

Abstract
Parasite growth within the erythrocyte causes dramatic alterations of host cell which on one hand facilitates nutrients acquisition from extracellular environment and on other hand contributes to the symptoms of severe malaria. The current paper focuses on interactions between the Plasmodium parasite and its metabolically highly reduced host cell, the natural selection of numerous polymorphisms in the genes encoding hemoglobin and other erythrocyte proteins.
1. Introduction
Malaria is caused by approximately 200 known Plasmodium species that infect particular lineages of primates, rodents, birds, and reptiles. The Plasmodium species exhibit strong specificity for certain host species while occasional jumps between host species have been reported, and two such jumps are thought to account for human origin of P. vivax 53,000–265,000 years ago when an ancestral parasite switched host from macaque monkeys to humans and for P. knowlesi recently [1]. At present, five members of Plasmodium species (P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi) have been shown to cause human malaria [2], and all develop through the same general life cycle which alternates between the human host and the anopheline mosquito. The cycle begins when a Plasmodium infected female anopheline mosquito probes for a blood meal and injects the sporozoites into the dermis. Sporozoites then migrate to the liver, infect hepatocytes, and remain in a clinically silent stage. Between 5 to 16 days later depending on the Plasmodium species, sporozoite undergoes a process of asexual replication releasing thousands of merozoites per infected hepatocytes into the blood. Some P. vivax and P. ovale sporozoites are predestined to develop into nondividing hepatocytic forms (hypnozoites) which can remain latent in the liver for months to years until they activate and cause relapse infections. Once inside the bloodstream each merozoite then invades an erythrocyte and resides in a self-created membrane-bound vacuole, and undergoes repetitive rounds of growth, division, and invasion in one-day (P. knowlesi), two-day (P. falciparum, P. vivax, and P. ovale), or three-day (P. malariae) periods leading to the symptoms and complications of malaria. A subset of developing merozoites differentiates into male and female gametocytes, and the Plasmodium life cycle continues when both of these gametocytes are taken up by the mosquitoes during blood meals. In the mosquito midgut, gametocytes undergo fertilization and maturation forming an infective ookinete, which migrates through the midgut into the hemocele and develops into the oocyst in which sporozoites are formed. When fully matured, the oocysts burst and release sporozoites, which migrate into the mosquito's salivary glands to initiate another life cycle.
The malaria-associated pathology only occurs during the blood stage of infection, and of the five, P. falciparum is the most deadly parasite species in humans as host erythrocytes infected with its mature forms avoid the splenic clearance by sequestering in capillaries and microvenules of the brain and other vital organs, a process that is not common with erythrocytes infected by other human malaria parasite species. Furthermore, the parasite displays a family of functionally redundant ligands to invade human cells and has the ability of antigen variation to counter host defenses that pose a great threat to control infections by this deadly parasite. In the course of parasite growth within the erythrocytes, the dramatic alterations of the host cell facilitate the acquisition of nutrients from the extracellular environment which are not provided by the host cell, while at the same time have the detrimental consequence of the symptoms of severe malaria [3]. The current paper focuses on interactions between the Plasmodium parasite and its metabolically highly reduced host cell, appearance of pathogen-induced novel physiological properties in a host cell, and finally the natural selection of numerous polymorphisms in the genes encoding hemoglobin and other erythrocyte proteins.
2. Parasite Invasion of Red Blood Cell
The malaria parasite P. falciparum infects mammalian erythrocytes which largely lack biosynthetic pathways and is metabolically sluggish, nutritionally deprived, and devoid of intracellular compartments. From the time of first contact, it takes about half a minute for Plasmodium merozoites to become internalized into the erythrocytes and can be divided into two distinct phases: (i) preinvasive and (ii) invasive phase [4]. In the preinvasive phase, waves of deformation on the erythrocyte plasma membrane initiated that soon ceased leaving the merozoite attached to the erythrocyte by its anterior end. The host cell's membrane deformation produced by erythrocytic cytoskeletal changes increases the area of contact between merozoite and the host to assist apical reorientation of the merozoite and is thought to be stimulated by a localised influx of calcium ions triggered by merozoite contact [5, 6]. The duration and degree of deformation may be dependent on closeness of the apical end to the erythrocyte surface during initial contact as the merozoite must reorientate and bring its penetrative apical end in contact with the RBC prior to invasion [4]. However, both the parasite ligands and RBC receptors involved in this process have not been defined. Murphy et al. [7] have shown the role of G protein-coupled β-adrenergic receptors on the RBC membrane in membrane deformability during merozoite invasion probably by cyclic AMP-induced changes in the rigidity of underlying cytoskeleton. After deformation ceases and the RBC resumes its normal shape, in the invasive phase merozoite rhoptries and micronemes are thought to secrete proteins like P. falciparum reticulocyte-binding protein homolog (PfRh) and erythrocyte-binding antigens (EBAs) that are involved in tight junction (TJ) formation [8]. Following the establishment of TJ between parasite and RBC, parasite entry to the host cell is mediated by the movement of TJ from the apical end to posterior end of the merozoite in a complex series of events powered by the parasite actin-myosin motor [9]. Invasion then triggers the host cell to undergo echinocytosis, possibly due to water loss from the erythrocyte stimulated by an efflux of potassium and chloride ions. The loss of potassium could be caused by the opening of Gardos channels in the erythrocytic membrane stimulated by an influx of calcium ions into the erythrocyte to reseal the erythrocyte membrane behind the invading merozoite [4]. The postinvasive phase takes several minutes for the iRBC to resume its normal shape depending on whether it is multiply infected, and during this phase merozoites transform into amoeboid ring-stage [4, 5]. It is the infection of and multiplication within the RBC that cause the severe symptoms of malaria, and a large part of the intraerythrocytic development takes place in the parasitophorous vacuole (PV), a compartment which forms an interface between the parasite and the cytoplasm of the host cell. Although the molecular mechanisms leading to the formation of PV remain unknown, the exclusion of major erythrocyte membrane proteins (such as the glycophorins or the anion transporter band 3) from PVM and the contents discharged of rhoptries and micronemes from the apical end of parasite during the process of erythrocyte invasion suggest that it is parasite derived [10, 11]. However, the observation of several erythrocyte membrane proteins in the PVM during or soon after parasite invasion by Bietz et al. [12] suggests its origin to be more complex. In the course of parasite growth the host cell undergoes dramatic alterations and includes establishment of nutrients transportation between host cell cytoplasm and the parasite within PV, across PVM and maintenance of the electrolyte balance of the host cell. Moreover, the iRBC needs surface modulation in order to import nutrients not readily provided by the RBCs (such as isoleucine) and engaged in the linked processes of sequestration and antigenic variation.
