ± 0.5 ms, n = 6; DCKA, 5.2 ± 0.5 ms, n = 6, p = 0.300, paired t test; Fig. 1A inset), the input resistance (in MΩ: control, 133.7 ± 15.2; DCKA, 132.9 ± 10.8 MΩ, p = 0.944, n = 6, paired t test), or the holding current (in pA: control, −251.2 ± 29.0; DCKA, −253.6 ± 28.3, p = 0.799, n = 6, paired t test).
Surprisingly, application of d-APV (50 μm), the NMDAR glutamate-binding site competitive antagonist, did not change either the frequency (F(4,43) = 31.65, one-way ANOVA; p = 1.000, Bonferroni post-test) or amplitude (F(4,43) = 35.15, one-way ANOVA; p = 1.000, Bonferroni post-test; Fig. 1) of mEPSCs. In addition, in the presence of d-APV (50–100 μm), the dose-dependent inhibitory effects of DCKA on the frequency (p < 0.0001, Bonferroni post-test; Fig. 1) and peak amplitude (p < 0.0001, Bonferroni post-test; Fig. 1) of mEPSCs remained intact. Furthermore, the effects of DCKA were mimicked by another NMDAR glycine-site antagonist 7-Cl (100 μm; frequency: 34.7 ± 4.1% of baseline, p < 0.01, n = 6; amplitude: 72.1 ± 3.6% of baseline, p < 0.01, n = 6, paired t tests; Fig. 1C). These results, although not supporting the hypothesized role of NMDARs, do not exclude this possibility either, because the glycine- and glutamate-binding sites may separately trigger distinct downstream signaling cascades (Nong et al., 2003).
Moreover, when the external Ca2+ was removed (bath containing 0 mm Ca2+, 1 mm EGTA, and 1 μm TTX), the effects of DCKA (100 μm) on mEPSCs remained intact (p < 0.01, DCKA vs baseline for both frequency and amplitude, n = 6; Fig. 1C). Thus, the results so far suggest that the DCKA-induced effects in the NAc occur either in the presynaptic release or postsynaptic responsiveness, or both, via a mechanism that can be independent of extracellular Ca2+.
Beyond the NAc, recordings from pyramidal neurons in the hippocampal CA1 region (Fig. 1C) and somatosensory cortex layer II/III (Fig. 1C) showed a similar decrease in the frequency (hippocampus, p < 0.001, n = 6; cortex, p < 0.001, n = 4, paired t test) and amplitude of mEPSCs (hippocampus, p < 0.05, n = 6; cortex, p < 0.05, n = 4, paired t test) in response to the application of DCKA (50 μm). In contrast, miniature IPSCs (mIPSCs) in NAc MSNs were not significantly altered by application of DCKA (100 μm) in either the frequency (p = 0.103, n = 11, paired t test) or peak amplitude (p = 0.060, n = 11, paired t test; Fig. 1C). Thus, the effects of DCKA appear to be specific for excitatory synapses and exist across brain regions.
Conversely, application of NMDAR glycine-site agonist glycine (10 μm; in the presence of 1 μm strychnine to block potential glycine receptors) significantly increased the frequency (p < 0.01, n = 11, paired t test), but not the amplitude, of mEPSCs in NAc MSNs (p = 0.110, n = 11; Fig. 1C). Thus far, the results show that pharmacological manipulations that influence the glycine-binding site of NMDARs bidirectionally regulate excitatory synaptic transmission to NAc MSNs via presynaptic and likely also postsynaptic mechanisms. These effects are not dependent on glutamate binding to NMDARs, nor mediated by strychnine-sensitive glycine receptors.
DCKA reduces evoked EPSCs by a presynaptic mechanism
We next examined whether the DCKA-induced effects on spontaneous synaptic activity also occur in action potential-dependent synaptic transmission. Under whole-cell voltage-clamp mode (at −70 mV), AMPAR-mediated EPSCs in NAc MSNs were evoked by local electrical stimulations and were isolated in the presence of PTX (100 μm) and d-APV (50 μm). The evoked EPSCs in NAc MSNs were strongly and reversibly inhibited by DCKA in a dose-dependent manner (F(3,25) = 19.73, p < 0.0001; Fig. 2A–C). Simultaneously, the paired-pulse ratio (PPR) of EPSC peaks (EPSC2/EPSC1) was increased by DCKA (at 20 μm, p < 0.05, n = 5; at 100 μm, p < 0.05, n = 12, paired t test; Fig. 2A,D), suggesting that presynaptic alterations contribute to the decrease in EPSCs.
Figure 2.

Application of DCKA reduces evoked EPSCs in NAc MSNs. A, Evoked (left) and scaled (right) EPSCs recorded from an example NAc MSN during the periods of control, perfusion of d-APV (50 μm), coperfusion of d-APV and DCKA (50 μm), and wash-out. B, The time course of DCKA-induced alterations in the peak amplitudes of first EPSCs in the example NAc neuron in A. Bars indicate the duration of drug applications. C, Summarized data showing the dose-dependent inhibition of EPSC peak amplitude by DCKA. D, Summarized data showing increases in the PPR concurrently occurred with the DCKA-induced inhibitions of EPSCs in NAc MSNs. *p < 0.05, **p < 0.01, ***p < 0.001.
Voltage control error, if prominent, also changes PPR as the peak amplitude changes; it tends to deviate PPR away from 1 as amplitude decreases. Specifically, if PPR is <1 at baseline conditions, a decrease in the peak amplitude would further decrease the PPR if the potential voltage control error is involved. However, this is not the case here because the majority of the recorded neurons that exhibited a PPR <1 showed an increase in PPR by DCKA (8 of 10 cells). Thus, the DCKA-induced increase in PPR likely reflects decreased release probability, which is consistent with its effect on the frequency of mEPSC (Fig. 1).
Presynaptic mechanisms are predominant
Although the above results suggest that both presynaptic and postsynaptic mechanisms are involved in DCKA-mediated synaptic regulation, the results are not unequivocal. For example, a decrease in the frequency of mEPSCs or an increase in the PPR may reflect presynaptic inhibition, but it can also be explained by desensitization of postsynaptic AMPARs (Heine et al., 2008). Meanwhile, a decrease in the amplitude of mEPSCs that is normally interpreted as a decrease in postsynaptic responsiveness can also be explained by a decrease in the quantal content during presynaptic release (Silver, 2003). To better evaluate the presynaptic and postsynaptic contributions to DCKA-mediated effects, we took two additional approaches, which are relatively more specific in detecting presynaptic and postsynaptic alterations, respectively.
To isolate postsynaptic responsiveness, we evoked postsynaptic AMPAR-mediated current by brief local pressure application of glutamate through a puffing pipette directed at the dendritic region of an MSN, thus by-passing presynaptic glutamate release. Puffing glutamate (500 μm) to the dendritic area of NAc MSNs (in the presence of 50 μm d-APV and 100 μm PTX; membrane held at −70 mV) elicited AMPAR-mediated currents, verified as the near-complete inhibition by NBQX (15 μm; 3.0 ± 0.4% of baseline, p < 0.001, n = 7; Fig. 3A), an AMPAR-selective antagonist. Since NBQX was bath-applied, the almost complete inhibition also indicates that bath-applied drugs have sufficient access to puff-activated AMPARs. Nonetheless, bath application of DCKA (50 μm) did not significantly affect the peak amplitude of puff-evoked currents, albeit a trend of inhibition was observed (90.4 ± 3.7% of baseline peak amplitude, p = 0.078, n = 7; Fig. 3A). This result suggests that the effect of DCKA at the concentrations of 50 μm or lower is not predominantly mediated by direct inhibition of postsynaptic AMPARs. One interpretational caveat is that the nonsynaptically located AMPARs, which can also be activated by puffed glutamate but are insensitive to DCKA, dilute the potential synaptic AMPAR-specific changes. This is not highly likely; given the preferential synaptic distribution of AMPARs (Zamanillo et al., 1999), such a robust reduction (∼50%; Fig. 2A–C) of synaptic transmission by DCKA, if mediated by postsynaptic AMPARs, should be reflected in the puff-induced current.
We then used presynaptically oriented approaches to examine the effect of DCKA. One potential mechanism is that application of DCKA decreases the Pr of glutamate from presynaptic terminals. In addition to Pr, we also intended to examine the Q of presynaptic glutamate vesicles because 7-Cl, an analog of DCKA, has been shown to inhibit presynaptic vesicular glutamate transporters (Bartlett et al., 1998). The first experiment took advantage of the pharmacological properties of Baf, which inhibits vacuolar-type proton pump (V-ATPase) and thus blocks glutamate uptake into synaptic vesicles. In the presence of Baf, neurotransmitter vesicles cannot be refilled once released, and therefore spontaneous or evoked EPSCs would gradually diminish as the transmitter-filled vesicles are consumed during transmission. In the presence of Baf, the time course of decrease in EPSCs depends on the Pr, but not the Q. More specifically, synapses with a decreased Pr should exhibit a prolonged diminishing time course, and vice versa. In contrast, synapses with different Q but similar Pr should exhibit a similar diminishing time course. As shown in Figure 3B, in the presence of Baf (2 μm), the peak amplitude of evoked EPSCs in MSNs gradually diminished over a period of 30 min (aCSF containing 4 mm K+ was used to accelerate vesicular release). Application of DCKA (50 μm) prolonged the diminishing time course of EPSCs (F(1,78) = 6.41, p < 0.05, two-way ANOVA; Fig. 3B), suggesting a decrease in the Pr by DCKA.
To quantify the DCKA-induced presynaptic alterations, we focused on three key presynaptic quantal parameters, the Pr, Q, and number of release sites (N), by performing the nonstationary V-M analysis (Clements and Silver, 2000; Silver, 2003; Saviane and Silver, 2007). Briefly, the V-M plot is generated by having the same set of synapses experience different release conditions so as to obtain the mean peak amplitude and the variance of the amplitudes under each condition (Meyer et al., 2001; Scheuss and Neher, 2001; Scheuss et al., 2002). This analysis is feasible only if the different presynaptic release sites behave uniformly as they go through different release conditions. However, in NAc glutamatergic synapses within different afferents exhibit different Prs (Pr = 0.18 ± 0.04, 0.57 ± 0.05, and 0.70 ± 0.03 in VTA, PFC, and amygdala afferents, respectively; see below; Fig. 4), likely resulting in distorted V-M plot. To overcome this, we dissected three glutamatergic afferents, the afferent from the infralimbic PFC, the afferent from the VTA, and the afferent from BLA, by confined expression of channel (ChR2) (Boyden et al., 2005) within each of these three brain regions (see Materials and Methods). The NAc slices were then prepared, with only presynaptic input from the intended brain region expressing ChR2. Thus, a light (at 473 nm) stimulation selectively activated the intended presynaptic input to NAc MSNs.