3. Transporting System of iRBC
The rapidly growing and replicating parasite has a high nutritional demand which cannot be satisfied by the host cell, and the seclusion of Plasmodium parasite within the PV raises the need to induce new permeability pathways (NPPs) for the acquisition of a variety of nutrients from the extracellular milieu and from the host cell cytosol. However, if the RBC membrane becomes very permeable, the high concentrations of haemoglobin inside would normally encourage fluid to move in, swelling and bursting the cell. To prevent early bursting of the invaded RBC, the parasite digests a large proportion of haemoglobin than it needs for its own proteins [13], mostly by cytostome, a large phagosome-like structure of the intraerythrocytic trophozoite stage of the parasite [14]. Although hemoglobin is the main source of amino acids for the parasite, the uptake of several other amino acids including isoleucine, pantothenic acid (the precursor of coenzyme A), and many other solutes must be made from the surrounding medium of the host cell [15, 16]. There are three proposed models (parasitophorous duct model, metabolic window model, and sequential uptake model) for the acquisition of nutrients from the extracellular milieu and in all models; the solutes are proposed to find access from the PV into the parasite via transporters within the parasite plasma membrane [3, 17]. Although, a unifying model explaining the uptake of variety of substances is a major challenge, the sequential uptake model has recently received considerable attention as working hypothesis [3, 17]. However, the nature of the proteins mediating the transport across the erythrocyte membrane is debated and has long been thought to be parasite derived owing to the lack of protein biosynthetic pathway in noninfected RBC. Recent studies have shown the involvement of endogenous host cell proteins within the erythrocyte membrane in solute transport after activation via infection-induced oxidative stress and are protease sensitive [3, 18, 19]. Studies have shown these channels as anion-selective and either the parasite exported kinases modulate the activity and/or substrate specificity of otherwise silent endogenous transport proteins [20] or auxiliary parasite proteins may be required for activation [19]. Thus, secreted parasite proteins contributing to transporter activation could be potential drug targets. As well as gaining access to the parasite many nutrients must further be transported into organelles such as the apicoplast, which contains several plant or bacteria-specific essential biochemical pathways, and rely on a constant delivery of raw materials from the parasite cytosol. However, the molecules mediating these transport processes remain largely uncharacterized, and further research on this may help finding a highly significant target for antimalarial drug development. Furthermore, the survival of parasite under low cytosolic Ca++ concentration in RBC poses an interesting problem to the parasite that depends on a Ca++ signaling system to carry out its vital functions. Gazarini et al. [21] propose that the Ca2+ ATPase on RBC membrane responsible for pumping Ca++ across the plasma membrane toward the extracellular space, after membrane invagination during erythrocyte invasion, faces the parasite and transports Ca++ from the host cytosol across the PVM and into the PV allowing a Ca++ gradient to form and makes Ca++ signaling within the malaria parasite possible. This raises the possibility that the RBC Ca++ ATPase could be a potential site of intervention in the control of malaria and needs modification in the PVM to be specifically targeted.