With this approach, we applied a 5-pulse train (20 Hz) of presynaptic stimuli to induce EPSCs in NAc MSNs (Fig. 4). The train of stimuli presumably activates the recorded synapses repetitively and consistently through different states of release probability, thus, allowing the V-M analysis (Meyer et al., 2001; Scheuss and Neher, 2001; Scheuss et al., 2002). EPSC V-M plot was generated by calculating the mean EPSC amplitude and variance of EPSC amplitudes under each release condition. The V-M plot was then fitted by a parabola function and interpreted assuming binomial statistics of transmitter release (Quastel, 1997; Silver, 2003). To eliminate AMPAR desensitization, which may occur during repeated stimulations and distort the V-M plot (Quastel, 1997; Silver, 2003), cyclothiazide (100 μm) was included in the bath in ∼50% of the recordings and results were combined. With this approach, we observed that application of DCKA (20 μm) decreased the Pr and/or N of excitatory synapses, but not Q. Specifically, synapses within the VTA projection exhibited a relatively low Pr; application of DCKA decreased the Pr (Pr of EPSC1: control, 0.18 ± 0.04; DCKA, 0.16 ± 0.03; p < 0.05, n = 6, paired t test) and N (control, 289 ± 37; DCKA, 205 ± 33; p < 0.05, n = 6, paired t test; Fig. 4A). Synapses within the PFC projection exhibited a medium Pr; application of DCKA did not significantly affect the Pr (Pr of EPSC1: control, 0.57 ± 0.05; DCKA, 0.49 ± 0.12; p = 0.500, n = 6, paired t test) but decreased the N (control, 15 ± 7; DCKA, 12 ± 7; p < 0.05, n = 5, paired t test; Fig. 4B). Synapses within the BLA projection exhibited a relatively high Pr; application of DCKA decreased both the Pr (Pr of EPSC1: control, 0.70 ± 0.03; DCKA, 0.64 ± 0.04; p < 0.01, n = 12, paired t test) and N (control, 20 ± 4; DCKA, 15 ± 4; p < 0.01, n = 12, paired t test; Fig. 4C). A decrease in either Pr or N suggests a presynaptic inhibition. In contrast to the Pr and N, the Q of synapses was not affected by application of DCKA in any of the examined afferent (in pA: VTA: control, 5.1 ± 0.3; DCKA, 5.4 ± 0.4; p = 0.193, n = 6; PFC: control, 20.7 ± 1.1; DCKA, 20.8 ± 2.8, p = 0.964, n = 5; BLA: control, 16.6 ± 3.6; DCKA, 18.5 ± 3.6; p = 0.105, n = 12; paired t test; Fig. 4). In the V-M analysis, the Q is measured as the postsynaptic response (in pA) induced by all glutamate released from a single vesicle (quantum). As such, the lack of change in the Q suggests no change in the vesicular content and postsynaptic AMPARs following application of DCKA. Note that the two effects of DCKA, namely the lack of change in Q and the decrease in the amplitude of mEPSCs, are at odds in terms of presynaptic or postsynaptic interpretation of the DCKA effects (see Discussion). Also note that the relatively small Q in the VTA afferent can be interpreted as smaller vesicular content, weaker postsynaptic AMPARs, or, more likely, relatively distally located synapses.
The effects of DCKA remain intact in NR3A or NR1 KO synapses
The above results suggest that the effects of DCKA and glycine are mediated through the glycine-binding site of NMDARs that are potentially located presynaptically. In an attempt to specify the subtypes of the mediating NMDARs, we first focused on NR3-containing NMDARs. NR3-containing NMDAR is a likely candidate because it has been recently shown to promote neurotransmission by presynaptic mechanisms (Larsen et al., 2011). Furthermore, NR3-containing NMDARs are activated upon agonist binding to their glycine-binding sites without the involvement of glutamate (Chatterton et al., 2002; Low and Wee, 2010). This property is also consistent with our observation that application of the competitive glutamate-binding site antagonists did not prevent the effects of DCKA or 7-Cl (Fig. 1C). To test this, we used NR3A KO mice and examined whether the established DCKA-mediated effects on mEPSCs of NAc MSNs were affected. Our results show that the inhibitory effects of DCKA (50 μm) were similar between the KO and WT mice (% of baseline in DCKA: frequency: WT, 27.5 ± 5.2, n = 5; KO, 21.4 ± 3.2, p = 0.221; n = 8; amplitude: WT, 71.3 ± 3.1, n = 5; KO, 66.1 ± 4.4, p = 0.197, n = 8, t test; Fig. 5A). These results suggest that NR3A-containing NMDA receptors are not involved in DCKA-induced inhibition of excitatory synaptic transmission to NAc MSNs.
Figure 5.
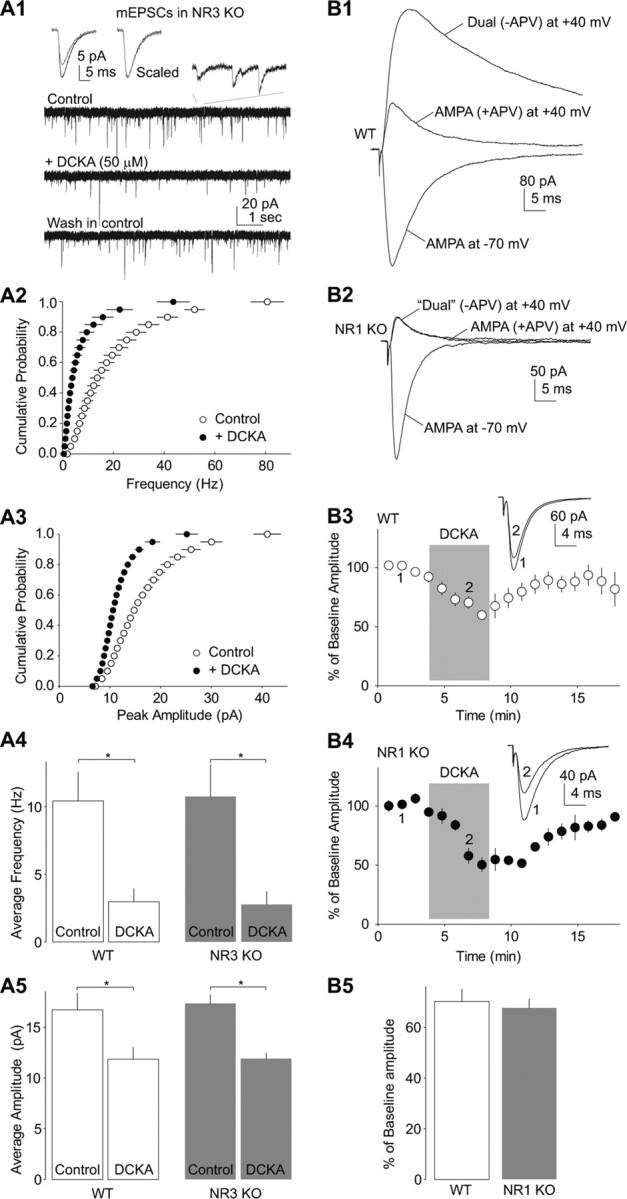
DCKA-induced synaptic inhibition is intact in NR1- or NR3-absent synapses. A, DCKA-induced inhibition of EPSCs in NAc MSNs was intact in NR3 KO mice. Example traces (A1) and summarized data in cumulative distribution plots (A2 and A3) show that both the frequency and amplitude of mEPSCs in NAc MSNs in NR3 KO mice were decreased by bath-application of 50 μm DCKA to a degree similar to that in WT mice (A4 and A5). Left insets in A1 show averaged mEPSCs recorded during control (black) and perfusion of DCKA (gray), and after being scaled to the same peak amplitude. Right inset in A1 shows a portion of mEPSCs in an extended time scale. B1, B2, Example evoked EPSCs from a somatosensory cortical layer II/III pyramidal neuron recorded at −70 and +40 mV from a WT (B1) or NR1 KO (B2) mouse. Note the absence of APV (50 μm)-sensitive component in the KO mouse. B3, B4, Summarized data showing the normalized peak amplitude of EPSCs was reversibly inhibited by application of DCKA (20 μm) in WT (B3) and NR1 KO (B4) mice. Shades indicate the duration of drug applications. B5, Summarized data showing that DCKA-induced inhibition of EPSCs was similar between NR1-containing and NR1-absent synapses. *p < 0.05.