4. Protein Trafficking
Owing to very restricted access of host cell proteins to the PV, there is an active unidirectional transport of parasite proteins beyond the confines of its own plasma membrane (PM), the PVM, and across the RBC cytosol to the host cell plasma membrane [22]. The parasite exports about 5% of its genome-encoded proteins into the host cell cytosol [23]. Although the function of most of these exported proteins is unknown, a recent gene knockout screen has shown about 25% of exported proteins to be implicated in essential blood-stage survival role [24], and some of which on erythrocyte surface appear to have virulence-associated roles, such as promoting infected cell adhesion and/or rigidity and are the major determinants of the unique pathogenicity of this parasite [23, 25]. Trafficking of proteins within the confines of the parasite appears to involve most of the elements of a classical vesicle-mediated secretory pathway [22]. However, trafficking of proteins beyond the confines of the parasitophorous vacuole membrane represents a major logistical challenge. Studies have shown that proteins destined to export from the parasite arrive at the PPM, released into the vacuolar lumen or trafficked across the PVM either in one step at contact points between PPM and PVM or in two steps by first delivering into the lumen of the vacuole followed by translocation across the PVM [26]. In the later case, there must be a protein conducting channel (PCC) to translocate the protein across the PVM, and a pentapeptide RxLxE/Q/D towards the N terminus of the protein referred to as the Plasmodium export element (PEXEL) or vacuolar translocation signal (VTS) directs traffic of proteins to locations beyond the PVM (reviewed in [27]). Since the PEXEL motif is cleaved within the endoplasmic reticulum (ER) of the parasite before export with processing at the conserved leucine (L) and acetylation of the new N-terminus that commences with xE/Q/D, how the PEXEL/VTS acts to export the protein to its final destination remains unclear. Recently, de-Koning-Ward et al. [28] have identified a translocon in the PVM called PTEX complex, which recognize, and translocates proteins deposited into the vacuolar space and destined for export. However, the absence of this protein translocon machinery even in closely related parasites proves the difficulty to trace its evolutionary origin and provides an “Achilles heel” for antimalaria drug developers. The existence of several PEXEL-negative exported proteins (PNEPs) like the skeleton binding protein 1 (SBP1), the membrane-associated histidine-rich protein 1 (MAHRP1), and ring-exported protein (REX 1&2) indicates that alternative export pathways might also exist. However, Spielmann and Gilberger [29] suggest that despite a lack of recognizable export motifs in PNEPs, there are similarities to mature PEXEL proteins downstream of PEXEL cleavage suggesting that the export pathway might accommodate both types of proteins. Clearly, experimental data are now required to provide evidence for this hypothesis. Although, mature RBC has no endogenous trafficking machinery, some exported proteins destined for the erythrocyte cytoplasm and membrane associate with P. falciparum-induced heterogeneous structures of convoluted flotillas of flattened discs that are tethered to the RBC membrane, called the maurer's clefts (MCs), and the later play an important role in the trafficking of parasite proteins to the surface of the host cell [30, 31]. The parasite displays remarkable versatility in the types of proteins exported to the MCs and in the functions of the proteins within the MCs. Two new pathways are suggested for export of MC resident proteins to the MCs: (i) a PEXEL-independent pathway, where PNEPs might accumulate or diffuse laterally into the MCs as MCs form and “bud” out of the PVM; however, additional experimental evidences are needed to verify protein entry into the forming MCs and the requirements for a “sorting signal” and (ii) a second pathway, where some PNEPs and PEXEL-positive exported proteins (PPEPs) cross the PVM through a transporter and become associated with MCs, where they translocate to the erythrocyte cytoplasm or the erythrocyte membrane [32]. Distinct and independent from the MCs, a tubovesicular membrane network (TVN) extending from the PVM is present within the infected erythrocyte, and whether interconnections exist between the TVN and MCs such that there is continuity between both compartments is another area of controversy [33, 34]. Furthermore, TVN possesses distinct markers (such as, erythrocyte vesicle protein 1, TVN junction protein, sphingomyelin synthase, etc.) not found in the MCs, and inhibition of TVN formation had no effect on protein export through the MCs. Tamez et al. [34] suggest that the parasite employs separate membrane networks for export and import of macromolecules, and unlike the MCs, TVN is speculated to be open to the outside of the erythrocyte surface and is suggested to be involved in macromolecular import. Since, the properties of the RBCs are modified by parasite secreted proteins, for therapeutic interventions to succeed, the crucial steps of variant antigen transport through the MCs will need to be identified.
5. Egress from RBC
During erythrocytic development the parasite stays in the PV through successive stages like ring, trophozoite, and schizont. By the time, most of the RBC hemoglobin is consumed, and after several rounds of asexual division the schizont produces 8–24 mature merozoites which must breech the PV and RBC membranes to escape from the host cell for another round of RBC invasion. The merozoite egress from host erythrocyte is a tightly regulated process mediated by multiple classes of proteolytic enzymes. Recently, Arastu-Kapur et al. [35] have identified two proteases, the subtilisin family serine protease PfSUB1 and the dipeptidyl peptidase 3 (DPAP3) as primary regulators of this process. The regulated secretion of PfSUB1 into the PV from the exoneme a novel organelle that is distinct from the apical organelles processes the abundant papain-like serine repeat antigen (SERAs), which in turn act on membrane components of the PV and RBC to initiate host cell rupture [35, 36]. The DPAP proteases are a class of cysteine proteases, and the DPAP3 is suggested to contribute to parasite egress by regulating the maturation of PfSUB1. Furthermore, the related DPAP1 protease has an essential role in end-stage hemoglobin degradation [37], and both class of proteases (DPAP and PfSUB1) could be a potential target for intervention.
5.1. Host-Parasite Interactions and Malaria Severity
The various clinical features of malaria such as impaired consciousness, coma, difficulty in breathing, severe anaemia, and multiorgan failure occur during the blood stage of infection and thought to occur because of a combination of a high parasite burden and the ability of parasitized erythrocytes to adhere to vascular endothelial cells (cytoadherence), uninfected erythrocytes (rosetting), and platelets (clumping or autoagglutination) [41, 42]. All these phenomena have been linked to interactions between parasite-encoded clonally variant antigens on the surface of parasitized erythrocytes and host receptors. Three families of variant genes have been characterized in P. falciparum: the var genes encoding P. falciparum erythrocyte membrane protein 1 (PfEMP1); the repetitive interspersed family (rif) of genes; the subtelomeric variant open reading frame (stevor) genes. Although, rif and stevor genes appear to be closely related, their receptors are yet to be identified, and in most cases adhesion to host receptors has been shown to be mediated by PfEMP1. The var genes are tightly regulated at the transcriptional level, expressed during the pigmented trophozoite and schizont stages and the resultant PfEMP1 traffic from the Maurer's clefts to the erythrocyte membrane where these are organized into knob-like structures [41]. Out of a family of 60 var genes in a parasite, only one is expressed at a time giving rise to an antigenically distinct PfEMP1 variant [43] and contains extracellular regions consisting of tandemly arranged cysteine-rich domains called duffy-binding-like (DBL), cysteine-rich interdomain regions (CIDR), and C2 domains [44]. The number, location, and type of DBL and CIDR domains vary among PfEMP1 variants, and on the basis of 5' conserved upstream regions, the PfEMP1 can be divided into three major groups (A, B, and C) [45] with different functional and clinical significance, and the binding domains for several host receptors, such as CD36, complement receptor 1, and ICAM1, have been mapped to individual DBL and CIDR domains (Figure 1). The switching of var gene expression can occur at each new asexual blood stage cycle and allows the parasite to modify the antigenic and functional properties of infected erythrocytes, thereby evading immunity and altering adhesion capabilities. The adhesion properties of parasite isolates are therefore not fixed, but can change in subsequent cycles as PfEMP1 expression changes and allow it to avoid the host's normal splenic clearance mechanisms. The capacity for phenotypic switching provides an extralevel of complexity, and the factors that determine which var gene is selected for transcription in each IE are currently unclear [41]. Moreover, although cell adhesive properties of IEs lead to microvascular obstruction, metabolic disturbances, and release of damaging inflammatory mediators which can combine to cause severe disease and death of the human host, out of the three major adhesion phenotypes (cytoadherence, resetting, and platelets clumping), which phenotype (s) contribute to life-threatening malaria has proved difficult. Although, all P. falciparum isolates sequester, yet not all infections lead to life threatening disease, and current data suggest the possibility of geographic variation in adhesion phenotypes causing severe malaria (Table 1) which might be due to differences in malaria transmission levels and host immunity. In addition, the ability of P. falciparum parasites to express selectively var2csa in pregnant hosts mediates attachment of IEs to chondroitin sulfate A (CSA) expressed on the syncytiotrophoblasts of the placenta and is associated with intrauterine growth restriction, miscarriage, and premature delivery [46, 47]. Mothers in their first pregnancy are particularly susceptible to these complications; however after several pregnancies acquired immunity to CSA-binding PfEMP-1 molecules protects the mothers as well as their fetuses and provides strong support for the contribution of variant antigens to the parasite's immune evasion and virulent mechanisms. IEs are also known to bind to a variety of immune system cells, which has important immunological consequences and are beyond the scope of this paper.
Figure 1.
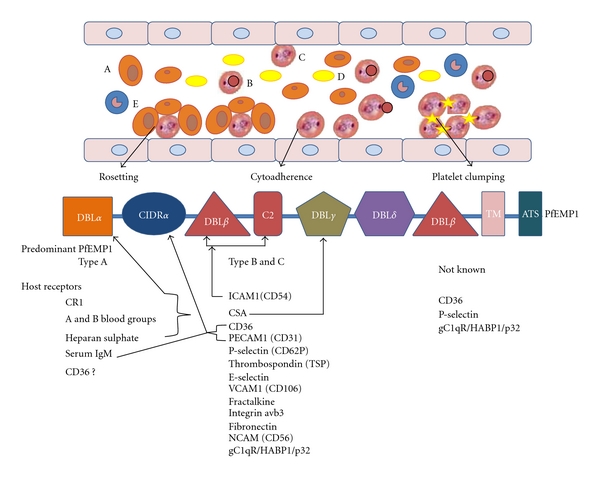
(i) Erythrocytes infected with mature forms of P. falciparum parasites (pigmented trophozoites and schizonts) have the ability to bind to vascular endothelium of various organs and tissues such as heart, lung, brain, muscle, and adipose tissue (sequestration), uninfected erythrocytes (rosetting), and platelets (platelet-mediated clumping) thus, allowing only young ring forms of the parasite to be detected in human peripheral blood samples. Rosetting and platelet-mediated clumping are phenotypes that are displayed by some but not all P. falciparum isolates, and the PfEMP1 variants that mediate rosetting are predominantly of the group A type, which do not adhere to CD36. Owing to the fact that heparan sulphate mediates binding of rosetting IEs to endothelial cells, it is unclear whether binding of IE to heparan sulphate on endothelial cells occurs independently of rosetting, or all parasites that bind heparan sulphate form rosettes. Although three receptors for cytoadherence have been identified for platelet clumping, the molecular mechanisms of the interaction of P. falciparum ligands with platelets are not fully understood, and PfEMP1 is thought to be a likely candidate molecule. Furthermore, these receptors do not form clump in all the cases and suggest the participation of distinct epitopes on these receptors or additional platelet receptors in clumping formation. (ii) The predicted domain organization and binding properties of PfEMP-1to different host receptors. Arrow from the receptors to PfEMP1 domain indicates their respective binding sites, and for other receptors, the binding domains are yet to be identified. A: Erythrocyte, B: Infected erythrocyte, C: Infected erythrocyte at pigmented trophozoite stage, D: Platelet, and E: Leukocyte.
Table 1.