We then attempted to determine whether presynaptic NMDARs are involved in the effects of DCKA in any manner. One way to do this is to deplete all NMDARs and examine whether this manipulation abolishes the effect of DCKA. In addition to bearing glycine-binding sites, NR1 subunits are also obligatory subunits for NMDARs. As such, genetically knocking out NR1 subunits should delete all functional NMDARs at excitatory synapses. Because NR1 KO mice are not viable (Forrest et al., 1994), we used cortical NR1-KO mice instead, in which cortical pyramidal neurons do not express NR1 subunits (Ultanir et al., 2007). In this mouse line, all four transmembrane domains and the C-terminal tail of NR1 subunit are removed; all eight known splice variants of NR1 are expected to be deleted simultaneously (Tsien et al., 1996).
We first confirmed the knock-out efficiency of NR1 in cortical neurons of this mouse line. At postnatal day 21, NMDAR EPSCs were absent in somatosensory cortical layer II/III pyramidal neurons (Fig. 5B), suggesting a complete deletion of NR1-containing, and thus likely all types of, NMDARs within cortical pyramidal neurons. Because layer II/III pyramidal neurons within the sensory cortex receive glutamate innervations almost exclusively from within the cortex (Schubert et al., 2007), monosynaptic EPSCs recorded by strictly confining the presynaptic stimulation and postsynaptic recording within layer II/III should be generated from excitatory synapses with no NMDARs on either presynaptic or postsynaptic membranes. Nonetheless, these excitatory synapses with presumably no presynaptic NMDARs were still sensitive to DCKA-induced suppression. In NR1 KO and WT mice, application of DCKA (20 μm) reduced the peak amplitude of evoked AMPAR EPSCs in layer II/III cortical neurons to a similar extent (% of baseline in DCKA: WT, 72.5 ± 5.1, n = 5; KO, 67.1 ± 3.2, n = 10, p = 0.208, t test; Fig. 5B). These quantitatively similar effects of DCKA between KO and WT mice suggest that presynaptic and postsynaptic NMDARs are not the mediators of DCKA-induced synaptic inhibition.
Glycine transporters and receptors?
If not NMDARs, DCKA must interact with other molecular substrates to achieve its synaptic effects. Presynaptically located glycine transporters, if inhibited, may decrease excitatory synaptic transmission (Raiteri et al., 2005). However, application of ALX 5407 (2 μm), the selective type I glycine transporter antagonist, did not change the frequency (F(2,18) = 13.34; p = 0.845, Bonferroni post-test) or amplitude (F(2,18) = 3.608; p = 0.189, Bonferroni post-test) of mEPSC in NAc MSNs. In the presence of ALX 5407 (2 μm), the inhibitory effects of DCKA (20 μm) on mEPSCs remained (frequency: 73.6 ± 2.4% of baseline, p < 0.001, Bonferroni post-test; amplitude: 91.5 ± 2.8% of baseline, p = 1.000, Bonferroni post-test; Fig. 6A,E,F). Thus, glycine transporter I is not likely the mediator of the effects of DCKA.
Figure 6.

An unidentified glycine-binding substrate regulates excitatory synaptic transmission. A, Example mEPSCs from a NAc MSN in the periods of control, perfusion of glycine transporter 1 antagonist ALX 5407 (ALX, 2 μm), coperfusion of ALX (2 μm) and DCKA (20 μm), and wash-out. B, Example mEPSCs from a NAc MSN in the periods of control, perfusion of d-serine (10 μm), and wash-out. C, Example mEPSCs from a NAc MSN in the periods of control (containing 50 μm d-APV), perfusion of glycine (100 μm), and wash-out. D, Example mEPSCs from a NAc MSN in the periods of control, perfusion of DCKA (50 μm), coperfusion of DCKA and glycine (100 μm), and wash-out. E, Summarized data showing that application of ALX did not prevent DCKA-induced inhibition of the frequency of mEPSCs, application of d-serine did not affect mEPSCs, and application of DCKA prevented the glycine-induced increase in the frequency of mEPSCs in NAc MSNs. F, Summarized data showing that none of the above manipulations significantly altered the amplitude of mEPSCs in NAc MSNs. *p < 0.05.
Another potential neuronal substrate for DCKA is the glycine receptor, which also contains the glycine-binding site and exists at presynaptic terminals (Turecek and Trussell, 2001; Kawa, 2003; Lee et al., 2009). Our results show that the effects of DCKA remained intact in the presence of the glycine receptor-selective antagonist strychnine (Fig. 1). Whereas most known glycine receptors are selectively inhibited by strychnine, strychnine-resistant glycine receptors do exist (Han et al., 1997). To explore the potential glycine receptors in DCKA-mediated effects, we took advantage of the pharmacological properties of glycine receptors.
First, d-serine is the endogenous glycine-site agonist of NMDARs. Our prior study shows that bath application of d-serine at 10 μm is sufficient to enhance NMDAR-mediated EPSCs in the slice preparation (Huang et al., 2008). However, d-serine does not interact with all known glycine receptors. Application of d-serine (10 μm) or depletion of d-serine in slices by d-amino acid oxidase (DAAO, 150 μg/ml, bath application for 30 min) did not significantly alter either the frequency (F(3,37) = 0.2113, p = 0.8879, one-way ANOVA) or amplitude (F(3,37) = 2.579, p = 0.0683) of mEPSCs in NAc MSNs (Fig. 6B,E,F; d-APV was not included in the bath). This result suggests that (1) the glycine-binding site compounds that do not affect glycine receptors do not significantly regulate mEPSCs, and (2) activating the glycine-sites of NMDARs by enhancing d-serine tone in the brain slice does not alter (enhance) mEPSCs in NAc MSNs.