Geographic variations in malaria pathogenesis.
| South East Asia | Africa | Ref |
|---|---|---|
| Low transmission in general | High transmission | [38] |
| Severe malaria affects all age group | Severe malaria affects mostly the children under 5 yrs of age | [39] |
| Multiorgan failure is common with seizures, respiratory distress, and anemia being more common in children whereas renal and hepatic failures are in adults. | Cerebral malaria, severe anemia, and respiratory distress are common | [39, 40] |
| Total parasite burden is associated with risk of severe malaria and deaths | This relationship is less clear as children tolerate high parasitemias without developing severe malaria. | [38] |
| Rosetting is not associated with severe malaria | Rosetting is associated with severe malaria | [41] |
| Platelet-mediated clumping is associated with severe malaria | Platelet-mediated clumping is associated with severe malaria or high parasitemia | [41] |
5.2. Red Blood Cell Polymorphisms and Malaria
Malaria has exerted great evolutionary pressure on the human genome resulting in the natural selection of numerous polymorphisms in the genes encoding some erythrocyte surface proteins, hemoglobin, and other effectors of immunity. In malaria-endemic areas of disease, the combined effects of very high incidence of infection and malaria-associated deaths mostly in young children favor selection and retention of infection-resistant genetic variants although in some instances homozygosity for these same variants can cause other diseases that might have otherwise disfavored their retention [57, 58]. Genetic variants of erythrocytes affecting invasion and replication within the RBC and/or elimination of iRBC have a major effect on infection. The existence of naturally occurring red cell variants that are deficient in specific surface proteins involved in parasite invasion gives protection against severe malaria and includes the red cell glycophorins (GYP), band 3, duffy negative, CR1 variants, and ABO group (Table 2). Of particular interest is the ABO Glycosyltransferase (GlycosylT), a branching enzyme that adds N-acetylgalactosamine, galactose, or neither tothe H precursor to create the major blood groups A, B, and O, respectively. Loss of ABO GlycosylT function results in “O” group and functional ABO enzyme (in particular the A haplotype) have been associated with greater risk of severe malarial anemia, implying that the O blood group offers a certain degree of protection against malaria [51].
Table 2.
Red blood cell polymorphisms and mechanism of protection against severe malaria.
| Name | Gene affected | Polymorphisms | Mechanism of protection | References |
|---|---|---|---|---|
| Invasion | ||||
| Membrane proteins | ||||
| Duffy negative | FY | GATA-1 motif | Duffy-negative RBCs fail to form an apical junction and prevents invasion of P. vivax and P. knowlesi | [3] |
| Glycophorin C deficiency | GYP C | Exon3 deletion | Protection against EBA-140-mediated invasion by P. falciparum parasites. Mechanism in common with other causes of ovalocytosis | [48, 49] |
| Band 3 | SLC4A1 | 27 bp deletion | Resistance to invasion. Increased adhesion of P. falciparum-infected ovalocytes to CD36, thus reducing neurovascular binding of iRBCs in the brain | [48] |
| CR proteins | CR1 | Sl2 or McCb | Reduced ability of P. falciparum—infected CR1—deficient red blood cells to form rosettes | [50] |
| ABO | ABO GlycosylT | Polymorphisms in exon 6 and 7 | Loss of ABO Glycosyltransferase function results in the O blood group which prevents rosette formation | [51] |
| Replication within the RBC and/or elimination of iRBC | ||||
| RBC enzymes | ||||
| G6PD deficiency | G6PD | A376G/G202A [G6PD(A–)] | Early phagocytosis of iRBCs | [52] |
| PK deficiency | PKLR | *About 200 variants | Reduced rate of parasite replication within RBC and enhanced phagocytosis | [53] |
| Hemoglobinopathies | ||||
| (i) Structural variants | ||||
| HbS | HBB | β6: glutamate to valine | Increased sickling of parasitized erythrocytes leading to enhanced clearance by the spleen. Reduced erythrocyte invasion, early phagocytosis, and inhibited parasite growth under low oxygen tension in venous microvessels. Altered PfEMP-1 display and reduced cytoadherence of parasitized erythrocytes Enhancement of innate and acquired immunity | [1, 54] |
| HbC | HBB | β6: glutamate to lysine | Altered PfEMP-1 display and reduced cytoadherence of parasitized erythrocytes | [55] |
| HbE | HBB | β26: glutamate to lysine | Unidentified membrane abnormality renders resistant to invasion | [56] |
| (ii) Thalassemia | ||||
| α-thalassemia | HBA1/HBA2 | 3.7-kb deletion | Reduced expression of CR1 reduces P. falciparum resetting and confers protection against severe malaria. Increased microerythrocyte count in homozygotes reduces the amount of hemoglobin lost for any given parasite density, thus protecting against severe anemia | [1] |
*Population distribution of variants are yet to be established.
The mature red blood cells depend on anaerobic metabolism of glucose due to the lack of mitochondria and deficiencies of two enzymes of glucose metabolism (Pyruvate kinase; PK and Glucose- 6-phosphate dehydrogenase; G6PD) have been shown to protect against malaria. Recent in vitro studies have shown that PK-deficient homozygotes had reduced rate of parasite replication and had increased phagocytosis by macrophages in both homo- and heterozygote PK-deficient mutant [53]. Although, the molecular basis of PK deficiency is diverse, its population distribution has yet to be established through systematic studies. Similarly, the G6PD deficiency has been shown to be protective against malaria probably due to the early phagocytosis of iRBCs and may result from accelerated oxidative membrane damage in deficient cells with impaired antioxidant defence. Like PK deficiency, the G6PD deficiency has diverse genetic origins and in some malaria-endemic areas was observed at increased frequency [52].