Second, glycine is the endogenous glycine-site agonist for both NMDARs and glycine receptors. Application of glycine significantly increased the frequency of mEPSCs in NAc MSNs in the presence of d-APV (50 μm) and strychnine (1 μm) (p < 0.05, n = 6; no change in amplitude, p = 0.468, n = 6; t test; Fig. 6C,E,F). Furthermore, although most known glycine receptors do not exhibit an affinity to DCKA (Popik et al., 1995; Han et al., 2004), application of DCKA (50 μm) prevented the enhancing effect of glycine on mEPSCs (frequency, p = 0.496, n = 8; amplitude, p = 0.471, n = 8, One-way ANOVA, Fig. 6D–F), Thus, if NMDARs, glycine transporters, and strychnine-sensitive glycine receptors are all excluded, this glycine-mediated enhancing effect can be explained to be mediated by an unidentified glycine-binding substrate; it is sensitive to DCKA and glycine, but not d-serine or strychnine.
Discussion
Using an approach combining electrophysiological, pharmacological, and genetic tools, the present study was originally set to define potential presynaptic NMDARs in the NAc. However, our results suggest that the seemingly presynaptic NMDAR-mediated effects were indeed mediated by an NMDAR-independent mechanism, possibly through unidentified, strychnine-resistant glycine-activated substrates. These results may help reinterpret some of the previous results related to presynaptic NMDARs.
Presynaptic versus postsynaptic mechanisms
An alteration in presynaptic release of neurotransmitters is often associated with alterations in the frequency of mEPSCs. Application of DCKA exerted a consistent inhibitory effect on the frequency of mEPSCs in neurons from three brain regions (Fig. 1). Furthermore, the presynaptic parameters, Pr or N, of excitatory synapses were also decreased by DCKA within the three examined afferents (Fig. 4). These observations suggest the involvement of presynaptic alterations in DCKA-mediated synaptic inhibition. Whereas the Pr and N are two relatively independent parameters, they also reciprocally influence each other in the V-M analysis. An extreme example is that when the Pr at some release sites decreases to 0 (thus these sites become inactive), the total N will decrease. In this case, if the Pr at active release sites is not altered, the total Pr will remain the same because the V-M analysis only measures active release sites to calculate the Pr. Nonetheless, although synapses within three glutamatergic afferents are inhibited by DCKA presynaptically, the inhibition exhibits distinct presynaptic patterns as the P and N were affected differentially (Fig. 4).
In addition to the frequency, application of DCKA also significantly decreased the amplitude of mEPSCs in NAc MSNs (Fig. 1), suggesting the involvement of postsynaptic mechanisms. This interpretation is at odds with the results from the V-M analysis, which show that application of DCKA did not affect the Q (Fig. 4). Specifically, a decrease in the amplitude of mESPC suggests reduced vesicular content of glutamate or reduced number/function of postsynaptic AMPARs, either of which should be reflected by a reduction in the Q. However, the Q was not affected by DCKA within the three examined afferents (Fig. 4). One possibility to reconcile these results is that DCKA-induced reduction in Q occurs not in the VTA-, PFC-, and BLA-to-NAc pathways, but in other glutamate afferents. A second possibility is that DCKA does not affect the Q, but preferentially decreases the Pr of synapses with high Q values. Thus, if the frequency of mEPSCs with large amplitudes is preferentially decreased by DCKA, it would result in a decrease in mean amplitude at the overall level. In the NAc, MSNs receive glutamatergic synaptic inputs from a variety of brain regions, and very often, these synaptic inputs converge at the single MSN level (O'Donnell and Grace, 1995; Finch, 1996; French and Totterdell, 2002, 2003). These glutamatergic inputs are thought to carry different emotional and motivational information and differentially influence the overall functional output of NAc MSNs (Kalivas and McFarland, 2003; Lüscher and Malenka, 2011). The sizes of presynaptic vesicles, and thus likely Q, at synapses from different pathways to NAc MSNs are substantially different (French and Totterdell, 2004). Thus, the Q, depending on its “size,” may influence the regulation susceptibility of different excitatory inputs to the NAc. A third possibility is that application of DCKA reduces basal multivesicular release. Multivesicular release may preferentially occur at high Pr conditions, and if reduced, the variance of EPSC amplitudes will also be reduced, and predictably with a more robust reduction in EPSCs within larger amplitudes (Fig. 4). If multivesicular release also occurs in mEPSCs (Wall and Usowicz, 1998), suppression of multivesicular release would yield a reduction in the amplitude of mEPSCs. It is not clear, however, how prevalent multivesicular release is in NAc MSNs, although it is the case for dorsal striatal MSNs upon single presynaptic stimulations in young rats (postnatal 15–18 d) (Higley et al., 2009). Nonetheless, these three possibilities suggest that the seemingly postsynaptic effects of DCKA (i.e., changes in the amplitude of mEPSCs) can be explained by presynaptic mechanisms.
It is noteworthy that our results cannot definitively rule out postsynaptic mechanisms. If the function of postsynaptic AMPARs is decreased by DCKA, the amplitude of mEPSCs will be decreased, and because of this decrease, some mEPSCs may become undetectable, resulting in a decrease in the frequency of mEPSCs. Thus, some of the seemingly presynaptic mechanisms can also be explained by postsynaptic mechanisms. However, postsynaptic mechanisms cannot readily explain several apparent presynaptically oriented alterations induced by DCKA, including the change in the Pr, the lack of change in Q, the decreased blockade time course of EPSCs by Baf, the change in the PPR of evoked EPSCs, and the lack of change in puff-induced AMPAR-mediated currents. Moreover, in the above presented NAc MSNs (Fig. 1), 3 of 18 neurons exhibited negligible DCKA-induced inhibition in mEPSC amplitude (by 3.7 ± 0.2%), but these neurons exhibited the same degree of decrease in mEPSC frequency as the population average (by 26.9 ± 2.9%). Thus, although postsynaptic mechanisms are possibly involved, presynaptic mechanisms likely predominate in DCKA-induced inhibition of excitatory synaptic transmission.
Molecular substrates for DCKA
It has been well documented that both DCKA and 7-Cl are highly selective for the glycine-binding site of NMDARs (Kemp et al., 1988; Baron et al., 1990; Leeson et al., 1991; Mugnaini et al., 1998). Using two transgenic mouse lines (Fig. 5), our experiments largely rule out presynaptic NMDARs in DCKA-induced inhibition of excitatory synaptic transmission. Rather, our results raise the possibility that DCKA and 7-Cl may also have other binding partners in the brain, possibly glycine-binding substrates.
Glycine receptor-mediated regulation of synaptic transmission has been reported at mammalian synapses (Turecek and Trussell, 2001; Kawa, 2003; Lee et al., 2009). For these characterized regulations, the involved glycine receptors are sensitive to strychnine, and activation of these receptors facilitates presynaptic release by inducing depolarization of presynaptic terminals, which, in turn, promotes Ca2+ entry through voltage-gated calcium channels (Turecek and Trussell, 2001; Kawa, 2003; Lee et al., 2009). However, these established typical glycine receptors are not likely the potential glycine/DCKA-bound substrates or receptors insinuated in the current study, because the latter ones were DCKA-sensitive but strychnine-resistant, and could function independent of voltage-gated sodium or calcium channels (Fig. 1). Thus, if these glycine/DCKA-bound substrates are glycine receptors, they are atypical receptors with pharmacological properties distinct from those identified previously; they appear tonically and partially active, allowing bidirectional regulations (by DCKA and glycine).
The atypical glycine receptor-oriented scenario is supported by several recent studies. In the salamander retina and HEK cells expressing glycine receptor subunits, application of DCKA inhibits glycine-induced currents in a strychnine-resistant manner (Han et al., 1997, 2004). Thus, the putative glycine receptors that mediate glycine-induced current are atypical as typical glycine receptors are strychnine-sensitive and DCKA-resistant (Han et al., 1997, 2004). Furthermore, in a screening test, the rat cortical tissues are shown to exhibit two clearly dissociable binding sites for either DCKA or 7-Cl; the high affinity site is identified as the glycine-binding site of NMDARs, whereas the low, micromolar-affinity site has not been specified (Mugnaini et al., 1998). This low-affinity site can be atypical glycine receptors suggested by Han et al. (1997, 2004) or other unidentified glycine-binding substrates. In the present study, although DCKA and 7-Cl at 20–100 μm (Kemp et al., 1988; Frankiewicz et al., 2000; Barria and Malinow, 2002; Krasteniakov et al., 2005) would primarily target the high affinity site, partial occupancy of the low affinity site is also possible. Thus, atypical glycine receptors are possible substrates that mediate the observed effect of DCKA. As such, a potential future study would be to identify whether these putative atypical glycine receptors are expressed in the NAc, hippocampus, cortex, and other related brain regions, and if so, whether they are located presynaptically.
Impact of the current findings
Pharmacological activation or inactivation of the glycine site of NMDARs becomes a common means to manipulate NMDAR function in vitro and in vivo. A PubMed search of “glycine site NMDA receptor” reaches >1500 hits (as of 2011), among which ∼1/3 involve using DCKA, 7-Cl, glycine, or strychnine in physiological, pharmacological, or behavioral experiments. Our current results indicate that in addition to NMDARs, these glycine-site tools may also interact with unidentified molecular substrates to regulate synaptic transmission in a manner similar to what is mediated by manipulating NMDARs. These findings provide an empirical base to reconsider related prior results with a new, NMDAR-independent interpretation. Furthermore, our results suggest the existence of a putative strychnine-resistant glycine-binding substrate, possibly a novel class of glycine receptors. This putative receptor, if identified, would introduce a new mechanism in regulating excitatory synaptic transmission.
Notes
Supplemental material for this article is available at http://www.vetmed.wsu.edu/research_vcapp/DongLab/publications/supplemental.aspx. Supplemental Materials: Additional data related to Figure 1 in Huang et al., Searching for Presynaptic NMDA Receptor in the Nucleus Accumbens. This material has not been peer reviewed.
Footnotes
The study was supported by the American Heart Association, the Humboldt Foundation, and NIH–NIDA Grants DA029565, DA028020, DA023206 and DA024570, and DA031551. We thank Drs. Roger Nicoll and Craig Jahr for constructive discussions on the data, Anirvan Ghosh, Susumu Tonegawa, and Armin-Klaus Nave for providing transgenic mouse lines, and Martin Schwarz and Karel Svoboda for plasmids of AAV vector and related suggestions.