Once inside the erythrocyte, Plasmodium parasites degrade hemoglobin (Hb) for nutritional needs; however, disorders of hemoglobin structure (HbS, HbC, HbE) and production due to deletions or point mutations in the noncoding portion of the globin genes causing inadequate synthesis of the α- and β-globin chains (α- and β-thalassaemias resp.) have been shown to protect against death from malaria. Although, HbAS is strongly protective against all forms of clinical malaria, HbC variants appear to be protective against relatively specific cerebral malaria, and both are associated with reduced parasite densities and increased phagocytosis of infected erythrocytes [48]. However, in HbAS, HbAC, and HbCC individuals who still display substantial parasite densities, the mechanism for protection against severe malaria was unknown. Recently, the protective effect of HbAS, HbAC, and HbCC variants is attributed to the altered expression of PfEMP-1 and reduction in binding of these iRBCs to endothelial monolayers and blood monocytes possibly interfering trafficking and anchoring of PfEMP‑1, mediated by elevated levels of membrane-bound, oxidized, and denatured hemoglobin molecules (hemichromes) in HbS and HbC iRBCs [54, 55]. In contrast to this, the α-thalassaemia appears to be protective against relatively specific severe malaria anaemia and has no effect on parasite densities suggesting a different protection mechanism. This is further supported by the study of Williams et al. [59] who showed the negative epistatic interactions between HbS and α-thalassaemia alleles when coinherited and lost their individual protective effect. Although, the mechanism of protection against severe anemia is unknown, the observation of reduced expression of CR1 in α-thalassaemia [60] attributes the reduction in CR1-mediated rosetting could have some roles and needs to be explored in further clinical studies. The protective effect of HbE variant is associated with reduced invasion of HbE erythrocytes by Plasmodium parasites, and the mutation frequency is very common in Southeast Asia with up to a 70% carrier in some endemic regions [56].
6. Conclusion
Efforts made towards the eradication of malaria have so far been unsuccessful. Owing to the confinement of malaria-associated pathology to the blood stage of infection, understanding of host-Plasmodium interactions at this stage is essential to design new intervention strategies against severe malaria. Understanding these interactions may lead to novel therapeutic approaches based on impairment in import of essential nutrients not readily provided by the RBCs from the extracellular milieu to Plasmodium, blocking of parasite trafficking of proteins to the host cell surface that are crucial for antigenic variations and evading host defense mechanisms or development of interventions to reverse adhesion of infected RBCs to other human cells and inhibitors of proteases that are likely to have significant effects on parasite metabolism and host cell egress. Furthermore, the continuing exploration of the RBC polymorphisms and the molecular basis by which these variants exert their protective effects will be an important source of information for the erythrocytic stage of the disease.
Acknowledgments
The authors thank Council of Scientific and Industrial Research, New Delhi and Indian Council of Medical Research, and New Delhi for intramural financial support. They would like to apologize to those authors whose published articles have not been cited in this paper due to space constraints and the huge body of literature available.
References
- 1.Wellems TE, Hayton K, Fairhurst RM. The impact of malaria parasitism: from corpuscles to communities. Journal of Clinical Investigation. 2009;119(9):2496–2505. doi: 10.1172/JCI38307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White NJ. Plasmodium knowlesi: the fifth human malaria parasite. Clinical Infectious Diseases. 2008;46(2):172–173. doi: 10.1086/524889. [DOI] [PubMed] [Google Scholar]
- 3.Gero AM, Kirk K. Nutrient transport pathways in Plasmodium-infected erythrocytes: what and where are they? Parasitology Today. 1994;10(10):395–399. doi: 10.1016/0169-4758(94)90231-3. [DOI] [PubMed] [Google Scholar]
- 4.Gilson PR, Crabb BS. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. International Journal for Parasitology. 2009;39(1):91–96. doi: 10.1016/j.ijpara.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Lew VL, Tiffert T. Is invasion efficiency in malaria controlled by pre-invasion events? Trends in Parasitology. 2007;23(10):481–484. doi: 10.1016/j.pt.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Vaid A, Thomas DC, Sharma P. Role of Ca2+/calmodulin-PfPKB signaling pathway in erythrocyte invasion by Plasmodium falciparum. Journal of Biological Chemistry. 2008;283(9):5589–5597. doi: 10.1074/jbc.M708465200. [DOI] [PubMed] [Google Scholar]
- 7.Murphy SC, Harrison T, Hamm HE, Lomasney JW, Mohandas N, Haldar K. Erythrocyte G protein as a novel target for malarial chemotherapy. PLoS Medicine. 2006;3(12):2403–2415. doi: 10.1371/journal.pmed.0030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duraisingh MT, Maier AG, Triglia T, Cowman AF. Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4796–4801. doi: 10.1073/pnas.0730883100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124(4):755–766. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Lingelbach K, Joiner KA. The parasitophorous vacuole membrane surrounding Plasmodium and Toxoplasma: an unusual compartment in infected cells. Journal of Cell Science. 1998;111(11):1467–1475. doi: 10.1242/jcs.111.11.1467. [DOI] [PubMed] [Google Scholar]