References
- Awobuluyi M, Yang J, Ye Y, Chatterton JE, Godzik A, Lipton SA, Zhang D. Subunit-specific roles of glycine-binding domains in activation of NR1/NR3 N-methyl-d-aspartate receptors. Mol Pharmacol. 2007;71:112–122. doi: 10.1124/mol.106.030700. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron BM, Harrison BL, Miller FP, McDonald IA, Salituro FG, Schmidt CJ, Sorensen SM, White HS, Palfreyman MG. Activity of 5,7-dichlorokynurenic acid, a potent antagonist at the N-methyl-d-aspartate receptor-associated glycine binding site. Mol Pharmacol. 1990;38:554–561. [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ. Substituted quinolines as inhibitors of l-glutamate transport into synaptic vesicles. Neuropharmacology. 1998;37:839–846. doi: 10.1016/s0028-3908(98)00080-x. [DOI] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Brasier DJ, Feldman DE. Synapse-specific expression of functional presynaptic NMDA receptors in rat somatosensory cortex. J Neurosci. 2008;28:2199–2211. doi: 10.1523/JNEUROSCI.3915-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Mu P, Ferguson D, Dietz D, Ohnishi YN, Lin Y, Suska A, Ishikawa M, Huang YH, Shen H, Kalivas PW, Sorg BA, Zukin RS, Nestler EJ, Dong Y, Schlüter OM. A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J Neurosci. 2011;31:8163–8174. doi: 10.1523/JNEUROSCI.0016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal RN, Everitt BJ. Neural and psychological mechanisms underlying appetitive learning: links to drug addiction. Curr Opin Neurobiol. 2004;14:156–162. doi: 10.1016/j.conb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002;415:793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- Clements JD, Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Brasier DJ, Feldman DE, Philpot BD. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist. 2008;14:609–625. doi: 10.1177/1073858408322675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Das S, Sasaki YF, Rothe T, Premkumar LS, Takasu M, Crandall JE, Dikkes P, Conner DA, Rayudu PV, Cheung W, Chen HS, Lipton SA, Nakanishi N. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature. 1998;393:377–381. doi: 10.1038/30748. [DOI] [PubMed] [Google Scholar]
- Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci. 2004;5:135–145. doi: 10.1038/nrn1297. [DOI] [PubMed] [Google Scholar]
- Finch DM. Neurophysiology of converging synaptic inputs from the rat prefrontal cortex, amygdala, midline thalamus, and hippocampal formation onto single neurons of the caudate/ putamen and nucleus accumbens. Hippocampus. 1996;6:495–512. doi: 10.1002/(SICI)1098-1063(1996)6:5<495::AID-HIPO3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Frankiewicz T, Pilc A, Parsons CG. Differential effects of NMDA-receptor antagonists on long-term potentiation and hypoxic/hypoglycaemic excitotoxicity in hippocampal slices. Neuropharmacology. 2000;39:631–642. doi: 10.1016/s0028-3908(99)00168-9. [DOI] [PubMed] [Google Scholar]
- French SJ, Totterdell S. Hippocampal and prefrontal cortical inputs monosynaptically converge with individual projection neurons of the nucleus accumbens. J Comp Neurol. 2002;446:151–165. doi: 10.1002/cne.10191. [DOI] [PubMed] [Google Scholar]
- French SJ, Totterdell S. Individual nucleus accumbens-projection neurons receive both basolateral amygdala and ventral subicular afferents in rats. Neuroscience. 2003;119:19–31. doi: 10.1016/s0306-4522(03)00150-7. [DOI] [PubMed] [Google Scholar]
- French SJ, Totterdell S. Quantification of morphological differences in boutons from different afferent populations to the nucleus accumbens. Brain Res. 2004;1007:167–177. doi: 10.1016/j.brainres.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Rasmusson AM, Bunney BS, Roth RH. The NMDA glycine site antagonist (+)-HA-966 selectively regulates conditioned stress-induced metabolic activation of the mesoprefrontal cortical dopamine but not serotonin systems: a behavioral, neuroendocrine, and neurochemical study in the rat. J Neurosci. 1994;14:4937–4950. doi: 10.1523/JNEUROSCI.14-08-04937.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Zhang J, Slaughter MM. Partition of transient and sustained inhibitory glycinergic input to retinal ganglion cells. J Neurosci. 1997;17:3392–3400. doi: 10.1523/JNEUROSCI.17-10-03392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Li P, Slaughter MM. Selective antagonism of rat inhibitory glycine receptor subunits. J Physiol. 2004;554:649–658. doi: 10.1113/jphysiol.2003.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Soler-Llavina GJ, Sabatini BL. Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat Neurosci. 2009;12:1121–1128. doi: 10.1038/nn.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Brown TE, Han MH, Saal DB, Neve RL, Zukin RS, Sorg BA, Nestler EJ, Malenka RC, Dong Y. CREB modulates the functional output of nucleus accumbens neurons: a critical role of N-methyl-d-aspartate glutamate receptor (NMDAR) receptors. J Biol Chem. 2008;283:2751–2760. doi: 10.1074/jbc.M706578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M, Dannhardt G. Antagonists and agonists at the glycine site of the NMDA receptor for therapeutic interventions. Eur J Med Chem. 2003;38:661–670. doi: 10.1016/s0223-5234(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Javitt DC. Is the glycine site half saturated or half unsaturated? Effects of glutamatergic drugs in schizophrenia patients. Curr Opin Psychiatry. 2006;19:151–157. doi: 10.1097/01.yco.0000214340.14131.bd. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, McFarland K. Brain circuitry and the reinstatement of cocaine-seeking behavior. Psychopharmacology (Berl) 2003;168:44–56. doi: 10.1007/s00213-003-1393-2. [DOI] [PubMed] [Google Scholar]
- Kawa K. Glycine facilitates transmitter release at developing synapses: a patch clamp study from Purkinje neurons of the newborn rat. Brain Res Dev Brain Res. 2003;144:57–71. doi: 10.1016/s0165-3806(03)00159-7. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–776. doi: 10.1016/j.neubiorev.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Smith-Roe SL, Holahan MR. Response-reinforcement learning is dependent on N-methyl-d-aspartate receptor activation in the nucleus accumbens core. Proc Natl Acad Sci U S A. 1997;94:12174–12179. doi: 10.1073/pnas.94.22.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JA, Foster AC, Leeson PD, Priestley T, Tridgett R, Iversen LL, Woodruff GN. 7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-d-aspartate receptor complex. Proc Natl Acad Sci U S A. 1988;85:6547–6550. doi: 10.1073/pnas.85.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasteniakov NV, Martina M, Bergeron R. Role of the glycine site of the N-methyl-d-aspartate receptor in synaptic plasticity induced by pairing. Eur J Neurosci. 2005;21:2782–2792. doi: 10.1111/j.1460-9568.2005.04099.x. [DOI] [PubMed] [Google Scholar]
- Kretschmer BD, Schmidt WJ. Behavioral effects mediated by the modulatory glycine site of the NMDA receptor in the anterodorsal striatum and nucleus accumbens. J Neurosci. 1996;16:1561–1569. doi: 10.1523/JNEUROSCI.16-04-01561.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda H, Kutner RH, Bazan NG, Reiser J. Simplified lentivirus vector production in protein-free media using polyethyleneimine-mediated transfection. J Virol Methods. 2009;157:113–121. doi: 10.1016/j.jviromet.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Larsen RS, Corlew RJ, Henson MA, Roberts AC, Mishina M, Watanabe M, Lipton SA, Nakanishi N, Pérez-Otaño I, Weinberg RJ, Philpot BD. NR3A-containing NMDARs promote neurotransmitter release and spike timing-dependent plasticity. Nat Neurosci. 2011;14:338–344. doi: 10.1038/nn.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lee EA, Cho JH, Choi IS, Nakamura M, Park HM, Lee JJ, Lee MG, Choi BJ, Jang IS. Presynaptic glycine receptors facilitate spontaneous glutamate release onto hilar neurons in the rat hippocampus. J Neurochem. 2009;109:275–286. doi: 10.1111/j.1471-4159.2009.05960.x. [DOI] [PubMed] [Google Scholar]
- Leeson PD, Baker R, Carling RW, Curtis NR, Moore KW, Williams BJ, Foster AC, Donald AE, Kemp JA, Marshall GR. Kynurenic acid derivatives. Structure-activity relationships for excitatory amino acid antagonism and identification of potent and selective antagonists at the glycine site on the N-methyl-d-aspartate receptor. J Med Chem. 1991;34:1243–1252. doi: 10.1021/jm00108a002. [DOI] [PubMed] [Google Scholar]
- Li Y, Krupa B, Kang JS, Bolshakov VY, Liu G. Glycine site of NMDA receptor serves as a spatiotemporal detector of synaptic activity patterns. J Neurophysiol. 2009;102:578–589. doi: 10.1152/jn.91342.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low CM, Wee KS. New insights into the not-so-new NR3 subunits of N-methyl-d-aspartate receptor: localization, structure, and function. Mol Pharmacol. 2010;78:1–11. doi: 10.1124/mol.110.064006. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C, Mesic I, Bartholomäus I, Nicke A, Betz H, Laube B. Principal role of NR3 subunits in NR1/NR3 excitatory glycine receptor function. Biochem Biophys Res Commun. 2007;354:102–108. doi: 10.1016/j.bbrc.2006.12.153. [DOI] [PubMed] [Google Scholar]
- Makinodan T, Hahn TJ, McDougall S, Yamaguchi DT, Fang M, Iida-Klein A. Cellular immunosenescence: an overview. Exp Gerontol. 1991;26:281–288. doi: 10.1016/0531-5565(91)90021-d. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnaini M, Antolini M, Corsi M, van Amsterdam FT. [3H]5,7-dichlorokynurenic acid recognizes two binding sites in rat cerebral cortex membranes. J Recept Signal Transduct Res. 1998;18:91–112. doi: 10.3109/10799899809047739. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilpel N, Landeck N, Klugmann M, Seeburg PH, Schwarz MK. Rapid, reproducible transduction of select forebrain regions by targeted recombinant virus injection into the neonatal mouse brain. J Neurosci Methods. 2009;182:55–63. doi: 10.1016/j.jneumeth.2009.05.020. [DOI] [PubMed] [Google Scholar]
- Popik P, Lewin A, Berrang B, Nowak G, Layer R, Skolnick P. [3H]1-aminocyclopropanecarboxylic acid, a novel probe for strychnine-insensitive glycine receptors. Eur J Pharmacol. 1995;291:221–227. doi: 10.1016/0922-4106(95)90061-6. [DOI] [PubMed] [Google Scholar]
- Quastel DM. The binomial model in fluctuation analysis of quantal neurotransmitter release. Biophys J. 1997;72:728–753. doi: 10.1016/s0006-3495(97)78709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiteri L, Stigliani S, Siri A, Passalacqua M, Melloni E, Raiteri M, Bonanno G. Glycine taken up through GLYT1 and GLYT2 heterotransporters into glutamatergic axon terminals of mouse spinal cord elicits release of glutamate by homotransporter reversal and through anion channels. Biochem Pharmacol. 2005;69:159–168. doi: 10.1016/j.bcp.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Banerjee A, Paulsen O. Presynaptic NMDA receptors and spike timing-dependent depression at cortical synapses. Front Synaptic Neurosci. 2010;2:18. doi: 10.3389/fnsyn.2010.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, Silver RA. Estimation of quantal parameters with multiple-probability fluctuation analysis. Methods Mol Biol. 2007;403:303–317. doi: 10.1007/978-1-59745-529-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Neher E. Estimating synaptic parameters from mean, variance, and covariance in trains of synaptic responses. Biophys J. 2001;81:1970–1989. doi: 10.1016/S0006-3495(01)75848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Kötter R, Staiger JF. Mapping functional connectivity in barrel-related columns reveals layer- and cell type-specific microcircuits. Brain Struct Funct. 2007;212:107–119. doi: 10.1007/s00429-007-0147-z. [DOI] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Tricklebank MD, Bristow LJ, Hutson PH, Leeson PD, Rowley M, Saywell K, Singh L, Tattersall FD, Thorn L, Williams BJ. The anticonvulsant and behavioural profile of L-687,414, a partial agonist acting at the glycine modulatory site on the N-methyl-d-aspartate (NMDA) receptor complex. Br J Pharmacol. 1994;113:729–736. doi: 10.1111/j.1476-5381.1994.tb17054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Ultanir SK, Kim JE, Hall BJ, Deerinck T, Ellisman M, Ghosh A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci U S A. 2007;104:19553–19558. doi: 10.1073/pnas.0704031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MJ, Usowicz MM. Development of the quantal properties of evoked and spontaneous synaptic currents at a brain synapse. Nat Neurosci. 1998;1:675–682. doi: 10.1038/3677. [DOI] [PubMed] [Google Scholar]
- Wilcox KS, Fitzsimonds RM, Johnson B, Dichter MA. Glycine regulation of synaptic NMDA receptors in hippocampal neurons. J Neurophysiol. 1996;76:3415–3424. doi: 10.1152/jn.1996.76.5.3415. [DOI] [PubMed] [Google Scholar]
- Wlaź P, Ebert U, Löscher W. Low doses of the glycine/NMDA receptor antagonist R-(+)-HA-966 but not d-cycloserine induce paroxysmal activity in limbic brain regions of kindled rats. Eur J Neurosci. 1994;6:1710–1719. doi: 10.1111/j.1460-9568.1994.tb00563.x. [DOI] [PubMed] [Google Scholar]
- Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci. 2006;26:406–410. doi: 10.1523/JNEUROSCI.4413-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, Lübke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]