- 11.Preiser P, Kaviratne M, Khan S, Bannister L, Jarra W. The apical organelles of malaria merozoites: host cell selection, invasion, host immunity and immune evasion. Microbes and Infection. 2000;2(12):1461–1477. doi: 10.1016/s1286-4579(00)01301-0. [DOI] [PubMed] [Google Scholar]
- 12.Bietz S, Montilla I, Külzer S, Przyborski JM, Lingelbach K. Recruitment of human aquaporin 3 to internal membranes in the Plasmodium falciparum infected erythrocyte. Molecular and Biochemical Parasitology. 2009;167(1):48–53. doi: 10.1016/j.molbiopara.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Lew VL, Tiffert T, Ginsburg H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood. 2003;101(10):4189–4194. doi: 10.1182/blood-2002-08-2654. [DOI] [PubMed] [Google Scholar]
- 14.Elliott DA, McIntosh MT, Hosgood HD, III, et al. Four distinct pathways of hemoglobin uptake in the malaria parasite Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2463–2468. doi: 10.1073/pnas.0711067105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin RE, Kirk K. Transport of the essential nutrient isoleucine in human erythrocytes infected with the malaria parasite Plasmodium falciparum. Blood. 2007;109(5):2217–2224. doi: 10.1182/blood-2005-11-026963. [DOI] [PubMed] [Google Scholar]
- 16.Kirk K, Saliba KJ. Targeting nutrient uptake mechanisms in Plasmodium. Current Drug Targets. 2007;8(1):75–88. doi: 10.2174/138945007779315560. [DOI] [PubMed] [Google Scholar]
- 17.Baumeister S, Winterberg M, Przyborski JM, Lingelbach K. The malaria parasite Plasmodium falciparum: cell biological peculiarities and nutritional consequences. Protoplasma. 2010;240(1–4):3–12. doi: 10.1007/s00709-009-0090-3. [DOI] [PubMed] [Google Scholar]
- 18.Huber SM, Uhlemann A-C, Gamper NL, Duranton C, Kremsner PG, Lang F. Plasmodium falciparum activates endogenous Cl− channels of human erythrocytes by membrane oxidation. EMBO Journal. 2002;21(1-2):22–30. doi: 10.1093/emboj/21.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumeister S, Winterberg M, Duranton C, et al. Evidence for the involvement of Plasmodium falciparum proteins in the formation of new permeability pathways in the erythrocyte membrane. Molecular Microbiology. 2006;60(2):493–504. doi: 10.1111/j.1365-2958.2006.05112.x. [DOI] [PubMed] [Google Scholar]
- 20.Merckx A, Bouyer G, Thomas SLY, Langsley G, Egée S. Anion channels in Plasmodium-falciparum-infected erythrocytes and protein kinase A. Trends in Parasitology. 2009;25(3):139–144. doi: 10.1016/j.pt.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Gazarini ML, Thomas AP, Pozzan T, Garcia CRS. Calcium signaling in a low calcium environment: how the intracellular malaria parasite solves the problem. Journal of Cell Biology. 2003;161(1):103–110. doi: 10.1083/jcb.200212130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooke BM, Lingelbach K, Bannister LH, Tilley L. Protein trafficking in Plasmodium falciparum-infected red blood cells. Trends in Parasitology. 2004;20(12):581–589. doi: 10.1016/j.pt.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Van Ooij C, Tamez P, Bhattacharjee S, et al. The malaria secretome: from algorithms to essential function in blood stage infection. PLoS Pathogens. 2008;4(6) doi: 10.1371/journal.ppat.1000084. Article ID e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier AG, Rug M, O’Neill MT, et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell. 2008;134(1):48–61. doi: 10.1016/j.cell.2008.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starnes GL, Waters AP. Home improvements: how the malaria parasite makes the red blood cell home sweet home. Journal of Molecular Cell Biology. 2010;2(1):11–13. doi: 10.1093/jmcb/mjp024. [DOI] [PubMed] [Google Scholar]
- 26.Charpian S, Przyborski JM. Protein transport across the parasitophorous vacuole of Plasmodium falciparum: into the great wide open. Traffic. 2008;9(2):157–165. doi: 10.1111/j.1600-0854.2007.00648.x. [DOI] [PubMed] [Google Scholar]
- 27.Boddey JA, Moritz RL, Simpson RJ, Cowman AF. Role of the Plasmodium export element in trafficking parasite proteins to the infected erythrocyte. Traffic. 2009;10(3):285–299. doi: 10.1111/j.1600-0854.2008.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Koning-Ward TF, Gilson PR, Boddey JA, et al. A newly discovered protein export machine in malaria parasites. Nature. 2009;459(7249):945–949. doi: 10.1038/nature08104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spielmann T, Gilberger T-W. Protein export in malaria parasites: do multiple export motifs add up to multiple export pathways? Trends in Parasitology. 2010;26(1):6–10. doi: 10.1016/j.pt.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Tilley L, Sougrat R, Lithgow T, Hanssen E. The twists and turns of Maurer’s cleft trafficking in P. falciparum-infected erythrocytes. Traffic. 2008;9(2):187–197. doi: 10.1111/j.1600-0854.2007.00684.x. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharjee S, Van Ooij C, Balu B, Adams JH, Haldar K. Maurer’s clefts of Plasmodium falciparum are secretory organelles that concentrate virulence protein reporters for delivery to the host erythrocyte. Blood. 2008;111(4):2418–2426. doi: 10.1182/blood-2007-09-115279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sam-Yellowe TY. The role of the Maurer’s clefts in protein transport in Plasmodium falciparum. Trends in Parasitology. 2009;25(6):277–284. doi: 10.1016/j.pt.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Wickert H, Krohne G. The complex morphology of Maurer’s clefts: from discovery to three-dimensional reconstructions. Trends in Parasitology. 2007;23(10):502–509. doi: 10.1016/j.pt.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Tamez PA, Bhattacharjee S, Van Ooij C, et al. An erythrocyte vesicle protein exported by the malaria parasite promotes tubovesicular lipid import from the host cell surface. PLoS Pathogens. 2008;4(8) doi: 10.1371/journal.ppat.1000118. Article ID e1000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arastu-Kapur S, Ponder EL, Fonović UP, et al. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nature Chemical Biology. 2008;4(3):203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- 36.Lee MCS, Fidock DA. Arresting malaria parasite egress from infected red blood cells. Nature Chemical Biology. 2008;4(3):161–162. doi: 10.1038/nchembio0308-161. [DOI] [PubMed] [Google Scholar]
- 37.Klemba M, Gluzman I, Goldberg DE. A Plasmodium falciparum dipeptidyl aminopeptidase I participates in vacuolar hemoglobin degradation. Journal of Biological Chemistry. 2004;279(41):43000–43007. doi: 10.1074/jbc.M408123200. [DOI] [PubMed] [Google Scholar]
- 38.Miller LH, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264(5167):1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 39.Dondorp AM, Lee SJ, Faiz MA, et al. The relationship between age and the manifestations of and mortality associated with severe malaria. Clinical Infectious Diseases. 2008;47(2):151–157. doi: 10.1086/589287. [DOI] [PubMed] [Google Scholar]
- 40.Marsh K, Forster D, Waruiru C, et al. Indicators of life-threatening malaria in African children. New England Journal of Medicine. 1995;332(21):1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
- 41.Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Reviews in Molecular Medicine. 2009;11:p. e16. doi: 10.1017/S1462399409001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415(6872):673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 43.Roberts DJ, Craig AG, Berendt AR, et al. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature. 1992;357(6380):689–692. doi: 10.1038/357689a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith JD, Subramanian G, Gamain B, Baruch DI, Miller LH. Classification of adhesive domains in the Plasmodium falciparum erythrocyte membrane protein 1 family. Molecular and Biochemical Parasitology. 2000;110(2):293–310. doi: 10.1016/s0166-6851(00)00279-6. [DOI] [PubMed] [Google Scholar]
- 45.Kyes SA, Kraemer SM, Smith JD. Antigenic variation in Plasmodium falciparum: gene organization and regulation of the var multigene family. Eukaryotic Cell. 2007;6(9):1511–1520. doi: 10.1128/EC.00173-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duffy PE. Plasmodium in the placenta: parasites, parity, protection, prevention and possibly preeclampsia. Parasitology. 2007;134(13):1877–1881. doi: 10.1017/S0031182007000170. [DOI] [PubMed] [Google Scholar]
- 47.Rogerson SJ, Boeuf P. New approaches to pathogenesis of malaria in pregnancy. Parasitology. 2007;134(13):1883–1893. doi: 10.1017/S003118200700011X. [DOI] [PubMed] [Google Scholar]
- 48.Williams TN. Red blood cell defects and malaria. Molecular and Biochemical Parasitology. 2006;149(2):121–127. doi: 10.1016/j.molbiopara.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Maier AG, Duraisingh MT, Reeder JC, et al. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nature Medicine. 2003;9(1):87–92. doi: 10.1038/nm807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stoute JA. Complement-regulatory proteins in severe malaria: too little or too much of a good thing? Trends in Parasitology. 2005;21(5):218–223. doi: 10.1016/j.pt.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 51.Fry AE, Griffiths MJ, Auburn S, et al. Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Human Molecular Genetics. 2008;17(4):567–576. doi: 10.1093/hmg/ddm331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cappellini M, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. The Lancet. 2008;371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 53.Ayi K, Min-Oo G, Serghides L, et al. Pyruvate kinase deficiency and malaria. New England Journal of Medicine. 2008;358(17):1805–1810. doi: 10.1056/NEJMoa072464. [DOI] [PubMed] [Google Scholar]
- 54.Cholera R, Brittain NJ, Gillrie MR, et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(3):991–996. doi: 10.1073/pnas.0711401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fairhurst RM, Baruch DI, Brittain NJ, et al. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature. 2005;435(7045):1117–1121. doi: 10.1038/nature03631. [DOI] [PubMed] [Google Scholar]
- 56.Olivieri NF, Muraca GM, O’Donnell A, Premawardhena A, Fisher C, Weatherall DJ. Studies in haemoglobin E beta-thalassaemia. British Journal of Haematology. 2008;141(3):388–397. doi: 10.1111/j.1365-2141.2008.07126.x. [DOI] [PubMed] [Google Scholar]
- 57.Bongfen SE, Laroque A, Berghout J, Gros P. Genetic and genomic analyses of host-pathogen interactions in malaria. Trends in Parasitology. 2009;25(9):417–422. doi: 10.1016/j.pt.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 58.Allison AC. Genetic control of resistance to human malaria. Current Opinion in Immunology. 2009;21(5):499–505. doi: 10.1016/j.coi.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 59.Williams TN, Mwangi TW, Wambua S, et al. Negative epistasis between the malaria-protective effects of α+-thalassemia and the sickle cell trait. Nature Genetics. 2005;37(11):1253–1257. doi: 10.1038/ng1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cockburn IA, Mackinnon MJ, O’Donnell A, et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(1):272–277. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]